
Abstract
B cells have been shown to be essential for Type 1 diabetes development in the non-obese diabetic mouse, where their contribution as antigen presenting cells has been emphasised. Other important functions for B cells include surface capture of immunoglobulins and transportation of immune complexes, with subsequent endocytosis, antigen processing and antigen presentation. We have previously demonstrated that NOD B cells capture IgM and IgG immune complexes through an unknown surface molecule. In this study, we revealed the presumptive immunoglobulin-binding molecule to be HSC70. Moreover, we detected increased levels of HSC70 on NOD B cells. HSC70 has been shown to play a role in antigen processing and presentation as well as being important in several autoimmune diseases, including rheumatoid arthritis and systemic lupus erythematosus. Due to its protein stabilising properties, increased HSC70 could contribute to enhanced self-antigen collection and presentation and thereby contribute to the development of Type 1 diabetes.
Introduction
B cells are essential in the humoral immune defense with a prime role to secrete antibodies that are distributed peripherally in order to neutralise and opsonise foreign antigens. In addition, the B cell subtypes follicular B cells and, in particular, marginal zone B cells can capture and transport antigen-immunoglobulin immune complexes (ICs) through surface expression of complement receptors [Citation1,Citation2]. Marginal zone B cells can further shuttle between the marginal zone and follicles in the spleen [Citation3] and deliver ICs to follicular dendritic cells for further activation of the immune system [Citation1]. Studies have also shown that B cells are involved in the endocytosis of foreign antigen-antibody complexes through an Fc(αμ)R-dependent mechanism [Citation4] and activated B cells can through membrane transfer share their B cell receptors with bystander B cells [Citation5]. Thus, the role of B cells in the immune system is undoubtedly versatile and indispensable for a balanced immune homeostasis.
Although clearly important in the host defense against foreign pathogens, B cells are also involved in the development of several autoimmune diseases, including Type 1 diabetes [Citation6]. This disease has been readily investigated in the non-obese diabetic (NOD) mouse strain, which spontaneously develops diabetes as a result of an auto-destruction of pancreatic β cells in the islets of Langerhans [Citation7]. B cells infiltrate the pancreatic islets of NOD mice early in the disease development [Citation8], and autoantibodies against islet antigens indicate an increased risk of disease [Citation9]. Depletion of B cells in NOD have a protective role in the development of Type 1 diabetes [Citation10–12] and B cells of NOD play a critical role as disease-promoting antigen-presenting cells in their interaction with T cells, which primes pro-inflammatory T cell responses to β cell antigens [Citation13–15]. Moreover, NOD mice display enhanced and prolonged immune responses [Citation16,Citation17] as well as an aberrant VH gene utilisation pattern [Citation18]. We have previously described an enhanced capture of IgG and IgM antibodies by B cells of NOD compared to those of C57BL/6 mice, a trait contributing to increased IC trapping by NOD B cells [Citation19]. We excluded that the binding was due to the poly-reactive immunoglobulin repertoire in NOD as well as potential receptors such as the FcR and complement receptors CR1/2 [Citation19]. The phenomenon was concluded to be restricted to the extracellular part of the B cell plasma membrane as an act of an unknown binding partner. In this study, we sought to identify the binding partner responsible for the increased IgG and IgM capturing on NOD B cells.
Materials and methods
Source of cells
NOD/ShiLtJ (NOD) and C57BL/6J mice used in this study were originally obtained from The Jackson Laboratory (Bar Harbour, ME, USA) and were bred and maintained in the general animal facility at Umeå University. Experimental procedures were performed in compliance with the relevant Swedish and Institutional laws and guidelines, and approved by the Umeå research animal ethic committee (diary number: A14-09, A44-12, A2-15, A39-2019, A2-2021). In this study, female NOD mice at an age of 6-33 weeks and age-matched female C57BL/6 mice were used.
B cell enrichment
Splenic single cell suspension was prepared by pressing spleens between glass slides. B cell enrichment was performed using the MACS technique. For mice experiments, splenic or blood derived cells were washed in PBS containing 0.5% BSA + 2 mM EDTA, incubated with CD19-labelled microbeads and collected using MACS MS separation columns (Miltenyi Biotec), according to the manufacturer’s descriptions. These purification steps collect all B cell subsets except plasma cells, which do not express CD19. The purity was determined by flow cytometry and was consistently >95% (data not shown).
Labelling of IgG1 with the Sulfo-SBED cross-linker
Following procedures were performed in a dark room: “No-Weigh” Sulfo-SBED (SSBED) (Pierce) was prepared according to the manufacturer’s recommendations and incubated for 30 min with 0.2 mg/ml of mouse isotype control IgG1κ antibodies (BD Pharmingen) dissolved in PBS. The reaction mixture was centrifuged at 10 000 g for 30 sec to remove any formed precipitate. The resulting IgG1-Sulfo-SBED complex was desalted in a D-salt dextran desalting column (Pierce) following the manufacturer’s recommendations except for the following changes: a total of 13 fractions were collected, each of a volume of 250 µl. Protein containing fractions were detected with a spectrophotometer (NanoDrop).
