
Abstract
Semaphorin 3A (Sema3A) is a secretory member of the semaphorin family of immune response regulators. This research focuses on its effects on inflammation and oxidative stress in acute respiratory distress syndrome (ARDS). By analysing the GEO dataset GSE57011, we obtained Sema3A as the most downregulated gene in ARDS samples. Lipopolysaccharide (LPS) was used to stimulate rat pulmonary microvascular endothelial cells (PMVECs) and rats to induce ARDS-like symptoms in vitro and in vivo, respectively. LPS induced severe damage in rat lung tissues, in which reduced immunohistochemical staining of Sema3A was detected. Sema3A overexpression reduced apoptosis and angiogenesis of LPS-induced PMVECs and alleviated lung injury and pulmonary edoema of rats. Moreover, ELISA results showed that Sema3A overexpression downregulated the levels of inflammatory cytokines and oxidative stress markers both in PMVECs and the rat lung. Activation of ERK/JNK signalling aggravated LPS-induced damage on PMVECs; however, the aggravation was partly blocked by Sema3A, which suppressed phosphorylation of ERK/JNK. Overall, this study demonstrates that Sema3A inactivates the ERK/JNK signalling to ameliorate inflammation and oxidative stress in LPS-induced ARDS models. Sema3A might therefore represent a candidate option for ARDS treatment.
Introduction
Since its first description in 1967 [Citation1], acute respiratory distress syndrome (ARDS) has been well recognised as a primary clinical issue in the global scale with high morbidity and mortality rates [Citation2]. It represents a syndrome of acute respiratory failure characterised by severe hypoxaemia resulted from excessive alveolocapillary permeability [Citation3]. An observational study involving 29,144 patients in 459 intensive care units (ICU) from 50 countries demonstrated that 10.4% of ICU patients and 23.4% of ventilation-requiring patients met the criteria for ARDS [Citation4]. ARDS involves secondary injury induced by systemic inflammation and oxidative stress [Citation5, Citation6]. Excess inflammation can result in alveolar damage, specifically increased epithelial and endothelial permeability, leading to accumulative protein-rich alveolar edoema fluid and hypoxaemia [Citation7]. Oxidative insult initiated following acute inflammatory injury evolves into chronic scarring and results in bronchopulmonary dysplasia [Citation8]. Novel strategies alleviating the excessive inflammation and oxidative stress have long being expected to help alleviate the lung injury and improve patients’ well-being.
To identify key molecules involved in the dysregulated inflammatory process during the ARDS progression, we investigated significantly aberrantly expressed genes in ARDS by analysing the GEO dataset GSE57011 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE57011), with semaphorin 3 A (Sema3A) identified as the most downregulated gene in ARDS samples. Sema3A is a secretory member of the semaphorin family which have been implicated in the regulation of T cell response and pathogenesis of many immune disorders [Citation9, Citation10] and cancers [Citation11, Citation12]. Sema3A has been reported to exert anti-inflammatory functions during atopic dermatitis [Citation13] and following myocardial infarction [Citation14]. Meanwhile, Sema3A has been found to mitigate lipopolysaccharide (LPS)-induced inflammation and apoptosis of chondrocytes [Citation15]. Considering the predicted poor expression profile of Sema3A in ARDS samples and LPS as a potent microbial mediator frequently used to induce acute lung inflammation and ARDS [Citation16], we conjectured that Sema3A might play specific roles, most likely anti-inflammatory, in ARDS as well.
Intriguingly, Sema3A has been reported to suppress activation of the extracellular signal-regulated kinase/c-Jun N-terminal kinase (ERK/JNK) signalling to alleviate apoptosis and inflammation in neonatal rats with bronchopulmonary dysplasia [Citation17]. Both ERK and JNK are classical downstream pathways of the mitogen-activated protein kinase (MAPK) signalling [Citation18, Citation19], which are well established key factors contributing to excess inflammation [Citation20]. Meanwhile, inactivation of the ERK/JNK pathways has been reported to alleviate oxidative stress in ARDS induced by neutrophil activation [Citation21]. As pulmonary endothelium mediates key processes implicated in lung homoeostasis, whose disruption is a key event leading to increased capillary permeability during ARDS [Citation22], we challenged rat pulmonary microvascular endothelial cells (PMVECs) and rats with LPS to establish cellular and animal models, respectively, to investigate the functions of Sema3A and ERK/JNK pathways in the inflammatory response and oxidative stress in ARDS.
