
Abstract
Nuclear receptor subfamily 3 group C member 2 (NR3C2) has been revealed to affect the progression of multiple inflammatory diseases, while NR3C2’s efficacy in coronary artery disease (CAD) remains largely unsolved. The study intended to elucidate the possible mechanisms of NR3C2 in oxidised low density lipoprotein (ox-LDL)-induced inflammation in human coronary endothelial cells (HCAECs) via regulating NACHT, LRR, and PYD domains-containing protein 3 (NLRP3). Patients who underwent CT angiography or coronary angiography for suspected CAD in our hospital were collected. The patients were divided into the CAD and the non-CAD (NCAD) groups. The expression of NR3C2 and NLRP3 in the peripheral blood of patients in both groups was examined by RT-qPCR. HCAECs were treated with ox-LDL to establish the model. The expression of NR3C2 and NLRP3 in ox-LDL-induced HCAECs was tested by RT-qPCR. The proliferation of HCAECs was measured using CCK-8 assay, the apoptosis of HCAECs was assessed by flow cytometry, and the levels of inflammation-related factors IL-1β and IL-18 in the cell supernatant were evaluated by ELISA. The molecular mechanisms of these factors in the proliferation and apoptosis of HCAECs and in the inflammatory response were further determined by knockdown and overexpression systems. The relationship between NR3C2 and NLRP3 was determined by ChIP and luciferase activity assays and bioinformatics analysis. NR3C2 and NLRP3 levels were elevated in the serum of CAD patients. The ox-LDL treatment elevated NR3C2 levels, evoked apoptosis and inflammation, and impeded cell viability in HCAECs, whereas downregulation of NR3C2 increased cell viability and reduced apoptosis and inflammatory response in ox-LDL-induced inflammation in HCAECs. NR3C2 levels were positively correlated with NLRP3, and NR3C2 elevated NLRP3 expression through transcription. Overexpression of NLRP3 counteracted the impacts of silencing NR3C2 on cell viability, cell apoptosis, and inflammatory response in ox-LDL-induced HCAECs. Our research stresses that NR3C2 transcription promotes NLRP3 to induce inflammatory responses in ox-LDL-induced HCAECs.
Introduction
Coronary artery disease (CAD) is a main cardiovascular disease afflicting the population worldwide [Citation1]. CAD is thought to be the root cause of about two-thirds of heart failure occurrences [Citation2]. Approximately 80% of these deaths from CAD can be attributed to alterable behaviours, like inadequate exercise and unhealthy diets [Citation3]. It is anticipated that the prevalence and mortality of CAD would reduce in high-income nations due to the advancements in its prevention and treatment. Lipid-lowering, antithrombotic, and anti-inflammatory medications are considered preventative measures [Citation4]. In addition, recent developments have begun to reveal the CAD’s genetic architecture, and approximately 60 genetic loci have been validated to connect to coronary risk [Citation5]. Therefore, continuing to explore gene therapeutic targets for CAD may provide promising treatment directions for CAD.
Nuclear receptor subfamily 3 group C member 2 (NR3C2) is located in the q31.1 region of human chromosome 4, and the gene mutations of NR3C2 are linked with multiple diseases [Citation6]. Specifically, NR3C2 has been revealed to be involved in inflammation-related pathways in different diseases [Citation7,Citation8]. For instance, in post-stroke cognitive impairment, it has been disclosed that silenced NR3C2 can relieve neuronal injury and inflammation through the interaction with microRNA [Citation7]. Another study has validated that NR3C2 serves as a pivotal regulator in cerebral ischemic/reperfusion damage [Citation9]. Yet, its roles in CAD remain largely unknown. As for inflammation, the NACHT, LRR, and PYD domains-containing protein 3 (NLRP3) inflammasome has also been demonstrated to be implicated in various auto-immune and auto-inflammatory diseases [Citation10]. In addition, many observations have manifested the link between NLRP3 inflammasome and cardiovascular diseases, such as atherosclerosis, ischemia/reperfusion, and heart failure [Citation11–13]. For example, NLRP3 inflammasome activation also contributes to the septic cardiomyopathy process [Citation14]. Furthermore, NLRP3 inflammasome pathway components (e.g. caspase1, NLRP3, and interleukin [IL]-1β) have been considered to be novel targets for cardiovascular diseases [Citation15]. Nevertheless, the efficacy of NR3C2 in CAD with the interaction with NLRP3 remains obscure. Based on this, this work was designed to probe the mechanism of NR3C2 in oxidised low-density lipoprotein (ox-LDL)-induced inflammation in human coronary artery endothelial cells (HCAECs) via modulating NLRP3, thus furnishing the treatment modalities for CAD.