Binding of isotype control Sulfo-SBED labelled IgG antibodies
Typically 1 × 106 splenic cells from NOD and C57BL/6 mice were re-suspended in PBS (Thermo Fisher scientific) containing 3% heat-inactivated FBS (Thermo Fisher scientific) and 0.01% sodium azide (Sigma Aldrich), i.e. FACS buffer, incubated with Sulfo-SBED labelled IgG1 isotype control antibodies in round-bottom 96-well plates for 30 min on ice. After washing, cells were stained with anti IgM-APC (clone II/41), CD19-PECy7 (clone 1D3) and Streptavidin-Alexa750 for 20 min on ice. Binding of the Sulfo-SBED labelled IgG1 by B cells was detected using a LSRII FACS machine and data analysed with the FACSDiva software (BD Biosciences). As control, samples where the Sulfo-SBED labelled IgG1 had been omitted was included.
Elisa
To verify that the Sulfo-SBED IgG1 biotin labelling was successful, the eluate from the D-salt dextran desalting column was tested in ELISA. For this procedure, plates were coated with 2.5 µg/ml unlabelled goat anti-mouse IgG (Southern Biotech, H + L specific, human adsorbed) diluted in PBS O/N at 4 °C. After washes, the sample wells were blocked in 1% milk-PBS at 37 °C for 20 min where after new washes were performed. Sulfo-SBED biotin labelled IgG1 diluted in 1% milk-PBS was added in a titration series and incubated for 2 h at 37 °C. After washes, Streptavidin-Alkaline phosphatase (BD Pharmingen) was added at a predetermined optimal concentration diluted in 1% milk-PBS and incubated for 1 h at 37 °C, where after p-nitrophenyl phosphatase (Sigma), diluted to 1 mg/ml in 1 M ethanol amine buffer (0.5 mM MgCl2, pH 9.8) was added and the absorbance was measured at 405 nm in a Molecular Devices, Thermo Max microplate reader. As positive control, biotinylated anti CD80 (clone 16-10A1) (BD Pharmingen) was used as a secondary antibody and as negative controls, the coating antibody (primary) and the labelled IgG (secondary) were excluded respectively.
Binding partner retagging and pulldown
Sulfo-SBED biotin labelled IgG1 (1.2 µg) diluted in 250 µl PBS was mixed with 20 × 106 splenic NOD B cells. The mixture was split into two wells of a round-bottom 96-well plate with 125 µl in each well, and incubated for 30 min on ice in the dark. Thereafter, cells were washed in PBS and transferred to a flat-bottom 96-well plate and exposed to UV-light of 360 nm at a distance of 5 cm for 30 min on ice. The UV-light activated the photoreactive group of the SSBED, which allowed the cross-linker to bind to the putative binding partner. Cells were centrifuged 5 min at 240 g and washed 2 × 5 min with 100 mM β-mercaptoethanol to cleave the amine reactive group of the SSBED, releasing IgG1 and leaving the binding partner retagged.
B cells were lysed in a buffer containing 100 mM HEPES (pH 7.4), 150 mM NaCl, 10% Triton X-100 and a protease inhibitor cocktail tablet (Roche, Switzerland) on ice for 30 min. The lysate was incubated in a monomeric Avidin kit (Pierce) for 1 h following the manufacturer’s recommendations except from the following change: 250 µl fractions were collected in a total of 30 fractions. The protein concentration in each fraction was measured by NanoDrop spectrophotometer. Success of isolated biotinylated binding partner was assayed by Western blot analysis as described below. A schematic picture of the labelling and crosslinking process is shown in .
Western blot analysis and MALDI-TOF
Twenty microgram of purified biotinylated protein from the respective fractions were separated in a 10% SDS-polyacrylamide gel (Laemmli), transferred onto a polyvinylidene fluoride membrane (Millipore), blocked with 5% milk in PBS (pH 7.4) overnight at 4 °C and incubated with 2 µg/ml of HRP conjugated Streptavidin (Millipore) for 1 h at room temperature. After washes, the bands of 70 kDa were detected using chemiluminescent HRP Plus substrate kit (Genscript Biotech). Consistently, the bands appeared in fractions seven to fourteen with peak luminescence in fraction nine. Samples from fractions nine, ten and eleven were pooled and proteins precipitated in ice cold acetone, run on 12% SDS-PAGE. A single band was detected using a silver staining kit (PageSilver kit, Fermentas). The band was extracted and sent for MALDI-TOF analysis (Alphalyse).