Materials and methods
Cells and treatment
Rat PMVECs (CP-R001) were procured from Procell Life Science & Technology Co., Ltd. (Wuhan, Hubei, China) and cultured in PMVEC-specific complete medium (CM-R001, Procell) at 37 °C with 5% CO2. Overexpression plasmid of Sema3A (oe-Sema3A) and the negative control (NC) plasmid were provided by GenePharma Co., Ltd. (Shanghai, China). Well-growing PMVECs were seeded into six-well plates. When the confluence reached around 50%, the cells were transfected with the plasmids using Lipofectamine® 2000 (Thermo Fisher Scientific, Rockford, IL, USA), followed by incubation at 37 °C with 5% CO2 for 48 h. To induce cell damage, the PMVECs were treated with 1 μg/mL LPS (ST1470, Beyotime Biotechnology Co., Ltd., Shanghai, China) for 4 h. For artificial activation of the ERK/JNK signalling pathways, 4 h after LPS treatment, the cells were treated with 0.1 μM ERK agonist LM22B-10 (HY-104047) or JNK agonist Juglanin (HY-N3442) (both from MedChemExpress, Monmouth Junction, NJ, USA) for 2 h. Overall, the cells were allocated into the following groups: Control (without treatment), LPS, LPS + oe-NC, LPS + oe-Sema3A, LPS+LM22B-10, LPS+LM22B-10+oe-NC, LPS+LM22B-10+oe-Sema3A, LPS+Juglanin, LPS+Juglanin+oe-NC, and LPS+Juglanin+oe-Sema3A.
Animal model of ARDS
Forty-eight male Wistar rats (eight weeks old; 276–300 g) were procured from Vital River Laboratory Animal Technology Co., Ltd. (Beijing, China) and used in compliance with the protocol ratified by the Institutional Review Board of Lishui People’s Hospital. All procedures were performed adhering to the Guide for the Care and Use of Laboratory Animals (NIH, Bethesda, MD, USA). The 48 rats were allocated into sham, ARDS, ARDS + oe-NC, and ARDS + oe-Sema3A groups, n = 12 in each. For ARDS induction, the rats were injected with 100 μL LPS (5 mg/kg) through tail vein. Rats in the sham group were injected with 100 μL phosphate-buffered saline (PBS) instead. For gene alteration, the rats were injected with 100 μL lentivirus-carried oe-NC or oe-Sema3A plasmids (1 × 108 TU/μL), followed by LPS injection 7 days later. Within 8 h after LPS injection, rats showed early respiratory distress and cyanosis. After 8 h, the respiratory rate of rats significantly increased (over 130–140 bpm), indicating successful induction of ARDS in rats. After 24 h, the rats were euthanized by excessive anaesthesia, followed by intratracheal injection of 0.5 mL PBS and 0.1 mM ethylene diamine tetraacetic acid (EDTA) to collect bronchoalveolar lavage fluid (BALF). The BALF was centrifuged to collect cell pellet. The cells were sorted, resuspended, and stained with Wright’s stain (C0135, Beyotime) to calculate the number of neutrophils.
Haematoxylin and eosin (HE) staining
Prepared rat lung tissue sections (5 μm) were de-paraffined and rehydrated in decreasing doses of ethanol. After that, the sections were stained with haematoxylin reagent for 5 min, differentiated in 0.5% hydrochloric ethanol for 20 s, and stained with 0.1–0.5% eosin reagent for 2 min. After staining, the sections were dehydrated in graded doses of ethanol, cleared in xylene, and sealed by neutral balsam for microscopy observation.
Cell counting kit-8 (CCK-8) method
Treated PMVECs were digested in trypsin, resuspended, and incubated in 96-well plates at 2,000 cells per well. Each well was then added with 10 μL diluted (10%) CCK-8 reagent (C0037, Beyotime) for 2 h of incubation in the dark. The optical density (OD) value at 450 nm was read by a microplate reader to evaluate the cell viability.
Terminal deoxynucleotidyl transferase (TdT)-mediated dUTP nick end labelling (TUNEL)
Apoptotic endothelial cells in rat lung tissue sections were examined following the instruction manual of the TUNEL kit (C1086, Beyotime). Labelled cells were observed under microscopy, and the TUNEL positive rate was calculated as follow: rate (%) = TUNEL-positive cells/total number of nuclei × 100%.
Detection of biochemical indicators
Concentrations of inflammatory cytokines tumour necrosis factor-α (TNF-α; RTA00, R&D Systems, Minneapolis, MN, USA), interleukin (IL)-1β (RLB00, R&D Systems) and IL-6 (R6000B, R&D Systems), angiogenesis marker vascular endothelial growth factor (VEGF; RRV00, R&D Systems), coagulation mediator tissue factor (TF; CSB-E07914r, CUSABIO Biotechnologies, Wuhan, Hubei, China), and thrombin-antithrombin complex (TAT; CSB-E08432r, CUSABIO) in the culture supernatant of PMVECs and rat BALF, and the concentrations of oxidative stress-related markers myeloperoxidase (MPO; ER0142, Fine Biotech Co., Ltd., Wuhan, Hubei, China), malondialdehyde (MDA; ER1878, Fine Biotech) and superoxidase dismutase (SOD; ER0649, Fine Biotech) in PMVECs and rat lung tissues were analysed in strict accordance with the protocols of the rat enzyme-linked immunosorbent assay (ELISA) kits . The OD value at 450 nm was detected by a microplate reader.