Material and methods
Ethical approval
The operations of this study got approval from the ethics committee of First Affiliated Hospital, School of Medicine, Zhejiang University (approval number: 20200728). All the participants provided written informed consent. The research was implemented following the Declaration of Helsinki.
Bioinformatics analysis
Through the gene expression omnibus database (https://www.ncbi.nlm.nih.gov/geo/), the coronary atherosclerosis expression chip GSE137578 was acquired. A total of 6 mRNA samples were divided into two groups: the oxLDL group and the normal control group (three samples in each group). The R language "limma" package was adopted for difference analysis with normal samples as a control. |logFC| > 3 and P value < 0.05 were utilised as the screening standard for differentially expressed genes; followed by the obtainment of the differentially expressed mRNAs. The first 1,000 “atherosclerosis”-related genes were selected through the Genecard database, and their intersection was taken. Information on protein-protein interaction was offered by the string database (https://string-db.org/) [Citation16]. The R language "Count" package was applied to sort according to the number of interaction nodes, and the key differentially expressed genes that may cause coronary atherosclerosis were screened out.
Clinical sample collection
The peripheral blood samples of patients who received computed tomography angiography or coronary angiography due to suspected CAD from October 2020 to December 2021 were collected. Patients were divided into the CAD (n = 48) group and the non-CAD group (NCAD, n = 10). If no less than coronary artery stenosis of any patient exceeded 50% and was confirmed as coronary heart disease by angiography, the diagnosis of CAD shall be made by a cardiologist. Exclusion criteria: diabetes, malignant tumour, infection, hematological disease, chronic renal or liver failure, history of inflammation, and use of immunosuppressive drugs [Citation17].
Cell cultivation and transfection
HCAECs (Lonza, Basel, Switzerland, CC-2585) were cultivated using endothelial cell growth medium EBM-2 (Lonza, CC-3156).
The NR3C2 interference was performed using both NR3C2 small interfering RNA (si-NR3C2) and the control sequence (si-NC). NLRP3 overexpression was performed using pCMV-NLRP3 (oe-NLRP3) and the control vector (oe-NC). The above siRNAs and plasmids were designed and synthesised by GenePharma (Shanghai, China). Concerning cell transfection, HCAECs were detached into single-cell suspension by trypsin and subsequently inoculated into 6-well plates at a certain density. The cell transfection was performed if reaching about 80% confluence with lipofectamine 2000 reagent (11668-019, Invitrogen, New York, California, USA). siRNAs or plasmids in each group with lipofectamine 2000 were subjected to a dilution with serum-free Opti MEM (Gibco, CA, USA), mixed thoroughly, followed by a 15-min stand-by, and added to a 6-well plate. After 6-h treatment, the medium containing transfection solution was removed and then replaced with EBM-2 medium.
Establishment of HCAECs inflammatory model
According to previous reports, HCAECs were exposed to ox-LDL (50 μg/mL; YB-002, Yiyuan Biology, Guangzhou, China) for 24 h to construct a CAD model in vitro [Citation18]. After 24-h transfection, the HCAECs were seeded. When the cell confusion reached 60-75%, the serum-free medium was synchronised for 6 h. HCAECs were exposed to 50 μg/mL ox-LDL for 24 h . The specific groups were as follows: control group (group A, HCAECs were exposed to 0 μg/mL ox-LDL), ox-LDL group (group B, HCAECs were exposed to 50 μg/mL ox-LDL), ox-LDL + si-NC group (group C, HCAECs were transfected with si-NC and exposed to 50 μg/mL ox-LDL), ox-LDL + si-NR3C2 group (group D, HCAECs were transfected with si-NR3C2 and exposed to 50 μg/mL ox-LDL), ox-LDL + si-NC + oe-NC group (group E, HCAECs were transfected with si-NC and oe-NC and exposed to 50 μg/mL ox-LDL), ox-LDL + si-NC + oe-NLRP3 group (group F, HCAECs were transfected with si-NC and oe-NLRP3 and exposed to 50 μg/mL ox-LDL), ox-LDL + si-NR3C2 + oe-NC group (group G, HCAECs were transfected with si-NR3C2 and oe-NC and exposed to 50 μg/mL ox-LDL), and ox-LDL + si-NR3C2 + oe-NLRP3 group (group H, HCAECs were transfected with si-NR3C2 and oe-NLRP3 and exposed to 50 μg/mL ox-LDL).