IgG1/IgM binding and HSC70 surface levels measured by flow cytometry
Spleens were collected from C57BL/6 and NOD female mice. Single cell suspensions were obtained by pressing organs between glass slides and then centrifuged at 300 g for 10 min. Cells were re-suspended in PBS and 1 × 106 − 2 × 106 cells were stained in a round-bottom 96-well plate. First, cells were stained with Zombie UV (BioLegend) for 20 min in RT. The cells were washed in FACS buffer and incubated with an unlabelled rat anti-mouse CD16/32 antibody (clone 2.4G2) for IgG1 and IgM binding experiment, or with an unlabelled rat anti-mouse CD16/32 antibody (clone 2.4G2) together with a rat IgG2aκ isotype control (clone eBR2a, Invitrogen) for HSC70 surface level experiment, for 20 min on ice. The cells were thereafter washed and stained for 20 min on ice with BV421 Hamster anti-mouse CD3e (clone 145-2C11), BB700 rat anti-mouse CD19 (clone 1D3), BV605 rat anti-mouse CD21/CD35 (clone 7G6), Biotin anti-mouse CD23 (FcεRII) (clone B3B4), Alexa Fluor 488 anti-mouse CD45 monoclonal antibody (clone 30-F11, Invitrogen), together with either a PE-conjugated IgG1κ isotype control (clone X40) or a PE-conjugated Mouse IgM κ isotype control (clone G155-228) for IgG1 and IgM binding experiment, or together with a PE conjugated monoclonal rat anti-hamster HSPA8/HSC70 antibody (clone 1B5, LSBio) or an appropriate PE conjugated IgG2a isotype control (Clone eBR2a, Invitrogen) for HSC70 surface level experiment. After wash, the cells were further stained with a streptavidin-APC conjugate (Invitrogen) for an additional 20 min on ice. All antibodies were purchased from BD Biosciences if not otherwise stated. Data was obtained by a ZE5 cell analyser (Bio-Rad Laboratories, Inc.) and analysed in FlowJo v10.4.2.
Testing IgM block capacity of HSC70 binding substances IgM on splenocytes
For the experiments testing HSC70 block prior to IgM binding, the same protocol as described above was followed, with an additional incubation after Fc-receptor block with either anti-HSC70 antibody (clone 1B5, Enzo Life Sciences), anti-HSC70 antibody (clone EP1531Y, Abcam), scP155 (GenScript Biotech) or P155 (GenScript Biotech) for 60 min on ice. In order to compare results from multiple flow cytometry experiments, data had to be normalised. Firstly, median fluorescence intensity (MFI) values obtained for PE were turned into logarithmic values. For each experiment, a mean value of the C57BL/6 samples for each cell subset was obtained (control intensity, Ic). For IgG1 and IgM binding experiments, values for IgG1 binding and IgM binding to C57BL/6 was used to calculate Ic, respectively. For the HSC70 surface level experiments, the values for IgG2a (i.e. isotype control) binding to C57BL/6 were used to calculate Ic. The median value of the different Ic obtained from each individual experiment was set as a reference value (reference intensity, Ir). The difference (Ir – Ic) was calculated for each individual experiment, and the obtained value was added to each sample within that experiment. Antilog was then applied on the normalised values. Negative samples (i.e. autofluorescence) obtained in the IgG1 and IgM binding experiments were not normalised, and all negative samples with a negative MFI for PE were set as 1.
Determining diabetes incidence
The diabetes incidence of female NOD mice was determined by analysing the presence of glucose in the urine by using Bayer Urine Dipstick tests (Diastix). The analysis was performed once every week from an age of 9–11 weeks until the mice reached an age of 40 weeks.
In vivo analysis of HSC70-binding substances
Ver-155008
Three-week old NOD mice were injected intraperitoneally (i.p.) with VER-155008 (10 µmol/kg per injection) dissolved in saline and 11% dimethyl sulfoxide (DMSO), three times per week for six weeks. As a control, vehicle solution (11% DMSO in saline) was used. VER-155008 was purchased from TargetMol.
P155
Three-week old NOD mice were injected with P155 in the tail vein (i.v.) three times a week for six weeks. As a control, scrambled P155 (scP155) was used. Either 50 µg per mouse (injection week 1–2) or 80 µg per mouse (injection week 3–6) was administered. P155 (MEMALMVAQTRKGKSVV) and scP155 (VMGMRVVAQKTESKMAL) were purchased from GenScript Biotech.
Statistical analysis
Statistical analysis was performed in GraphPad Prism 8. For comparison between mice strains, unpaired t-test was used. In the experiments where different conditions were applied on cells from individual mice, paired t-test was used. Log-rank (Mantel-Cox) test was applied to compare the effect of treatments with HSC70 binding molecules on diabetes development. Statistical significance was set as *p < 0.05, **p < 0.01, ***p < 0.001 and ****p < 0.0001.
Results
Enhanced capture of isotype control IgG1 and IgM by NOD lymphocytes
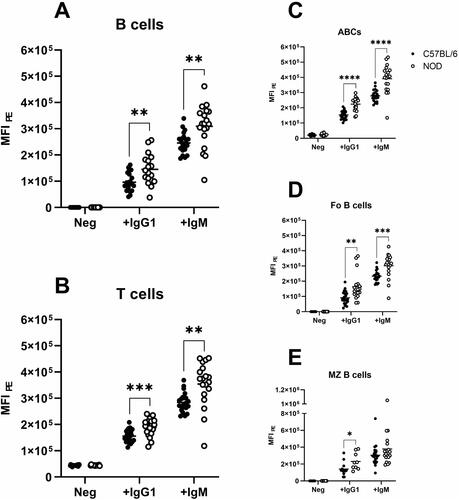
We have previously shown that NOD B cells display an enhanced capability of capturing IgG and IgM antibodies on their plasma membrane surface. To define if this was due to particular B cell subsets in the spleen, cells were incubated with PE-labelled IgG1 and IgM isotype control antibodies together with antibodies to separate the cell subsets. Increased binding of both IgG1 and IgM to B and T lymphocytes from NOD compared to B6 was confirmed (). B cell subset analysis revealed that the increased binding was present in all subsets but mostly pronounced on the CD19+CD21/35−CD23− age-associated B cell (ABC) subset ().