Western blot analysis
PMVECs and rat lung tissue samples were lysed in radio-immunoprecipitation assay buffer containing protease inhibitor for protein extraction. After concentration analysis using a bicinchoninic acid kit (C503021, Sangon Biotech Co., Ltd. (Shanghai, China), protein samples were wet-separated by SDS-PAGE and loaded onto polyvinylidene fluoride membranes, which were blocked by non-fat milk. Proteins on membranes were probed by the diluted antibodies including Sema3A (1:500, PA5-75452, Thermo Fisher Scientific), VE-cadherin (1:50, 36-1900, Thermo Fisher Scientific), α-E-catenin (1:1,000, 12831-1-AP, Proteintech Group, Inc., Hubei, China), β-actin (1:1,000, AF5003, Beyotime), ERK (1:500, 11257-1-AP, Proteintech), P-ERK (1:1,000, 28733-1-AP, Proteintech), JNK (1:2,000, 24164-1-AP, Proteintech) and p-JNK (1:1000, 80024-1-RR, Proteintech) at 4 °C overnight. On the next day, the membranes were incubated with horseradish peroxidase-conjugated goat anti-rabbit IgG (1:1000, A0208, Beyotime) at room temperature for 1 h. The protein blots were developed by enhanced chemiluminescence reagent. Relative gray value of target protein bands to the internal loading β-actin was detected by Image J to evaluate protein expression.
Flow cytometry
Apoptosis of PMVECs was determined following the protocol of the Annexin V-fluorescein isothiocyanate (FITC)/propidium iodide (PI) kit (P-CA-201, Procell). The PMVECs were centrifuged at 300 g for 5 min to discard supernatant. The cells were resuspended in diluted 1× Annexin V binding buffer working solution, to which 5 μL Annexin V-FITC and 5 μL PI were then added for 15–20 min of dark incubation at room temperature. The apoptosis rate of PMVECs were then analysed by the flow cytometer.
Angiogenesis assay
Prepared Matrigel was added to 96-well plate and mixed with the PMVEC complete medium until a gel state. The PMVECs were rinsed and digested in EDTA-trypsin to prepare cell suspension, which was then mixed with the culture medium for a 12-h incubation. The newly formed tubes were observed under microscopy.
Immunohistochemistry (IHC)
Prepared rat lung tissue sections were de-paraffined, rehydrated, heated in citrate buffer, treated with 3% hydrogen peroxide and blocked with 5% foetal bovine serum for 30 min. Thereafter, the sections were incubated with diluted anti-Sema3A (1:200, PA5-75452, Thermo Fisher Scientific) at 4 °C overnight and with IgG (1:1,000, A0208, Beyotime) at room temperature for 1 h. The DAB kit (ab64238, Abcam) was used for colour development. The sections were counter-stained with haematoxylin, dehydrated, cleared, and sealed for microscopy observation.
Lung permeability detection
Six rats in each group were intravenously injected with 20 mg/kg Evans blue (E2129, Sigma-Aldrich, Merck KGaA, Darfmstadt, Germany) at 1 h before euthanasia. The right ventricular was punctured for transcardial perfusion of the lung with PBS. After dried at 56 °C for 48 h, the lungs were harvested, weighed, and immersed in 1 mL/100 mg formamide (F7503, Sigma-Aldrich) for 24 h of incubation at 60 °C. The OD value of supernatant was then measured by the microplate reader.
Wet/dry (W/D) weight ratio
The rat left lungs were weighed after the surface blood was blotted up as the wet weight. Thereafter, the lungs were dried at 60 °C for 72 h and reweighed to calculate the W/D weight ratio.
Statistical analysis
All data analyses and histogram drawing were performed by GraphPad Prism 8 (GraphPad, La Jolla, CA, USA). All data were expressed as the mean ± SD. Intergroup differences were analysed by the unpaired t test. When more than two groups were concerned, one-way analysis of variance (ANOVA) was performed, followed by Tukey’s post-hoc test. The cut-off threshold for statistical significance was set at p < 0.05.
