
Abstract
Lupus nephritis (LN) is the most severe end-organ pathology in Systemic Lupus Erythematosus (SLE). Research has enhanced our understanding of immune effectors and inflammatory pathways in LN. However, even with the best available therapy, the rate of complete remission for proliferative LN remains below 50%. A deeper understanding of the resistance or susceptibility of renal cells to injury during the progression of SLE is critical for identifying new targets and developing effective long-term therapies. The complex and heterogeneous nature of LN, combined with the limitations of clinical research, make it challenging to investigate the aetiology of this disease directly in patients. Hence, multiple murine models resembling SLE-driven nephritis are utilised to dissect LN's cellular and genetic mechanisms, identify therapeutic targets, and screen novel compounds. This review discusses commonly used spontaneous and inducible mouse models that have provided insights into pathogenic mechanisms and long-term maintenance therapies in LN.
1. Pathophysiology of SLE and LN
SLE is a prototypic systemic autoimmune disease characterised by the loss of tolerance to self-DNA, histones, and ribonucleoproteins [Citation1,Citation2]. Hallmark features of SLE include immune system activation and the production of autoantibodies, resulting in the formation of immune complexes (IC) that induce local inflammation and pathology in multiple organs [Citation2,Citation3]. The aetiology of SLE is incompletely understood, but disease initiation results from a combination of genetic susceptibility, hormonal changes, and numerous external factors (e.g. viral infection, exposure to heavy metals) [Citation4]. The kidneys receive about 25% of the cardiac output and are highly vascular, rendering them susceptible to IC-mediated pathology.
Nephritis is a common and severe complication of SLE and can lead to end-stage kidney disease (ESKD) [Citation5,Citation6]. LN can present in multiple forms in the kidneys, including proliferative lesions with crescent formation, sclerosis, necrosis, and podocyte foot process effacement. The patterns of glomerular IC include mesangial deposits (classes I, II), subendothelial deposits with endocapillary hypercellularity or prominent glomerular basement membranes (GBMs) with necrotising and crescentic lesions (classes III and IV, focal and diffuse LN), or membranous forms (class V). Focal LN (class III) is present in about ∼30% of biopsied SLE patients [Citation7]. A simplified cross-section and description of a glomerular capillary with areas of IC deposits are depicted in .
Figure 1. A simplified cross section of the glomerulus is depicted. The glomerular assembly comprises of a fenestrated endothelium (pink), which is contiguous with the mesangium that houses the mesangial cells (green). As such, mesangial cells are constantly exposed to blood components. The fenestrated endothelium is lined by the glomerular basement membrane (cyan). The other side of the basement membrane is lined by a specialised type of epithelial cells called podocytes and their foot processes (cyan with pink blebs).
During the evolution of lupus nephritis (LN), circulating or in-situ formed immune complexes can deposit in the mesangial (orange, class I, II LN), sub-endothelial (blue, class III and IV) or sub-epithelial (red, class V) regions and induce cellular pathology and inflammation. Some of the common pathological features include (but are not restricted to) mesangial proliferation, basement membrane thickening, endocapillary proliferation, sclerosis, crescent formation with loss of podocyte foot processes.
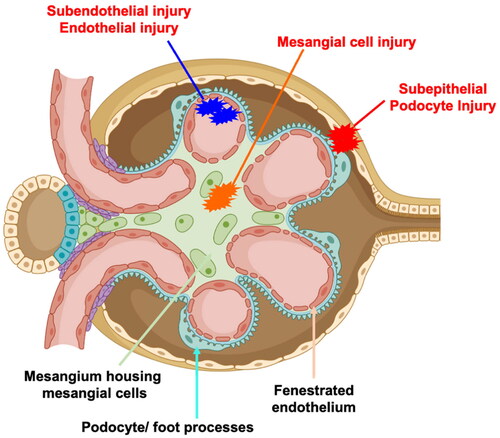
Traditionally, LN is thought to initiate in the glomeruli. However, tubulointerstitial inflammation, fibrosis, and atrophy strongly correlate with renal dysfunction, independently of the extent of glomerular damage, and predict worse outcomes [Citation8–11]. Genome-wide association studies (GWAS) indicated that genes driving autoimmunity and nephritis are distinct [Citation12], suggesting that the progression to LN in SLE patients results from genetic susceptibility to systemic autoimmunity and increased end-organ sensitivity to inflammatory stimuli.