Figure 1. Binding of IgG1 and IgM to the surface of (A) splenic CD19+ B cells, (B) CD3+ T cells, and the different B cell subsets (C) CD19+CD21/35−CD23− age-associated B cells, (D) CD19+CD21/35loCD23+ follicular B cells and (E) CD19+CD21/35+CD23− marginal zone B cells in C57BL/6 (black circles) and NOD (white circles) mice. Obtained MFI was standardised as described in Materials and Methods. Neg = negative samples, i.e. autofluorescence. ** p < 0.01, *** p < 0.001, **** p < 0.0001.
Isotype control IgG1κ antibodies bind HSC70
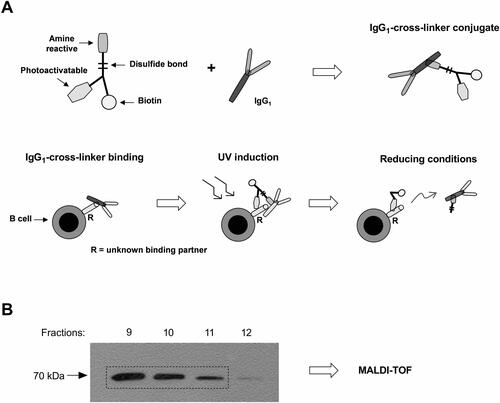
Next, to identify the binding partner responsible for capture of immunoglobulins to NOD B cells, a cross-linker was conjugated to an IgG1 isotype control antibody. Successful conjugation of the IgG1 antibody was tested by ELISA for the presence of biotin, as well as by flow cytometry for the ability of the conjugated IgG1 antibody to still bind NOD B cells (data not shown). Next, the conjugated IgG1 antibody was incubated with NOD B cells, and after photo activation by UV-light the B cells were lysed. The biotinylated binding partner was enriched in a monomeric avidin column and analysed by Western blot (). As shown in , the potential binding partner was eluted in several fractions, and consisted of solely one detectable band. MALDI-TOF analysis revealed Heat shock cognate 70 (HSC70, gene ID: 15481) as the only protein present in this band.
Figure 2. The surface binding partner that capture immunoglobulins was revealed by retagging. (A) Principle outline of the method applied. (B) Western blot analysis of fraction collected.
Increased presence of HSC70 on NOD B cells and age-associated B cells
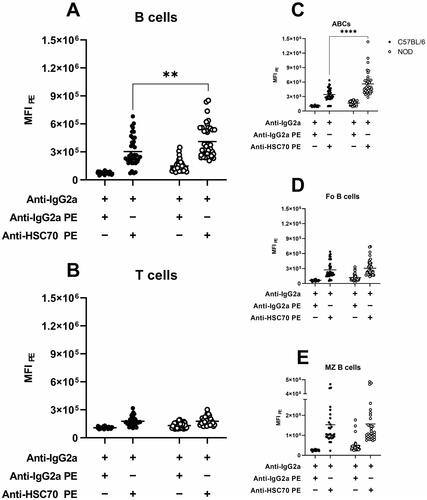
To investigate whether NOD B cells displayed an increased presence of HSC70 on the cell surface, the level of HSC70 on different populations of lymphocytes was determined by flow cytometry. To reassure that the staining was only targeting HSC70, and not representing non-epitope based capture of the anti-HSC70 antibody, cells were pre-incubated in non-labelled isotype control prior to the staining. As depicted in , NOD splenic CD19+ B cells displayed higher levels of HSC70 compared to C57BL/6. Although CD3+ T cells also showed HSC70 staining (), no significant difference between the two strains for this particular subset was observed. Further analysis of HSC70 expression on various B cell subsets revealed a significant increase for the ABC subset in NOD (p < 0.0001, ). For follicular B cells as well as for marginal zone B cells no significant difference was observed ().
Figure 3. Levels of PE-conjugated isotype control and anti-HSC70 antibodies on the cell surface of splenic (A) B cells, (B) T cells, and the different B cell subsets (C) age-associated B cells, (D) follicular B cells and (E) marginal zone B cells in C57BL/6 (black circles) and NOD (white circles) mice. MFI was standardised as described in Materials and Methods. MFI = median fluorescence intensity for PE. ** p < 0.01, **** p < 0.0001.