Cell viability assay
The CCK-8 kit (E606335, Sangon Biotech, Shanghai, China) was adopted for examining cell viability. CCK-8 reagent (10 μL) was appended per well after the cultivation of 0, 24, 48, and 72 h. After 2-h incubation, the absorbance at 450 nm was measured with the microplate reader (Bio Tek, Winooski, VT, USA).
Cell apoptosis assay
The cells were detached with 0.25% trypsin (without ethylene-diamine tetra-acetic acid) (YB15050057, YuBo Biotech, Shanghai, China) and harvested in a flow tube, followed by the removal of the supernatant post centrifugation. Annexin-V-FITC cell apoptosis detection kit (K201-100, Biovision, USA) was utilised for cell apoptosis measurement. Then, 1 × 106 cells were resuspended in 100 μL dye solution, and then appended with 1 mL HEPES buffer solution. Lastly, the fluorescence of FITC and PI was evaluated by excitation of 525 nm and 620 nm bandpass filters at 488 nm to assess the apoptosis respectively [Citation19].
Enzyme-linked immunosorbent assay (ELISA)
Cells were harvested and subsequently centrifuged at 1000 × g for 10 min for the harvest of the supernatant. The inflammation-related factors (IL-1β and IL-18) in the HCAECs culture medium were tested with the ELISA kit (H002/H015, Nanjing Jiancheng Bioengineering Institute, Nanjing, China) [Citation20].
RT-qPCR
Total RNA from peripheral blood and cells was conducted under the TRIzol kit (10296010, Invitrogen), and RNA concentration was measured. The primers were synthesised by Shanghai Sangon Biotech (Supplementary Table 1). Reverse transcription was implemented with an RNA reverse transcription kit (KR116, Tiangen Biotechnology Co., Ltd., Beijing, China). SYBR Green Master Mix (4367659, Applied Biosystems, CA, USA) was employed for fluorescent quantitative PCR detection. PCR was conducted with the Bio-Rad CFX system, which was used for data analysis. The 2-ΔΔCt method was adopted to evaluate gene expression, which was normalised to GAPDH.
Western blot
The total protein of cells was extracted with a highly efficient radio-immunoprecipitation assay cell lysis buffer (R0010, Solarbio, Beijing, China). The bicinchoninic acid kit (P0011, Beyotime, Shanghai, China) was employed to evaluate the protein concentration. Afterward, a protein of equal quality was taken for sodium dodecyl sulphate polyacrylamide gel electrophoresis. Thereafter, the protein was transferred to the membrane, and 5% skimmed milk powder was sealed for 1 h. The primary antibody: NR3C2 (21854-1-AP, 1:2000, Proteintech, Chicago, USA), NLRP3 (15101, 1:1000, Cell Signalling Technology, Danvers, MA, USA), and GAPDH (2118, 1:1000, Cell Signalling Technology) were cultivated overnight at 4 °C, followed by 1-h incubation with secondary antibody, and developing with the developer [Citation21].
Chromatin immunoprecipitation (ChIP) assay
Upon reaching 70-80% HCAEC confusion, 1% formaldehyde was added for fixation to cross-link the DNA and protein. After cross-linking, cells were randomly broken by ultrasound for 10 s at 10 s intervals for 15 cycles to break them into appropriate fragments. Samples were then centrifuged (12 000 g; 4 °C) for the harvest of the supernatant, which was assigned into two tubes, and the two tubes were respectively cultivated with anti-IgG (ab109489, Abcam) and anti-NR3C2 (21854-1-AP, Proteintech) at 4 °C overnight. The precipitation of the endogenous DNA-protein complexes was realised using Protein Agarose/Sepharose; the supernatant was removed with brief centrifugation. After that, the non-specific complexes were rinsed, followed by the purification of the DNA fragments with phenol/chloroform extraction overnight at 65 °C. The ChIP-qPCR products were qualitatively analysed using 3% agarose gel electrophoresis [Citation22,Citation23].
Dual luciferase report gene assay
HCAECs were cultivated in 48-well plates for 24 h. NLRP3 wild-type (WT) or mutant (Mut) plasmids were constructed with the application of the pGL3-promoter (E1761, Promega, Beijing, China). NLRP3 WT or MUT was co-transfected into HCAECs for 48 h with sh-NR3C2 or sh-NC. The Pierce™ Sea Kidney-Firefly Luciferase Dual Assay Kit (16186, Thermo Fisher Scientific, MA, USA) was implemented for the evaluation of renilla luciferase (Rluc) and firefly luciferase (Luc). The relative luciferase activity was counted as the ratio of Luc to Rluc [Citation22,Citation24].