Given the high clinical heterogeneity in SLE and LN patients and the complexity of the kidney in which multiple cell types are involved, it is challenging to recapitulate human SLE and LN in cell culture models. Furthermore, more recent studies have implicated novel contributors to LN, such as immunometabolism [Citation13], gut dysbiosis [Citation14], and ferroptosis [Citation15], and the fine mechanisms of these processes are incompletely understood. Hence, multiple animal models of spontaneous and induced SLE and LN that reproduce one or multiple aspects of human disease have been described and continue to evolve. These animal models are an indispensable tool for investigating the aetiology and genetics of SLE/LN, as well as identifying drug targets and validating therapeutics. Here, we discuss the principal murine models of SLE relevant to human LN, their characteristics, and significant contributions that have led to a better understanding of human disease. summarises the discussed models and their specific advantages and disadvantages.
Table 1. Summary of the discussed models and their specific advantages and disadvantages.
The spontaneous murine models of SLE vary from polygenic to single-gene knockout or transgenic expression, resulting in lupus-like phenotypes [Citation16,Citation17]. None of the spontaneous or induced mouse models of lupus represent the entire clinical spectrum observed in SLE patients. However, these models have delineated functional pathways in SLE and identified specific genes and genetic loci critical in maintaining systemic immune tolerance and IC-glomerulonephritis and increasing susceptibility to SLE. Multiple reviews on murine models of SLE have focused on genetic links between human and mouse SLE [Citation16,Citation18] or the mechanisms leading to systemic autoimmunity and clinical lupus [Citation19]. In addition, some of these models have provided insights into novel contributors to SLE, such as the microbiome [Citation20], immunometabolism [Citation21], or iron metabolism [Citation22,Citation23] as relevant to human SLE. As in humans, the kidney is the primary target organ in multiple murine models of SLE with a clear female gender bias (except for MRL/lpr and BXSB mice, discussed below). Each model represents an overlapping subset of the clinical features of the human disease and offers unique features of interest to address specific preclinical questions. In this review, we briefly summarise the most common mouse models of SLE, stressing their unique features, provide an update on the significant advances they have contributed to the field, and discuss the advantage of using one model over the others when testing fine mechanisms and specific hypotheses.
2. Spontaneous polygenic models
2.1. MRL/lpr
The Murphy-Ross Large (MRL) strain was developed by crossing several strains, including C3H/Di, LG/J, C57BL/6, and AKR/J [Citation24,Citation25]. One of the MRL sub-strains carries a spontaneous mutation named lpr for lymphoproliferation and develops a severe SLE-like phenotype characterised at the cellular level by the accumulation of double negative (DN: CD4 − CD8−) B220+ T cells. Like in SLE patients [Citation26], the DN T cells are autoreactive and pathogenic [Citation27], making this model relevant to SLE pathogenesis. Lpr corresponds to non-functional transcripts of the Fas gene, a key regulator of apoptosis in immune cells [Citation28]. In this strain, male and female mice are affected (though females show a more pronounced phenotype) and produce autoantibodies against dsDNA and Sm, leading to large amounts of IC that induce renal and skin pathology [Citation29]. Unlike SLE patients, these mice develop a massive lymphadenopathy. The data generated from the MRL/lpr strain has led to, among others, a better understanding of the role of TRL7 and TLR9 in lupus [Citation30], differences in TLR activation in B cells and dendritic cells [Citation31], and were used for the first time to report the critical role of the extrafollicular pathway for the development of autoreactive B cells [Citation32]. Additionally, these mice have shown that the kidney infiltrating T cells in LN have an exhausted metabolic and functional phenotype [Citation33]. Complement plays a critical role in nephritis in these mice, as a deficiency in the regulatory factor H accelerates kidney pathology [Citation34,Citation35]. MRL/lpr mice become azotaemic, leading to 70% mortality by 14 weeks of age. Unlike other lupus models (e.g. NZB/NZW F1 discussed below), nephritis in MRLlpr mice is independent of Type I interferons and FcγRs, which are two major drivers of SLE pathogenesis in general, including LN. This represents clear limitations to the relevance of the MRL/lpr model to human LN, in addition to the confounding massive lymphoproliferation [Citation36]. C57BL/6-Faslpr mice expressing the lpr mutation on a non-autoimmune C57BL/6 (B6) background develop systemic autoimmunity without clinical pathology and reduced lymphadenopathy [Citation37]. These B6.lpr mice have been used to investigate the involvement of IL-17 in lupus [Citation38,Citation39]. Finally, the MRL/lpr model has the major advantage of being the most rapid, with the highest incidence and severity of disease amongst the spontaneous models. Hence, it has been extensively used as a pre-clinical model of LN to test novel therapeutics [Citation23,Citation40]. For example, the MRL/lpr mice expressing human CD20 on their B cells were used to demonstrate the superiority of obinutuzumab, a type II anti-CD20 monoclonal antibody over rituximab [Citation41]. These mice were also utilised to highlight the efficacy of idebenone, an antioxidant that bypasses complex I activity and enhances ATP synthesis [Citation42].