HSC70-binding peptide P155 or anti-HSC70 antibodies do not block IgM capture
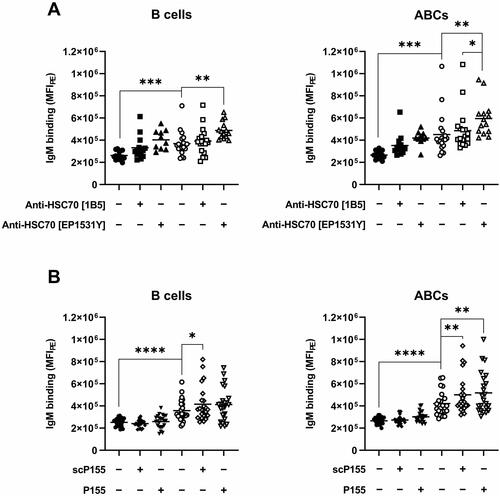
To determine whether the capturing of IgM by NOD B cells could be blocked with anti-HSC70 antibodies or HSC70-binding substances, splenocytes were incubated with two different clones of anti-HSC70 antibodies (clone 1B5 or EP1531Y), or the HSC70-binding peptide P155 prior to the addition of IgM-PE. Statistical analysis was performed comparing C57BL/6 and NOD regarding IgM binding without addition of HSC70-binding substances, as well as within the NOD group after addition of HSC70-binding substances. As shown in , no blocking effect by anti-HSC70 antibodies () or P155 () was observed. Rather an increase in IgM capture was noted, in particular when applying the anti-HSC70 clone EP1531Y or the P155 peptide. This enhancement in IgM capture was also noted after application of scrambled P155 (scP155).
Figure 4. Binding of IgM to splenocytes after incubation with HSC70-binding substances. (A) IgM binding to CD19+ B cells or the CD19+CD21/35−CD23− ABC subset of C57BL/6 mice (black) or NOD (white) after incubation with only Fc block (circles) or with the addition of anti-HSC70 antibody clone 1B5 (squares) or anti-HSC70 antibody clone EP1531Y (triangles). (B) IgM binding to CD19+ B cells or the CD19+CD21/35−CD23− ABC subset of C57BL/6 mice (black) or NOD (white) after incubation with only Fc block (circles) or with the addition of scP155 (squares) or P155 (triangles). Statistical significance was calculated with unpaired t-test between strains, and with paired t-test within strains. *p < 0.05, **p < 0.01, ***p < 0.001 and ****p < 0.0001.
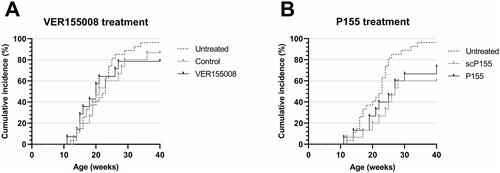
Treatment in vivo with P155 and VER-155008
To evaluate the potential role of HSC70 in the development of Type 1 diabetes, two groups of NOD mice were injected with two different HSC70-inhibiting agents, namely VER-155008 or P155. As depicted in , no difference was observed between the VER-155008 treatment group (black solid line), the vehicle treatment group (gray solid line) or untreated individuals (black dashed line). For the P155 treatment group, a significant reduction in diabetes was seen compared to the untreated individuals (p < 0.05, ). However, this trait could also be observed in the control group scP155 (p < 0.01, ). There was no difference in diabetes incidence between the P155 and the scP155 treatment groups.
Figure 5. Cumulative incidence of diabetes in our untreated colony of female NOD mice (dashed line, n = 27) and (A) Female NOD mice treated with the HSC70 binding agent VER155008 (black solid line, n = 14) or vehicle solution (gray solid line, n = 15). (B) Female NOD mice treated with the HSC70 binding agent P155 (black solid line, n = 15) or scP155 (gray solid line, n = 15).
Discussion
We have previously described an increased capability of NOD B cells to bind IgG and IgM antibodies to their cell surface compared to C57BL/6, through an unknown binding partner [Citation19]. These results could be reproduced in the current study. An increased antibody binding capacity was also observed for NOD T cells, which was not found to be significant in our previous study [Citation19]. We believe that this is a power effect where we in this study included many more individuals and thus could demonstrate statistical significance.
When investigating the antibody binding capacity of different B cell subsets in spleen, the most substantial binding was observed among the marginal zone B cell subset in both mouse strains. It is known that ICs can be captured on the surface of B cells in a complement receptor-dependent manner, which could be one explanation to the high binding capacity of CD21high expressing marginal zone B cells [Citation1]. Also, NOD mice have an increased marginal zone/follicular B cell ratio compared to C57BL/6 [Citation20], which suggests that the increased Ig capturing by NOD B cells could be explained by the marginal zone B cells. However, based on our data we rule out this possibility, as the most pronounced difference in antibody capture between the strains could be observed for the ABC (CD19+CD21/35−CD23−) subset.
Applying a bait-based isolation of the presumptive immunoglobulin binding partner on splenocytes we found HSC70 as the sole candidate. HSC70, also referred to as HSP73 or HSPA8, is a constitutively expressed, highly versatile protein belonging to the heat shock 70 protein family mainly localised in the cytosol and nucleus [Citation21]. In the cytosol, HSC70 is involved in several processes including protein folding, translocation, clathrin-mediated endocytosis as well as in multiple autophagy pathways, where it plays a key role in chaperone-mediated autophagy (CMA) [Citation21–23]. HSC70 is also involved in the presentation of peptides by major histocompatibility complex (MHC) class II on antigen presenting cells affecting availability and/or loading of peptides [Citation24,Citation25]. HSC70 has further been found on the cell surface of various cell types [Citation26,Citation27], including mouse lymphocytes [Citation28]. Additionally, HSC70 can act as a plasma membrane receptor for cell surface binding and infection of multiple viruses [Citation26,Citation29,Citation30] where blockage of HSC70 by antibodies or by HSC70 binding molecules reduce viral infection [Citation26,Citation30,Citation31]. It has also been suggested that secreted HSC70 can function as an immunomodulator [Citation32,Citation33].