Results
Sema3A is downregulated in the lungs of rats with ARDS
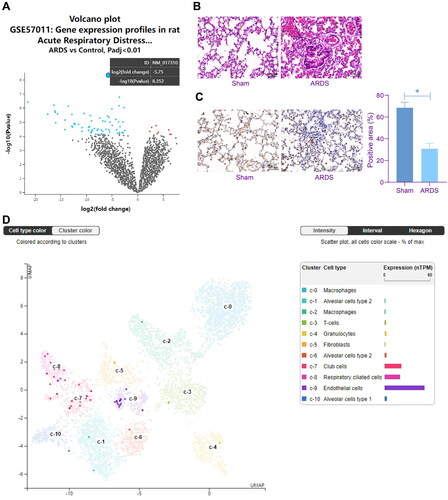
The GEO dataset GSE57011 comprising seven ARDS lung biospecimens (GSM1372904 ∼ GSM1372910) and five control lung biospecimens (GSM1372911 ∼ GSM1372915) was downloaded to analyse differential gene expression profiles between ARDS and control lungs, with p < 0.01 as the screening threshold. Sema3A (NM_017310) was identified as the one with the greatest degree of differential expression, which is downregulated in ARDS lungs (). We established a rat model of ARDS by LPS treatment. During the treatment process, the LPS stimulation led to several symptoms such as increased respiratory rate, respiratory distress and cyanosis. After animal euthanasia, conspicuous injury was observed in the lung tissues, including uneven alveolar size and increased alveolar septum thickening (). The IHC assay showed that the ARDS rats had increased intensity of Sema3A staining in their lung tissues compared to the sham-operated ones (). According to the single cell sequencing analysis in The Human Protein Atlas (https://www. proteinatlas.org/), Sema3A is mainly localised in endothelial cells in the lung (). This evidence indicates that Sema3A downregulation might have specific links to LPS-induced lung injury.
Figure 1. Sema3A is downregulated in the lungs of rats with ARDS. A, Sema3A (NM_017310) as the most significantly differentially expressed gene between ARDS and control lungs in the GSE57011 dataset; B, morphological changes in rat lung tissues observed by HE staining; C, Sema3A expression in rat lung tissues examined by IHC; D, single cell sequencing analysis of Sema3A localisation in the lung predicted in The Human Protein Atlas. In each group, n = 6. Differences were analysed by the unpaired t test (C). * p < 0.05.
Sema3A elevation alleviates inflammation and oxidative stress in rat PMVECs
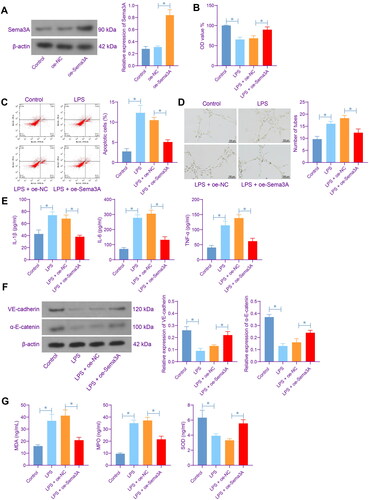
To specify the function of Sema3A in ARDS, we first induced Sema3A overexpression in LPS-treated PMVECs. Successful Sema3A upregulation in cells was validated by WB analysis (). The CCK-8 assay showed that the Sema3A overexpression in the PMVECs restored the viability of cells suppressed by LPS (). The LPS also induced apoptosis of cells, which was alleviated upon Sema3A overexpression (). The angiogenesis of PMVECs was stimulated by LPS as well but weakened by Sema3A overexpression (). In the culture supernatant of LPS-treated PMVECs, elevated levels of inflammatory cytokines including IL-1β, IL-6 and TNF-α were detected. However, this elevation was partly negated in the setting of Sema3A overexpression (). WB analysis the levels of VE-cadherin and α-E-catenin, two junction proteins of endothelial cells, were decreased in PMVECs by LPS treatment but rescued by Sema3A overexpression (). ELISA results also showed that the contents of MDA and MPO were elevated whereas the SOD content was reduced in PMVECs after LPS treatment, which are manifestations of significant oxidative stress. However, the oxidative stress was significantly alleviated by Sema3A overexpression ().
Figure 2. Sema3A elevation alleviates inflammation and oxidative stress in rat PMVECs. A, protein level of Sema3A in PMVECs after oe-Sema3A administration determined by WB analysis; B, viability of PMVECs examined by CCK-8 assay; C, apoptosis of PMVECs examined by flow cytometry; D, angiogenesis of PMVECs analysed by tube formation assay; E, concentrations of inflammatory cytokines IL-1β, IL-6 and TNF-α in the culture supernatant of PMVECs detected by ELISA kits; F, protein levels of VE-cadherin and α-E-catenin in PMVECs determined by WB analysis; G, concentrations of oxidative stress-related markers MDA, MPO and SOD in PMVECs detected by ELISA kits. Three biological replicates were performed. Differences were analysed by the one-way ANOVA (A-G). * p < 0.05.