Statistics
All data, expressed as mean ± standard deviation, were processed with SPSS 21.0 statistical software (SPSS, NY, USA). Comparison between two groups was performed using an unpaired t-test and among multiple groups by one-way analysis of variance followed by Tukey’s post hoc test. Pearson test was implemented for the correlation analysis of clinical samples. p < 0.05 was regarded to be statistically significant.
Results
NR3C2 is a key gene involved in CAD
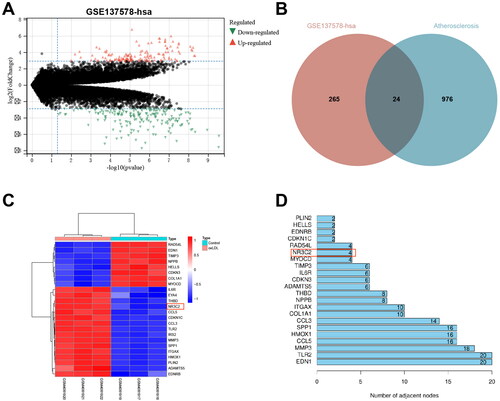
Since atherosclerosis plays a pivotal role in CAD, we screened the differentially expressed genes for possible involvement of atherosclerotic via inflammatory and apoptotic pathways from the GSE137578 microarray with adj.P.Val < 0.05 and |LogFoldChange| > 3. In sum, 289 differentially expressed genes were obtained, consisting of 124 upregulated genes and 165 down-regulated genes (). In addition, we selected the top 1,000 “atherosclerosis”-related genes from the Genecard database and took their intersections to obtain a total of 24 intersecting genes (). The gene expression heat maps were shown in . Based on the protein-protein interaction information offered by the String database, these 24 differentially expressed genes were sorted according to the number of interaction nodes, and the possible key differentially expressed genes for CAD were screened out (). It was revealed that NR3C2 exhibited a high level in the GSE137578 microarray after treated with ox-LDL. Moreover, NR3C2 has been revealed to participate in inflammation-related pathways in various diseases [Citation7,Citation8]. It is suggested that NR3C2 may be a key gene involved in CAD.
Figure 1. NR3C2 is a key gene involved in CAD. A, volcano map of GSE137578: red referred to up-regulated genes, and green referred to down-regulated genes; B, the intersection of the differentially expressed genes in GSE137578 and the top 1,000 “atherosclerosis”-related genes were selected from Genecard database; C, heat map of 24 intersecting differentially expressed genes in GSE137578: the abscissa indicated the sample number, the ordinate referred to the differentially expressed genes; the histogram at the top right was the colour scale; each rectangle in the figure corresponded to a sample expression value; red indicated high expression and blue indicated low expression; D, protein-protein interaction graph was offered by the String database.
NR3C2 is involved in the NLRP3/caspase1 signalling pathway-mediated inflammatory response
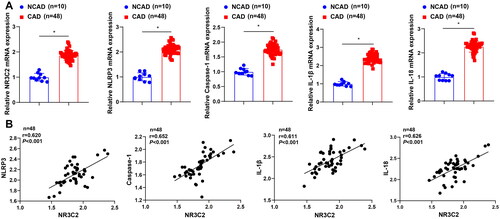
We further collected clinical blood samples from CAD patients. Results showed that NR3C2 expression was high in CAD patients (). The expression levels of the NLRP3/caspase-1 signalling pathway-related factors (NLRP3, caspase-1, IL-1β, and IL-18) were also elevated in CAD patients () NR3C2 expression in CAD was positively linked to the expression levels of NLRP3, caspase-1, IL-1β, and IL-18 (). The results uncover that NR3C2 may participate in the NLRP3/caspase-1 signalling pathway-mediated inflammatory response.
Figure 2. NR3C2 participates in the NLRP3/caspase-1 signalling pathway-mediated inflammatory response. A, expression of NR3C2, NLRP3, caspase-1, IL-1β, and IL-18 in serum from the non-CAD group (NCAD, n = 10) and CAD group (n = 48) was examined by RT-qPCR; B, the correlations of NR3C2 with NLRP3, caspase-1, IL-1β, and IL-18 was analysed by Pearson test. * p < 0.05.