To support the identification of HSC70 as the binding partner of immunoglobulins on NOD B cells, we tested to block IgM binding with anti-HSC70 antibodies or the HSC70 binding peptide P155 [Citation34]. Interestingly, addition of P155 or anti-HSC70 antibodies rather enhanced than inhibited the binding of IgM to the surface of B cells. Previous studies have demonstrated that HSC70 extracellular epitopes may differ in different cell types [Citation27,Citation35,Citation36], and epitope availability may vary depending on the localisation of the protein [Citation27,Citation36]. In addition, HSC70 is able to change conformation and several structures of the protein has been identified [Citation22]. Thus, the fact that not all epitopes of HSC70 are available on the cell surface could be one explanation why we were unable to block the IgM binding with anti-HSC70 antibodies or the HSC70 binding agent P155. Another explanation for the inability to block IgM binding could be that IgM and the HSC70 binding molecules used in this study do not share epitope. The observation that HSC70 binding agents enhance the binding of IgM to splenocytes rather suggests that the addition of either anti-HSC70 antibodies or either of the two peptides modify the binding properties, possibly through conformational change of HSC70. The most pronounced effect of the anti-HSC70 antibodies and the peptides was observed in the ABC subset, which is in line with that this subset displayed high expression HSC70. This fact also argues against the involvement of other immune complex binding molecules such as CR1 and CR2 as the ABC subset by definition express low levels of these molecules (i.e. CD21 and CD35).
In this study, we observe an increased level of HSC70 on the cell surface of splenic B cells in NOD compared to C57BL/6. Altered expression of HSC70 has previously been identified in numerous conditions including autoimmune diseases [Citation28,Citation37,Citation38], but also in cancer [Citation39,Citation40] and neurodegenerative diseases [Citation41,Citation42]. In the systemic lupus erythematosus (SLE) mouse model MLR/lpr, leukocytes express increased HSC70 both intra- and extracellularly [Citation28]. Interaction between HSC70 and the rheumatoid arthritis (RA)-susceptible HLA-DRB1*0401 allele has been suggested to contribute to disease development [Citation24]. HLA-DR positive synovial lining cells, involved in production and secretion of many factors as well as antigen presentation in RA patients, display an enhanced expression of HSC70 [Citation38]. Moreover. HSC70 is involved in endocytic targeting of the invariant chain-MHC class II complex [Citation43]. Taken together, this suggests that increased extracellular level of HSC70 could contribute to altered antigen presentation by B cells.
Previous studies have demonstrated upregulated RNA expression pattern of the HSC70 encoding gene Hspa8 in NOD mice compared to C57BL/6 [Citation44], from six weeks of age. It was also found that NOD mice had a significantly higher expression of Hspa8 compared to the congenic NOD mouse with no or little incidence of insulitis or diabetes [Citation44]. Based on this, HSC70 candidates as a potential drug target, and multiple HSC70 interacting molecules that affect its activity have already been described [Citation22]. For instance, the immunosuppressant 15-Deoxyspergualin (15-DSG) has shown to bind to the C-terminal of HSC70 (as well as to HSC90) [Citation45,Citation46]. In a study by Haberstroh et al. [Citation47], injection of 15-DSG protected NOD mice from diabetes. Other HSC70-interacting molecules include VER155008 and the peptide P155, which both interfere with the ATPase activity of HSC70, localised on the N-terminal domain [Citation22,Citation34]. In our study, in vivo i.p. administration of VER155008 showed no effect on diabetes incidence in our NOD mice colony. However, i.v. injection of P155 significantly decreased diabetes incidence. Interestingly, this was also observed in the control group scP155, which challenge the hypothesis of a specific effect on HSC70 activity. On the other hand – as discussed above scP155 also enhance the binding of IgM to B cells in vitro to the same extent as P155, suggesting that both peptides may share features that are similar, and that in due course may give effect on diabetes development.
In conclusion, we have observed an enhanced immunoglobulin binding capacity on the cell surface of splenocytes in NOD compared to C57BL/6 mice, mainly on the B cell subset ABCs. The potential binding partner was identified as HSC70 and in line with this increased HSC70 expression was detected on the cell surface, in particular on the ABCs in NOD. Treatment with a HSC70 targeting molecule decreased incidence of Type 1 diabetes, suggesting that that HSC70 may be involved in Type 1 diabetes development. Our report of HSC70 as a novel interaction partner for immunoglobulins on B cells contributes to a better understanding of the intricate nature of B cells. The increased potential of HSC70 in NOD to trap ICs could provide new insights into developing therapeutic drugs against Type 1 diabetes. Nevertheless, it indicates a new mechanism of which B cells use to direct immune responses during the autoimmune process.
Acknowledgements
We thank Johanna Rönnqvist, Radha Thyagarajan and Viqar Banday for their contribution to this project.