Sema3A restoration alleviates inflammation and oxidative stress in rat lung tissues
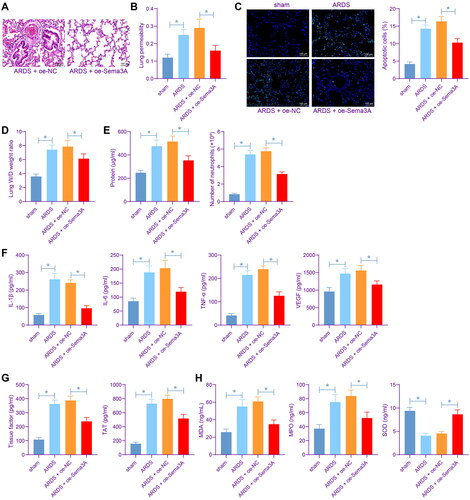
To examine the effects of Sema3A on lung injury in ARDS rats, the rats were pre-injected with lentivirus-carried oe-Sema3A prior to LPS injection. The pre-injection of oe-NC/oe-Sema3A lentivirus did not result in significant adverse effects on rats. Instead, rats pre-injected with oe-Sema3A showed alleviated ARDS symptoms. HE and Evans blue staining showed that the Sema3A restoration partly alleviated lung injury and reduced lung permeability induced by LPS (). TUNEL assay showed that Sema3A upregulation also alleviated LPS-induced endothelial cell apoptosis in vivo (). Compared to sham-operated mice, the ARDS rats had an ∼50% increase in the W/D weight ratio of the lung, and this increase was blocked by Sema3A overexpression as well (). The BALF of ARDS rats showed increased protein concentration and elevated number of neutrophils, and these indicators of lung injury were conspicuously reduced by Sema3A overexpression (). In line with the findings in vitro, elevated levels of IL-1β, IL-6 and TNF-α as well as the angiogenesis marker VEGF were detected in the BALF of LPS-induced rats but reduced after Sema3A upregulation (). Meanwhile, the LPS treatment resulted in elevated TF and TAT levels in the BALF, which were suppressed by Sema3A overexpression as well (). Similarly, by detecting the MDA. MPO and SOD contents, we identified that the oxidative stress in rat lung tissues were aggravated by LPS but ameliorated by oe-Sema3A ().
Figure 3. Sema3A restoration alleviates inflammation and oxidative stress in rat lung tissues. A, lung injury in rats determined by HE staining; B, lung permeability analysed by Evans blue staining; C, apoptosis of lung endothelial cells examined by TUNEL assay; D, pulmonary edoema evaluated by the W/D weight ratio; E, protein concentration in rat BALF examined by bicinchoninic acid and the number of neutrophils in rat BALF examined by Wright’s staining; F, concentrations of IL-1β, IL-6, TNF-α and VEGF in rat BALF determined by ELISA kits; G, concentrations of TF and TAT in rat BALF determined by ELISA kits; H, levels of MDA, MPO, and SOD rat BALF determined by ELISA kits. In each group, n = 6. Differences were analysed by the one-way ANOVA (B-H). * p < 0.05.
Sema3A suppresses the ERK/JNK signalling pathways
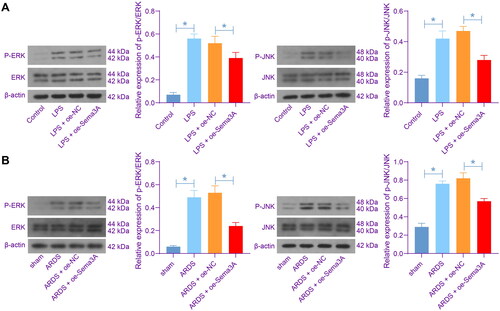
To verify if Sema3A has an effect on the ERK/JNK signalling pathways, we examined the activities of ERK and JNK in the PMVECs. The LPS significantly stimulated the phosphorylation of both ERK and JNK in cells; which was, however, blocked by Sema3A overexpression (). Similar trends were found in vivo that the phosphorylation of ERK and JNK in rat lung tissues was triggered by LPS treatment but reduced by Sema3A overexpression ().
Figure 4. Sema3A suppresses the ERK/JNK signalling pathways. A, phosphorylation of ERK and JNK in PMVECs detected by WB analysis; B, phosphorylation of ERK and JNK in rat lung tissues detected by WB analysis. For cellular experiments, three biological replicates were performed. For animal experiments, n = 6 in each group. Differences were analysed by the one-way ANOVA (A-B). * p < 0.05.