Interfering with NR3C2 inhibits ox-LDL-induced inflammation in HCAECs
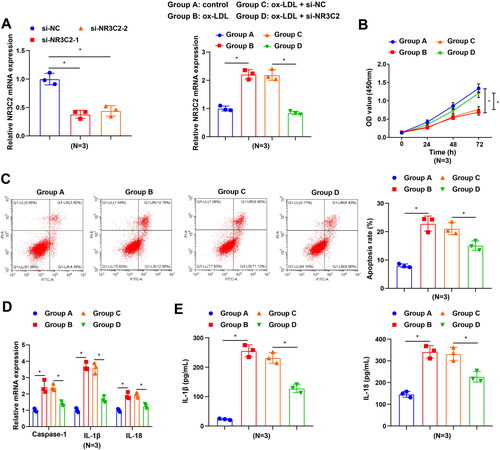
To further probe NR3C2 efficacy in ox-LDL-induced damage in HCAEC models, we downregulated the expression of NR3C2 in HCAECs using siRNA. RT-qPCR displayed that the expression of NR3C2 was decreased in cells transfected with either si-NR3C2-1 or si-NR3C2-2 compared with cells transfected with si-NC, and si-NR3C2-1 was selected for the subsequent study due to its higher silencing efficiency. Then HCAECs were exposed to ox-LDL, and the expression of NR3C2 was measured by RT-qPCR. The results unveiled that NR3C2 expression was augmented in HCAECs after ox-LDL treatment, whereas NR3C2 expression was depleted in response to siRNA-mediated silencing of NR3C2 (). Restrained cell viability, strengthened cell apoptosis, as well as elevated levels of the inflammation-related factors were witnessed in HCAECs in response to ox-LDL treatment, while all these trends were opposite in ox-LDL-induced HCAECs upon treatment with si-NR3C2 (). These findings all unearth that interference with NR3C2 impeded the ox-LDL-induced inflammatory response.
Figure 3. Interfering with NR3C2 diminishes ox-LDL-induced inflammation in HCAECs. A, NR3C2 level in ox-LDL-induced HCAECs were assessed by RT-qPCR in response to si-NR3C2 treatment; B, cell viability of ox-LDL-induced HCAECs was tested by CCK-8 assay in response to si-NR3C2 treatment; C. cell apoptosis of ox-LDL-induced HCAECs was tested by flow cytometry in response to si-NR3C2 treatment; D, caspase-1, IL-18, and IL-1β expression in ox-LDL-induced HCAECs was assessed by RT-qPCR in response to si-NR3C2 treatment; E, IL-1β and IL-18 levels in cell culture supernatant were detected by ELISA in response to si-NR3C2 treatment. All experiments were repeated three times (N = 3). * p < 0.05. Group A (control group): HCAECs were exposed to 0 μg/mL ox-LDL; Group B (ox-LDL group): HCAECs were exposed to 50 μg/mL ox-LDL; Group C (ox-LDL + si-NC group): HCAECs were transfected with si-NC and exposed to 50 μg/mL ox-LDL; Group D (ox-LDL + si-NR3C2 group): HCAECs were transfected with si-NR3C2 and exposed to 50 μg/mL ox-LDL.
NR3C2 transcription promotes NLRP3 expression
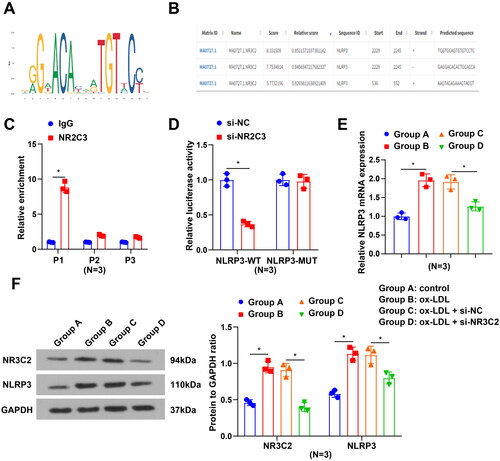
The JASPAR database indicated that NR3C2 had binding sites in the NLRP3 promoter region (). To further address whether NR3C2 might be involved in the transcriptional modulation of NLRP3, we chose three putative NR3C2 binding sites (P1-P3, ) in the NLRP3 promoter by JASPAR, followed by the performance of the ChIP experiment. Results from ChIP-qPCR revealed a higher enrichment of NR3C2 in the P1 region of the NLRP3 promoter region (). We further mutated the P1 region locus to probe the relationship between NR3C2 and NLRP3 via a luciferase assay, which disclosed that interference with NR3C2 diminished NLRP3-WT luciferase activity (). In vitro cell experiments further demonstrated that NR3C2 and NLRP3 levels were elevated after ox-LDL treatment, whereas the levels of which were decreased after interference with NR3C2 (). It is summarised that NR3C2 may transcriptionally promote NLRP3 expression.