2.2. The BXSB model
The BXSB strain is a recombinant inbred strain developed in the 1970s by crossing of C57BL/6 and SB/Le mice ((B6 X SB/Le) F1 to SB/Le, hence BXSB) [Citation43,Citation44]. The BXSB male mice develop an accelerated disease compared to females due to the Y chromosome-linked autoimmune accelerator (Yaa) [Citation44,Citation45], which has been identified as the translocation of a telomeric region of the X chromosome to the Y chromosome, resulting in the duplication of the toll-like receptor 7 (Tlr7) gene [Citation46,Citation47]. This increased TLR7 expression activates the Type I interferon cascade, which is critical in SLE pathogenesis [Citation48]. BXSB.Yaa mice have a relatively low titre of autoantibodies reactive to DNA and histones but produce a higher titre of anti-ribonucleoproteins (RNP) IgG with typical speckled patterns [Citation49]. Male BXSB mice develop exudative, proliferative nephritis with neutrophilic infiltrates and severe proteinuria by three months of age [Citation29,Citation50]. The kidneys show prominent IgG and C3 deposits in the glomerular mesangium and along the capillary walls, with 50% of the mice developing fatal nephritis starting around five months and 90% at around eight months of age [Citation29,Citation49]. Female mice develop a weaker disease phenotype with around 50% mortality around 15 months of age. Therefore, despite its predominant presentation in males, the BXSB/Yaa strain models the consequences of an overreactive TLR7/Type 1 IFN pathway and associated renal failure.
Hybrid (NZW × BXSB)F1 male mice carry the Yaa-associated two copies of Tlr7 and develop accelerated lupus with antiphospholipid syndrome (APS) [Citation51] as well as proliferative glomerulonephritis [Citation46]. Female (NZW × BXSB)F1 mice with only one active copy of Tlr7 develop late-onset glomerulonephritis but not APS [Citation51]. In addition to inflammatory glomerulonephritis, the males develop immune-mediated thrombocytopenia and a thrombotic vasculopathy with myocardial infarcts, myocardial fibrosis, and dilated cardiomyopathy [Citation52,Citation53]. Given the importance of antiphospholipid antibodies in lupus [Citation54], this model is a valuable tool to dissect the mechanisms and interventions in SLE-associated antiphospholipid syndrome [Citation53].
2.3. The NZB/NZW F1 model
The NZB/NZW F1 hybrid (NZB/W F1) is a spontaneous model of SLE and LN [Citation55,Citation56] and it is phenotypically a close approximation of human SLE and LN. As in humans, female mice are more severely affected, and anti-dsDNA antibodies are detected at around three months of age [Citation57]. Female mice develop splenomegaly and chronic polyclonal B cell activation [Citation29,Citation50]. The kidney pathology presents as sclerosis with proteinaceous deposits in the mesangium, diffuse proliferation of glomerular cells, and tubular casts. As in LN patients, prominent glomerular crescents are observed, and large periglomerular and interstitial inflammatory infiltrate [Citation29,Citation55]. A few critical contributions of the NZB/W F1 model to the field include the identification of the therapeutic effect of BAFF inhibition [Citation58], the role of type 1 IFN [Citation59], and the identification of LN biomarkers [Citation60]. We have shown that early treatment with a combination of metformin and CTLA4Ig decreases the development of LN in this model without attenuation of autoantibody production [Citation61].