Data availability statement
No additional data set is associated with this paper.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Ferguson AR, Youd ME, Corley RB. Marginal zone B cells transport and deposit IgM-containing immune complexes onto follicular dendritic cells. Int Immunol. 2004;16(10):1411–1422.
- Phan TG, Grigorova I, Okada T, et al. Subcapsular encounter and complement-dependent transport of immune complexes by lymph node B cells. Nat Immunol. 2007;8(9):992–1000.
- Cinamon G, Zachariah MA, Lam OM, et al. Follicular shuttling of marginal zone B cells facilitates antigen transport. Nat Immunol. 2008;9(1):54–62.
- Shibuya A, Sakamoto N, Shimizu Y, et al. Fcα/μ receptor mediates endocytosis of IgM-coated microbes. Nat Immunol. 2000;1(5):441–446.
- Quah BJ, Barlow VP, McPhun V, et al. Bystander B cells rapidly acquire antigen receptors from activated B cells by membrane transfer. Proc Natl Acad Sci U S A. 2008;105(11):4259–4264.
- Silveira PA, Grey ST. B cells in the spotlight: innocent bystanders or major players in the pathogenesis of type 1 diabetes. Trends Endocrinol Metab. 2006;17(4):128–135.
- Makino S, Kunimoto K, Muraoka Y, et al. Breeding of a non-obese, diabetic strain of mice. Jikken Dobutsu. 1980;29(1):1–13.
- Magnuson AM, Thurber GM, Kohler RH, et al. Population dynamics of islet-infiltrating cells in autoimmune diabetes. Proc Natl Acad Sci U S A. 2015;112(5):1511–1516.
- Ziegler AG, Nepom GT. Prediction and pathogenesis in type 1 diabetes. Immunity. 2010;32(4):468–478.
- Akashi T, Nagafuchi S, Anzai K, et al. Direct evidence for the contribution of B cells to the progression of insulitis and the development of diabetes in non-obese diabetic mice. Int Immunol. 1997;9(8):1159–1164.
- Forsgren S, Andersson A, Hillorn V, et al. Immunoglobulin-mediated prevention of autoimmune diabetes in the non-obese diabetic (NOD) mouse. Scand J Immunol. 1991;34(4):445–451.
- Xiu Y, Wong CP, Bouaziz JD, et al. B lymphocyte depletion by CD20 monoclonal antibody prevents diabetes in nonobese diabetic mice despite isotype-specific differences in Fc gamma R effector functions. J Immunol. 2008;180(5):2863–2875.
- Falcone M, Lee J, Patstone G, et al. B lymphocytes are crucial antigen-presenting cells in the pathogenic autoimmune response to GAD65 antigen in nonobese diabetic mice. J Immunol. 1998;161(3):1163–1168.
- Noorchashm H, Lieu YK, Noorchashm N, et al. I-Ag7-mediated antigen presentation by B lymphocytes is critical in overcoming a checkpoint in T cell tolerance to islet beta cells of nonobese diabetic mice. J Immunol. 1999;163(2):743–750.
- Serreze DV, Silveira PA. The role of B lymphocytes as key antigen-presenting cells in the development of T cell-mediated autoimmune type 1 diabetes. Curr Dir Autoimmun. 2003;6:212–227.
- Leijon K, Hammarstrom B, Holmberg D. Non-obese diabetic (NOD) mice display enhanced immune responses and prolonged survival of lymphoid cells. Int Immunol. 1994;6(2):339–345.
- Pedotti R, Sanna M, Tsai M, et al. Severe anaphylactic reactions to glutamic acid decarboxylase (GAD) self peptides in NOD mice that spontaneously develop autoimmune type 1 diabetes mellitus. BMC Immunol. 2003;4:2.
- Leijon K, Freitas A, Holmberg D. Analysis of VH gene utilisation in the non-obese diabetic mouse. Autoimmunity. 1993;15(1):11–18.
- Ekici R, Sundstrom M, Thay B, et al. Enhanced capture of extramembranous IgM and IgG on B cells in the NOD mouse–implications for immune complex trapping. Int Immunol. 2009 May;21(5):533–541.
- Stolp J, Marino E, Batten M, et al. Intrinsic molecular factors cause aberrant expansion of the splenic marginal zone B cell population in nonobese diabetic mice. J Immunol. 2013;191(1):97–109.
- Stricher F, Macri C, Ruff M, et al. HSPA8/HSC70 chaperone protein: structure, function, and chemical targeting. Autophagy. 2013;9(12):1937–1954.
- Bonam SR, Ruff M, Muller S. HSPA8/HSC70 in immune disorders: a molecular rheostat that adjusts chaperone-mediated autophagy substrates. Cells. 2019;8(8):849.
- Eisenberg E, Greene LE. Multiple roles of auxilin and hsc70 in clathrin-mediated endocytosis. Traffic. 2007;8(6):640–646.
- Auger I, Escola JM, Gorvel JP, et al. HLA-DR4 and HLA-DR10 motifs that carry susceptibility to rheumatoid arthritis bind 70-kD heat shock proteins. Nat Med. 1996;2(3):306–310.