Figure 4. NR3C2 transcription promotes NLRP3 expression A, the logo for the NR3C2 transcriptional regulatory site; B, NR3C2 binding sites in the NLRP3 promoter region were forecasted by Jaspar; C, the binding relation of NR3C2 and NLRP3 was validated by ChIP-qPCR assay; D, interactions between NR3C2 and NLRP3 were confirmed by dual luciferase reporter gene assay; E, NLRP3 mRNA expression was examined by RT-qPCR; F, protein expression of NR3C2 and NLRP3 was assessed by Western blot assay. All experiments were repeated three times (N = 3). * p < 0.05. Group A (control group): HCAECs were exposed to 0 μg/mL ox-LDL; Group B (ox-LDL group): HCAECs were exposed to 50 μg/mL ox-LDL; Group C (ox-LDL + si-NC group): HCAECs were transfected with si-NC and exposed to 50 μg/mL ox-LDL; Group D (ox-LDL + si-NR3C2 group): HCAECs were transfected with si-NR3C2 and exposed to 50 μg/mL ox-LDL.
NR3C2 transcription promotes NLRP3 to exacerbate ox-LDL-induced inflammation in HCAECs
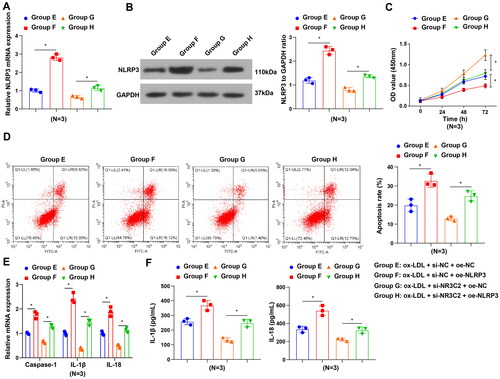
We further explored whether NR3C2 exacerbated ox-LDL-induced inflammatory responses by upregulating NLRP3. NLRP3 expression was elevated in the ox-LDL + si-NC + oe-NLRP3 group (group F) in contrast to the ox-LDL + si-NC + oe-NC group (group E), and the ox-LDL + si-NR3C2 + oe-NLRP3 group (group H) in contrast to the ox-LDL + si-NR3C2 + oe-NC group (group G) (). In response to NLRP3 augmentation, there exhibited restrained cell viability, strengthened cell apoptosis, as well as elevated levels of the inflammation-related factors in HCAECs after ox-LDL treatment. Moreover, overexpression of NLRP3 neutralised the impacts of silenced NR3C2 on cell viability and inflammatory response (). To conclude, interference with NR3C2 may restrict ox-LDL-induced inflammatory responses via the downregulation of NLRP3.
Figure 5. NR3C2 transcription promotes NLRP3 to exacerbate ox-LDL-induced inflammation in HCAECs. A/B, NLRP3 expression in HCAECs was assessed by RT-qPCR and Western blot assay in the rescue assay; C, cell viability of HCAECs was tested in the rescue assay; D, cell apoptosis of HCAECs was measured in the rescue assay E, caspase-1, IL-18, and IL-1β expression in HCAECs was evaluated by RT-qPCR in the rescue assay; F, IL-1β and IL-18 levels in cell culture supernatant were evaluated by ELISA in the rescue assay. All experiments were repeated three times (N = 3). * p < 0.05. Group E (ox-LDL + si-NC + oe-NC group): HCAECs were transfected with si-NC and oe-NC and exposed to 50 μg/mL ox-LDL; Group F (ox-LDL + si-NC + oe-NLRP3 group): HCAECs were transfected with si-NC and oe-NLRP3 and exposed to 50 μg/mL ox-LDL; Group G (ox-LDL + si-NR3C2 + oe-NC group): HCAECs were transfected with si-NR3C2 and oe-NC and exposed to 50 μg/mL ox-LDL; Group H (ox-LDL + si-NR3C2 + oe-NLRP3 group): HCAECs were transfected with si-NR3C2 and oe-NLRP3 and exposed to 50 μg/mL ox-LDL.
Discussion
Though advancement in CAD has been made recently, CAD is still related to high mortality and morbidity rates [Citation25]. Therefore, it is significant to further explore novel therapeutic targets for CAD therapy. Collectively, by focussing on the regulatory mechanism of NR3C2 in ox-LDL-induced inflammation in HCAECs, the study has manifested that silencing NR3C2 contributes to repressing the inflammatory responses in ox-LDL-treated HCAECs via modulating NLRP3 expression.