The serendipitous breeding between B/W and NZW mice led to the development of a panel of inbred New Zealand Mixed (NZM) mouse strains, several of which showed a highly penetrant SLE and LN resembling disease in parental NZB/W F1mice [Citation56,Citation62]. Pioneering studies on identifying the SLE susceptibility loci in murine SLE were conducted with the NZM2410 strain [Citation62,Citation63]. Identifying three lupus susceptibility loci (Sle1, Sle2, and Sle3 on chromosomes 1, 4, and 7, respectively) in the NZM2410 strain has also led to the development of congenic strains with unique advantages. Intercrossing the three Sle1, Sle2, and Sle3 susceptibility loci was sufficient to develop a lupus phenotype on the non-autoimmune B6 genetically resistant strain [Citation63,Citation64]. The B6.NZM2410.Sle1.Sle2.Sle3 is unique as it shares 95% of its genome with B6 and has provided original insight into the metabolic control of the disease [Citation65–67]. The corresponding single (Sle1) and bi-congenic (Sle1.Sle3) are also well suited to breed with B6-based gene knockouts. For instance, deletion of the plasmacytoid dendritic cells (pDC)-specific transcription factor Tcf4 in B6.Sle1.Sle3 mice provided genetic evidence that pDCs are critically involved in developing SLE [Citation68]. Furthermore, the transfer of bone marrow cells from B6.Sle1 mice into irradiated B6 recipients was sufficient to recapitulate the autoimmune phenotypes of B6.Sle1 mice, demonstrating that they are entirely accounted for by haematopoietic cells [Citation69]. Bone marrow transfer from these NZM2410-derived congenics into mice with specific gene knockout (e.g. in the kidney) provides a powerful tool for studying end-organ resistance or susceptibility to lupus.
A notable mention in the NZM sub-strains is NZM2328 [Citation70,Citation71], which develops nephritis in two stages: acute glomerulonephritis followed by more chronic nephritis [Citation72,Citation73]. These mice were vital in showing that autoantibodies, especially anti-dsDNA antibodies, are not required to develop renal disease in SLE [Citation74,Citation75].
3. Inducible models of SLE and LN
While the spontaneous models of lupus have greatly enhanced our understanding of human SLE and LN, these models exhibit delayed and variable onset of SLE-related symptoms and renal pathology, and most have complex genetic backgrounds. These factors limit the application of these models for testing therapeutics, investigating environmental factors, and the contribution of individual genes in the pathogenesis of SLE and LN. Thus, exposing wild-type non-autoimmune mice or mice with specific gene deficiencies in individual cell types to chemical agents and toxins is an alternative valuable approach to induce an SLE-like disease. These agents mimic the impact of environmental, genetic, and non-genetic factors on susceptibility or resistance to SLE and LN and in evaluating the efficacy of therapeutic drugs. Below, we discuss two commonly used approaches to induce lupus and immune-complex mediated glomerulonephritis.
3.1. Pristane-induced lupus
Initially used for priming mice to obtain ascite fluid to produce monoclonal antibodies, an intraperitoneal injection of pristane (2,6,10,14 tetramethylpentadecane, a major component of mineral oil) leads to the development of anti-RNP, anti-DNA and anti-histone antibodies in BALB/c mice [Citation76,Citation77]. In this inducible model, the BALB/c and SJL strains present glomerular IgG and complement deposits with cellular proliferation and develop proteinuria, whereas B6 mice develop milder (Class II like) disease [Citation78]. In contrast to most spontaneous models of SLE, the anti-RNP antibodies class switches to IgG, while anti-DNA antibodies are almost entirely IgM [Citation77]. These mice gradually develop nephritis six months after injection with focal to diffuse proliferative glomerulonephritis and moderate proteinuria [Citation77]. Pristane-treated BALB/c mice also develop lung haemorrhagic, capillary damage-associated pathology, and TNFα-driven arthritis that may resemble arthralgia and capillaritis occurring in SLE patients [Citation79–81]. In the SJL strain, pristane-induced lupus is more severe in females than males [Citation82]. Compared to males, the kidneys of pristane-injected SJL female mice present with significantly more glomerular hypercellularity and necrosis [Citation82]. Pristane-induced lupus is driven by type 1 IFN [Citation83] and thus, it is a valuable tool for investigating type 1 IFN-induced pathology, a critical pathogenic pathway in human lupus. This model was used to define the protective role of TLR9 in lupus as BALB/c mice with TLR9 deficiency developed more severe lupus than their WT TLR9-sufficient mice [Citation84]. Pristane-induced lupus is an excellent example of how an environmental factor can induce lupus-like disease in a non-autoimmune background.