Activation of ERK/JNK increases inflammation and oxidative stress in PMVECs
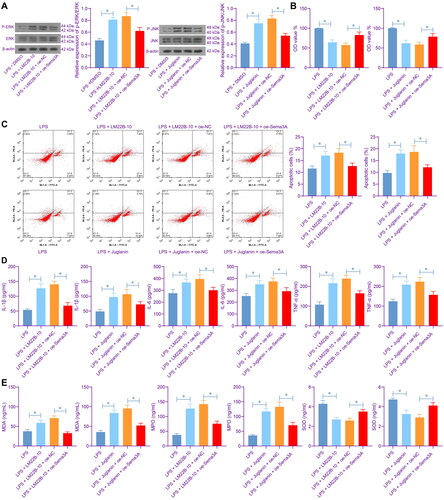
To validate the function of ERK/JNK signalling pathways in ARDS progression, we treated the LPS-stimulated PMVECs (with or without oe-Sema3A transfection) with ERK- and JNK-specific agonists LM22B-10 and Juglanin, respectively. The two agonists, as expected, enhanced the phosphorylation of ERK and JNK in the PMVECs. Yet the presence of Sema3A overexpression reduced the ERK/JNK activation (). It was observed that the LM22B-10 or Juglanin treatment suppressed viability of PMVECs () but increased cell apoptosis (). Meanwhile, it increased the concentrations of IL-1β, IL-6 and TNF-α in the culture supernatant of LPS-treated PMVECs () and aggravated the oxidative stress level (). Of note, the oe-Sema3A transfection partly negated the effects of LM22B-10 or Juglanin, as it rescued the viability of and suppressed apoptosis of PMVECs (), and alleviated inflammation as well as oxidative stress (). This ample evidence suggests that Sema3A can suppress the ERK/JNK signalling pathways to alleviate inflammation and oxidative stress in PMVECs.
Figure 5. Activation of ERK/JNK increases inflammation and oxidative stress in PMVECs. A, phosphorylation of ERK and JNK in PMVECs after LM22B-10 or Juglanin treatment; B, viability of PMVECs examined by CCK-8 assay; C, apoptosis of PMVECs examined by flow cytometry; D, concentrations of IL-1β, IL-6 and TNF-α in the culture supernatant of PMVECs detected by ELISA kits; E, concentrations of oxidative stress-related markers MDA, MPO and SOD in PMVECs detected by ELISA kits. Three biological replicates were performed. Differences were analysed by the one-way ANOVA (A-E). * p < 0.05.
Discussion
The lack of effective drugs and the high mortality of ARDS, especially when the coronavirus virus-19 pandemic has been causing an elevation in the disease morbidity [Citation3], challenges scientists and clinicians to discover new therapeutic approaches. Identification of novel key molecules might offer new insights into this field. In this work, the authors identified conspicuously downregulated Sema3A in LPS-induced ARDS samples and confirmed that the Sema3A restoration induced resolution of inflammatory response and oxidative stress both in vitro and in vivo.
Initially known for their regulatory roles in neurodevelopment, angiogenesis, tumour progression, and metastasis, semaphorin family members are increasingly recognised for their involvement in almost all phases of physiological and pathological immune responses [Citation23]. Genetic deletion of Sema3A was reportedly linked to thickened septae, reduced airspace size, attenuated, and increased death of postnatal alveolar septal cells in mice [Citation24]. However, there has no evidence elucidating the link of Sema3A to the pathogenesis of ARDS or acute lung injury. Nevertheless, evidence exists in terms of the anti-inflammatory properties of Sema3A in several human disorders. For instance, it alleviated LPS-induced inflammation and apoptosis of chondrocytes by binding to its receptor neuropilin-1 [Citation15, Citation25]. Moreover, in an experimental mouse model of bronchial asthma, Sema3A reduced inflammation and angiogenesis [Citation26], which is a key pathologic event induced by tissue infiltrating inflammatory cells, especially in hypoxic conditions [Citation27, Citation28]. Similar anti-inflammatory role of Sema3A has been witnessed in allergic rhinitis [Citation29] and systemic sclerosis [Citation30]. Here in this work, we identified reduced Sema3A expression in the lung tissues of LPS-challenged rats, which was in concert with the bioinformatics prediction results. Of note, the artificial Sema3A restoration reduced apoptosis and angiogenesis of LPS-induced PMVECs and alleviated lung injury and pulmonary edoema of rats. Moreover, it downregulated the levels of inflammatory cytokines and oxidative stress markers both in vitro and in vivo. Therefore, upregulation of Sema3A might help ameliorate lung injury, pulmonary edoema, inflammation, oxidative stress, and endothelial cell loss during ARDS progression.