NR3C2 has been found to exert effects on various diseases. For instance, NR3C2 has been found to exhibit a high level in hepatocellular carcinoma cells [Citation26]. It has been uncovered that overexpression of NR3C2 impedes the viability, migration, and invasion of cancer cells [Citation27]. Similar findings have also been validated in our work. It was demonstrated that NR3C2 displayed a high level in CAD patients and ox-LDL-treated HCAECs. Functionally, silencing NR3C2 relieves the ox-LDL-induced inflammatory response in HCAECs through the reduction of the inflammatory-related factor levels. As reported, the NR3C2 genotype has a correlation with the response to spironolactone in diastolic heart failure patients [Citation28]. Wang et al. have elucidated that miR-135a-5p-mediated NR3C2 depletion contributes to mitigating cerebral ischemia-reperfusion injury [Citation8]. Furthermore, evidence has shown that miR-135b-5p mitigates neuronal damage and inflammation in post-stroke cognitive impairment through the modulation of NR3C2 [7]. These findings confirm the association between NR3C2 and cardiovascular diseases. Nevertheless, further functional experiments are warranted to unveil the roles of NR3C2 in CAD.
The further step was to address whether NR3C2 might be involved in the transcriptional regulation of NLRP3, and it was summarised that NR3C2 may transcriptionally promote NLRP3 expression. Pharmacological depletion of NLRP3 activation leads to potent therapeutic impacts in a wide range of inflammatory diseases, as reflected by the effects of genetic suppression of NLRP3 on inflammatory disease [Citation29]. Inflammatory factors (IL-1β and IL-18) possess cardioprotective properties, which offer short-term adaptive reactions to tissue injury [Citation30]. NLRP3 inflammasome pathway components (e.g. caspase1, NLRP3, and IL-1β) could be novel targets for cardiovascular diseases [Citation15]. It is reported that NLRP3, IL-1β, and IL-18 levels are elevated in cardiac tissue post-myocardial infarction [Citation11], which is in accord with our findings: the inflammation-related factors were witnessed in HCAECs after ox-LDL treatment. In addition, NLRP3 inflammasome-induced generation of IL-1β and IL-18 can strengthen inflammation and plaque formation, thus further contributing to the atherosclerosis process via modulating diverse cellular and molecular targets [Citation31]. Mechanically, NR3C2 up-regulation can invert the repressive impacts of SOX2OT deficiency on oxidative stress, apoptosis, as well as inflammation [Citation8]. Partly in line with the previous findings, the study has manifested that NLRP3 also exhibited high levels in CAD patients and ox-LDL-induced HCAECs, and NLRP3 overexpression could neutralise the influences of NR3C2 silencing on cell viability and inflammatory response.
In summary, this work underlines that ox-LDL treatment increases NR3C2 level and induces apoptosis and inflammation in HCAECs, whereas silencing NR3C2 suppresses ox-LDL-induced inflammation and apoptosis in HCAECs. This study illustrates the properties of NLRP3 and NLRP3 in ox-LDL-induced inflammation in HCAECs and stresses their values in the inflammatory response, which is helpful for further research on the potential of NLRP3 in CAD. Nevertheless, more samples should be implemented in future works to diminish potential data errors. In addition, animal experiments should be supplemented to further validate the findings of our research.
Supplemental Material
Download MS Word (30.5 KB)Data availability statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Malakar AK, Choudhury D, Halder B, et al. A review on coronary artery disease, its risk factors, and therapeutics. J Cell Physiol. 2019;234(10):1–8.
- Lala A, Desai AS. The role of coronary artery disease in heart failure. Heart Fail Clin. 2014;10(2):353–365.
- Bruning RS, Sturek M. Benefits of exercise training on coronary blood flow in coronary artery disease patients. Prog Cardiovasc Dis. 2015;57(5):443–453.
- Steg PG, Ducrocq G. Future of the prevention and treatment of coronary artery disease. Circ J. 2016;80(5):1067–1072.
- Khera AV, Kathiresan S. Genetics of coronary artery disease: discovery, biology and clinical translation. Nat Rev Genet. 2017;18(6):331–344.
- Cui Z, Xu J, Jiang W. NR3C2 gene polymorphism is associated with risk of gestational hypertension in Han Chinese women. Medicine. 2019;98(50):e18215.
- Huang Y, Wang Y, Ouyang Y. Elevated microRNA-135b-5p relieves neuronal injury and inflammation in post-stroke cognitive impairment by targeting NR3C2. Int J Neurosci. 2022;132(1):58–66.
- Wang C, Hu F. Long noncoding RNA SOX2OT silencing alleviates cerebral ischemia-reperfusion injury via miR-135a-5p-mediated NR3C2 inhibition. Brain Res Bull. 2021;173:193–202.
- Cai J, Shangguan S, Li G, et al. Knockdown of lncRNA Gm11974 protect against cerebral ischemic reperfusion through miR-766-3p/NR3C2 axis. Artif Cells Nanomed Biotechnol. 2019;47(1):3847–3853.