- Panjwani N, Akbari O, Garcia S, et al. The HSC73 molecular chaperone: involvement in MHC class II antigen presentation. J Immunol. 1999;163(4):1936–1942.
- Guerrero CA, Bouyssounade D, Zarate S, et al. Heat shock cognate protein 70 is involved in rotavirus cell entry. J Virol. 2002;76(8):4096–4102.
- Mills DR, Haskell MD, Callanan HM, et al. Monoclonal antibody to novel cell surface epitope on Hsc70 promotes morphogenesis of bile ducts in newborn rat liver. Cell Stress Chaperones. 2010;15(1):39–53.
- Page N, Gros F, Schall N, et al. HSC70 blockade by the therapeutic peptide P140 affects autophagic processes and endogenous MHCII presentation in murine lupus. Ann Rheum Dis. 2011 May;70(5):837–843.
- Perez-Vargas J, Romero P, Lopez S, et al. The peptide-binding and ATPase domains of recombinant hsc70 are required to interact with rotavirus and reduce its infectivity. J Virol. 2006;80(7):3322–3331.
- Vega-Almeida TO, Salas-Benito M, De Nova-Ocampo MA, et al. Surface proteins of C6/36 cells involved in dengue virus 4 binding and entry. Arch Virol. 2013;158(6):1189–1207.
- Gutierrez M, Isa P, Sanchez-San Martin C, et al. Different rotavirus strains enter MA104 cells through different endocytic pathways: the role of clathrin-mediated endocytosis. J Virol. 2010;84(18):9161–9169.
- Rodriguez LS, Barreto A, Franco MA, et al. Immunomodulators released during rotavirus infection of polarized caco-2 cells. Viral Immunol. 2009;22(3):163–172.
- Zou N, Ao L, Cleveland JC, Jr., et al. Critical role of extracellular heat shock cognate protein 70 in the myocardial inflammatory response and cardiac dysfunction after global ischemia-reperfusion. Am J Physiol Heart Circ Physiol. 2008;294(6):H2805–H2813.
- Niu L, Lou F, Sun Y, et al. A micropeptide encoded by lncRNA MIR155HG suppresses autoimmune inflammation via modulating antigen presentation. Sci Adv. 2020 May;6(21):eaaz2059.
- Fishelson Z, Hochman I, Greene LE, et al. Contribution of heat shock proteins to cell protection from complement-mediated lysis. Int Immunol. 2001;13(8):983–991.
- Hirai I, Sato N, Qi W, et al. Localization of pNT22 70 kDa heat shock cognate-like protein in the plasma membrane. Cell Struct Funct. 1998;23(3):153–158.
- Aquino DA, Capello E, Weisstein J, et al. Multiple sclerosis: altered expression of 70- and 27-kDa heat shock proteins in lesions and myelin. J Neuropathol Exp Neurol. 1997;56(6):664–672.
- Schick C, Arbogast M, Lowka K, et al. Continuous enhanced expression of Hsc70 but not Hsp70 in rheumatoid arthritis synovial tissue. Arthritis Rheum. 2004;50(1):88–93.
- Kubota H, Yamamoto S, Itoh E, et al. Increased expression of co-chaperone HOP with HSP90 and HSC70 and complex formation in human colonic carcinoma. Cell Stress Chaperones. 2010;15(6):1003–1011.
- Sun G, Cao Y, Xu Y, et al. Overexpression of Hsc70 promotes proliferation, migration, and invasion of human glioma cells. J Cell Biochem. 2019;120(6):10707–10714.
- Arosio A, Cristofani R, Pansarasa O, et al. HSC70 expression is reduced in lymphomonocytes of sporadic ALS patients and contributes to TDP-43 accumulation. Amyotroph Lateral Scler Frontotemporal Degener. 2020;21(1–2):51–62.
- Piedrahita D, Castro-Alvarez JF, Boudreau RL, et al. β-secretase 1’s targeting reduces hyperphosphorilated tau, implying autophagy actors in 3xTg-AD mice. Front Cell Neurosci. 2015;9:498.
- Lagaudriere-Gesbert C, Newmyer SL, Gregers TF, et al. Uncoating ATPase Hsc70 is recruited by invariant chain and controls the size of endocytic compartments. Proc Natl Acad Sci U S A. 2002;99(3):1515–1520.
- Eckenrode SE, Ruan Q, Yang P, et al. Gene expression profiles define a key checkpoint for type 1 diabetes in NOD mice. Diabetes. 2004;53(2):366–375.
- Nadeau K, Nadler SG, Saulnier M, et al. Quantitation of the interaction of the immunosuppressant deoxyspergualin and analogs with Hsc70 and Hsp90. Biochemistry. 1994;33(9):2561–2567.
- Nadler SG, Dischino DD, Malacko AR, et al. Identification of a binding site on Hsc70 for the immunosuppressant 15-deoxyspergualin. Biochem Biophys Res Commun. 1998;253(1):176–180.
- Haberstroh J, Wilhelm T, Schulte-Monting J, et al. Prevention of type I diabetes in the non-obese diabetic (NOD) mouse with 15-deoxyspergualin (15-DS) or 15-DS + cyclosporin A (CyA). Immunol Lett. 1995;48(2):117–121.