As mentioned above, Sema3A has been reported to suppress the ERK/JNK signalling pathways to alleviate apoptosis and inflammation in rats with bronchopulmonary dysplasia [Citation17]. Likewise, suppression of the ERK signalling has also been implicated in Sema3A-mediated inflammation resolution in mechanical stress-stimulated chondrocytes [Citation25]. The ERK has been reported as the critical pathway responsible for IL-17A-mediated inflammation, chemokines and adhesion molecules in endothelial cells and vascular damage [Citation31]. The JNK pathway exerts a critical function in the cell response to proinflammatory cytokines, oxidative stress, and other stimuli, therefore affecting gene expression, cell death, and cellular senescence [Citation32]. In fact, both ERK and JNK pathways are frequently responsible for oxidative stress and inflammation and the consequential tissue injury induced by irritations [Citation33, Citation34]. Interestingly, the ERK/JNK could reportedly suppress the nuclear factor-kappa B signalling pathway, another master regulator of inflammatory responses [Citation33, Citation35]. Here in this work, indeed, we found that the Sema3A overexpression significantly suppressed the phosphorylation of ERK and JNK in rat lung tissues and PMVECs. The ERK or JNK agonists, as expected, aggravated LPS-induced cell apoptosis and the production of inflammatory cytokines and oxidative stress markers. These changes were partly negated by Sema3A overexpression. In a mouse model of LPS-induced ARDS, inhibition of the MAPK/ERK/JNK signalling has been found to reduce oxidative stress as well [Citation36]. These results indicate that suppression of Sema3A on endothelial cell death, lung injury, inflammation and oxidative stress in ARDS is due in part to the suppression of the ERK/JNK signalling pathways.
Conclusion
Collectively, this work demonstrates that downregulation of Sema3A might be responsible for excessive inflammation, oxidative stress, and tissue injury during ARDS progression. Restoration of Sema3a inactivates the ERK/JNK signalling pathways and alleviates the ARDS-like symptoms, therefore might represent a promising strategy for the management of ARDS.
Data availability statement
All data generated or analysed during this study are included in this published article.
Disclosure statement
The authors declare that they have no competing interests.
Additional information
Funding
References
- Ashbaugh DG, Bigelow DB, Petty TL, et al. Acute respiratory distress in adults. Lancet. 1967;2(7511):1–10.
- Confalonieri M, Salton F, Fabiano F. Acute respiratory distress syndrome. Eur Respir Rev. 2017;26(144):160116.
- Meyer NJ, Gattinoni L, Calfee CS. Acute respiratory distress syndrome. Lancet. 2021;398(10300):622–637.
- Bellani G, Laffey JG, Pham T, et al. Epidemiology, patterns of care, and mortality for patients with acute respiratory distress syndrome in intensive care units in 50 countries. JAMA. 2016;315(8):788–800.
- Marseglia L, D’Angelo G, Granese R, et al. Role of oxidative stress in neonatal respiratory distress syndrome. Free Radic Biol Med. 2019;142:132–137.
- Zhai Z, Ouyang W, Yao Y, et al. Dexamethasone-loaded ROS-responsive poly(thioketal) nanoparticles suppress inflammation and oxidative stress of acute lung injury. Bioact Mater. 2022;14:430–442.
- Huppert LA, Matthay MA, Ware LB. Pathogenesis of acute respiratory distress syndrome. Semin Respir Crit Care Med. 2019;40(1):31–39.
- Cannavo L, Perrone S, Viola V, et al. Oxidative stress and respiratory diseases in preterm newborns. Int J Mol Sci. 2021;22(22):12504.
- Avouac J, Pezet S, Vandebeuque E, et al. Semaphorins: from angiogenesis to inflammation in rheumatoid arthritis. Arthritis Rheumatol. 2021;73(9):1579–1588.
- Liu L, Watanabe M, Minami N, et al. Semaphorin 3A: a potential target for prevention and treatment of nickel allergy. Commun Biol. 2022;5(1):671.
- Imoto T, Kondo S, Wakisaka N, et al. Overexpression of semaphorin 3A is a marker associated with poor prognosis in patients with nasopharyngeal carcinoma. Microorganisms. 2020;8(3):423.
- Rivera LB, Bergers G. Location, location, location: macrophage positioning within tumors determines pro- or antitumor activity. Cancer Cell. 2013;24(6):687–689.
- Negi O, Tominaga M, Tengara S, et al. Topically applied semaphorin 3A ointment inhibits scratching behavior and improves skin inflammation in NC/nga mice with atopic dermatitis. J Dermatol Sci. 2012;66(1):37–43.
- Rienks M, Carai P, Bitsch N, et al. Sema3A promotes the resolution of cardiac inflammation after myocardial infarction. Basic Res Cardiol. 2017;112(4):42.