- Shao BZ, et al. NLRP3 inflammasome and its inhibitors: a review. Front Pharmacol. 2015;6:262.
- Sandanger Ø, Ranheim T, Vinge LE, et al. The NLRP3 inflammasome is up-regulated in cardiac fibroblasts and mediates myocardial ischaemia-reperfusion injury. Cardiovasc Res. 2013;99(1):164–174.
- Li P, Zhong X, Li J, et al. MicroRNA-30c-5p inhibits NLRP3 inflammasome-mediated endothelial cell pyroptosis through FOXO3 down-regulation in atherosclerosis. Biochem Biophys Res Commun. 2018;503(4):2833–2840.
- Sano S, Oshima K, Wang Y, et al. Tet2-Mediated clonal hematopoiesis accelerates heart failure through a mechanism involving the IL-1beta/NLRP3 inflammasome. J Am Coll Cardiol. 2018;71(8):875–886.
- Zhang W, Xu X, Kao R, et al. Cardiac fibroblasts contribute to myocardial dysfunction in mice with sepsis: the role of NLRP3 inflammasome activation. PLoS One. 2014;9(9):e107639.
- Wang Y, Liu X, Shi H, et al. NLRP3 inflammasome, an immune-inflammatory target in pathogenesis and treatment of cardiovascular diseases. Clin Transl Med. 2020;10(1):91–106.
- Szklarczyk D, Franceschini A, Wyder S, et al. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015;43(D1):D447–D452.
- Nowrouzi-Sohrabi P, Seghatoleslam A, Kalani M, et al. Up-regulated lncRNA-PVT1 expression in peripheral blood mononuclear cells of patients with coronary artery disease is correlated with decreased interleukin-10 production. Mol Biol Rep. 2022;49(5):3453–3459.
- Li P, Xing J, Zhang J, et al. Inhibition of long noncoding RNA HIF1A-AS2 confers protection against atherosclerosis via ATF2 downregulation. J Adv Res. 2020;26:123–135.
- Diaz D, Prieto A, Reyes E, et al. Flow cytometry enumeration of apoptotic cancer cells by apoptotic rate. Methods Mol Biol. 2015;1219:11–20.
- Li Y, Yang W, Quinones-Hinojosa A, et al. Interference with protease-activated receptor 1 alleviates neuronal cell death induced by lipopolysaccharide-stimulated microglial cells through the PI3K/Akt pathway. Sci Rep. 2016;6(1):38247.
- Sato H, et al. A monogenic dominant mutation in Rom1 generated by N-ethyl-N-nitrosourea mutagenesis causes retinal degeneration in mice. Mol Vis. 2010;16:378–391.
- Song C, Zhou C. HOXA10 mediates epithelial-mesenchymal transition to promote gastric cancer metastasis partly via modulation of TGFB2/smad/METTL3 signaling axis. J Exp Clin Cancer Res. 2021;40(1):62.
- Nelson JD. Fast chromatin immunoprecipitation assay. Nucleic Acids Res. 2006;34(1):e2–e2.
- Yang Q, Guo X-P, Cheng Y-L, et al. MicroRNA-143-5p targeting eEF2 gene mediates intervertebral disc degeneration through the AMPK signaling pathway. Arthritis Res Ther. 2019;21(1):97.
- Pyxaras SA, Wijns W, Reiber JHC, et al. Invasive assessment of coronary artery disease. J Nucl Cardiol. 2018;25(3):860–871.
- Yang C, Ma X, Guan G, et al. MicroRNA-766 promotes cancer progression by targeting NR3C2 in hepatocellular carcinoma. FASEB J. 2019;33(1):1456–1467.
- Fan Y, Li Y, Zhu Y, et al. Mir-301b-3p regulates breast cancer cell proliferation, migration, and invasion by targeting nr3c2. J Oncol. 2021;2021:8810517.
- Dumeny L, Vardeny O, Edelmann F, et al. NR3C2 genotype is associated with response to spironolactone in diastolic heart failure patients from the Aldo-DHF trial. Pharmacotherapy. 2021;41(12):978–987.
- Mangan MSJ, Olhava EJ, Roush WR, et al. Targeting the NLRP3 inflammasome in inflammatory diseases. Nat Rev Drug Discov. 2018;17(8):588–606.
- Chao W. Toll-like receptor signaling: a critical modulator of cell survival and ischemic injury in the heart. Am J Physiol Heart Circ Physiol. 2009;296(1):H1–12.
- Hoseini Z, Sepahvand F, Rashidi B, et al. NLRP3 inflammasome: its regulation and involvement in atherosclerosis. J Cell Physiol. 2018;233(3):2116–2132.