- Zhang H, Lu Y, Wu B, et al. Semaphorin 3A mitigates lipopolysaccharide-induced chondrocyte inflammation, apoptosis and extracellular matrix degradation by binding to neuropilin-1. Bioengineered. 2021;12(2):9641–9654.
- D’Alessio FR. Mouse models of acute lung injury and ARDS. Methods Mol Biol. 2018;1809:341–350.
- Liang Z, Zhang X, Liu Y, et al. SEMA3A protects against hyperoxia-induced lung injury in a bronchopulmonary dysplasia model of newborn rat by inhibiting ERK pathway. Allergol Immunopathol (Madr). 2021;49(6):8–15.
- Liu F, Yang X, Geng M, et al. Targeting ERK, an achilles’ heel of the MAPK pathway, in cancer therapy. Acta Pharm Sin B. 2018;8(4):552–562.
- Sui X, Kong N, Ye L, et al. p38 and JNK MAPK pathways control the balance of apoptosis and autophagy in response to chemotherapeutic agents. Cancer Lett. 2014;344(2):174–179.
- Lai JL, Liu YH, Liu C, et al. Indirubin inhibits LPS-Induced inflammation via TLR4 abrogation mediated by the NF-kB and MAPK signaling pathways. Inflammation. 2017;40(1):1–12.
- Tsai YF, Chen CY, Yang SC, et al. Apremilast ameliorates acute respiratory distress syndrome by inhibiting neutrophil-induced oxidative stress. Biomed J. 2022 [Sept 11]. DOI: 10.1016/j.bj.2022.09.001
- Vassiliou AG, Kotanidou A, Dimopoulou I, et al. Endothelial damage in acute respiratory distress syndrome. Int J Mol Sci. 2020;21(22):8793.
- Vadasz Z, Toubi E. Semaphorins: their dual role in regulating immune-mediated diseases. Clin Rev Allergy Immunol. 2014;47(1):17–25.
- Becker PM, Tran TS, Delannoy MJ, et al. Semaphorin 3A contributes to distal pulmonary epithelial cell differentiation and lung morphogenesis. PLoS One. 2011;6(11):e27449.
- Sumi C, Hirose N, Yanoshita M, et al. Semaphorin 3A inhibits inflammation in chondrocytes under excessive mechanical stress. Mediators Inflamm. 2018;2018:5703651.
- Adi SD, Eiza N, Bejar J, et al. Semaphorin 3A is effective in reducing both inflammation and angiogenesis in a mouse model of bronchial asthma. Front Immunol. 2019;10:550.
- Liu S, Xiong X, Thomas SV, et al. Analysis for carom complex, signaling and function by database mining. Front Biosci (Landmark Ed). 2016;21(4):856–872.
- Szade A, Grochot-Przeczek A, Florczyk U, et al. Cellular and molecular mechanisms of inflammation-induced angiogenesis. IUBMB Life. 2015;67(3):145–159.
- Xiang R, Xu Y, Zhang W, et al. Semaphorin 3A inhibits allergic inflammation by regulating immune responses in a mouse model of allergic rhinitis. Int Forum Allergy Rhinol. 2019;9(5):528–537.
- Rimar D, Nov Y, Rosner I, et al. Semaphorin 3A: an immunoregulator in systemic sclerosis. Rheumatol Int. 2015;35(10):1625–1630.
- Xing X, Yang J, Yang X, et al. IL-17A induces endothelial inflammation in systemic sclerosis via the ERK signaling pathway. PLoS One. 2013;8(12):e85032.
- Klimontov VV, Saik OV, Korbut AI. Glucose variability: how does it work? Int J Mol Sci. 2021;22(15):7783.
- El-Dessouki AM, El Fattah MA, Awad AS, et al. Zafirlukast and vincamine ameliorate tamoxifen-induced oxidative stress and inflammation: role of the JNK/ERK pathway. Life Sci. 2018;202:78–88.
- Zhu H, Zhang L, Jia H, et al. Tetrahydrocurcumin improves lipopolysaccharide-induced myocardial dysfunction by inhibiting oxidative stress and inflammation via JNK/ERK signaling pathway regulation. Phytomedicine. 2022;104:154283.
- Wang J, Huang J, Wang L, et al. Urban particulate matter triggers lung inflammation via the ROS-MAPK-NF-kappaB signaling pathway. J Thorac Dis. 2017;9(11):4398–4412.
- Huang X, Kong G, Li Y, et al. Decitabine and 5-azacitidine both alleviate LPS induced ARDS through anti-inflammatory/antioxidant activity and protection of glycocalyx and inhibition of MAPK pathways in mice. Biomed Pharmacother. 2016;84:447–453.