3.2. Nephrotoxic serum nephritis
The nephrotoxic serum nephritis (NTN), also known as anti-glomerular basement nephritis, is an inducible IC-mediated model of glomerulonephritis that resembles human Goodpasture’s Disease [Citation85,Citation86]. The antigen (anti-mouse glomerular basement membrane antibodies raised commonly in rabbits or sheep (donor) after immunisation with mouse glomerular preparation) binds the mouse glomerular basement membrane (GBM), leading to IC deposition and inflammatory response. Most nephrotoxic serum preparations are crude as they contain immunoglobulins against other structural elements of the kidney. Administration of the heterologous donor anti-GBM antiserum (NTS) leads to complement-dependent, transient renal injury (day 0–4) [Citation87], followed by the development of mouse anti–rabbit or sheep antibodies in the later, autologous stage (day 7–14) during which IC deposits are observed in the glomeruli, resulting in glomerulonephritis with renal failure [Citation88]. A modified accelerated model of NTS has also been developed wherein mice are pre-sensitized to the heterologous anti-GBM serum by preimmunization with normal, donor heterologous serum a few days (Day −4) before the introduction of the heterologous anti-GBM serum (Day 0), thus initiating the autologous phase of disease concomitant with the introduction of the anti-GBM serum [Citation88,Citation89].
The binding of polyclonal antibodies to the glomerular endothelial wall and GBM antigens triggers local immune activation and the secretion of proinflammatory chemokines and cytokines. In the heterologous phase, glomerular injury depends largely on complement activation and neutrophil-dominated inflammation [Citation90–92]. During the autologous phase, the renal dendritic cells promote nephritogenic T-cell responses [Citation93]. At this stage, a Th1 response stimulates the macrophages to mediate renal damage in a classical type IV hypersensitivity reaction [Citation94], though a pathogenic Th17 response has also been reported [Citation95]. Using this model, we have recently shown the importance of iron-mediated tubulointerstitial injury in the pathogenesis of LN [Citation96]. Since the phenotype of NTN involves changes in the renal functional parameters, glomerular and secondary tubulointerstitial injury, renal leukocyte infiltration, and adaptive cellular and humoral immune responses [Citation96], it serves as an all-encompassing tool to tease out delicate mechanisms associated with LN-like chronic IC-induced GN. The NTN model allows investigations into the pathogenesis of IC-mediated glomerulonephritis independent of genetic background and gender and is highly synchronised.
3.3. Chronic graft-versus-host model of systemic lupus erythematosus
Graft versus host disease (cGVHD) models are generated by transferring major histocompatibility complex (MHC) mismatched immune cells into a recipient/host. Compared to the acute GVDH model [Citation97], the chronic GVHD (cGVHD) model, where I-Abm12B6(C)-H2-Ab1bm12/KhEgJ (bm12) splenocytes are transferred into B6 mice produce symptoms closely resembling SLE. In this model, the adoptive transfer of immune cells from bm12 mice, a strain identical to B6 except for three amino acid substitutions on the MHC class II _I-A chain, into I-Ab B6 mice leads to alloactivation of donor CD4+ T cells by recipient APCs [Citation98]. The activated alloreactive donor CD4+ T cells then provide help to host self-reactive B cells [Citation99,Citation100]. Some of the key features include the expansion of donor-derived T follicular helper (Tfh) cells, recipient-derived germinal centre (GC) B cells and plasma cells, and the production of anti-dsDNA, anti-ssDNA, anti-chromatin, and anti-RBC antibodies [Citation98]. During the later phase, the recipient mice develop glomerulonephritis associated with IgG deposits in the kidneys’ glomerular, interstitial, and vascular segments [Citation101]. Host CD4+ T cells do not participate in the cGVHD induction, but their presence is essential for nurturing B cells through IL-4 and/or CD40 stimulation [Citation101,Citation102].
Like the NTN model, there are several advantages of this model. Genetically modified B6 mice can serve as either donors or recipients. This allows dissecting the contribution of specific genes and modified cells to disease progression. Furthermore, disease development in the bm12 model requires only two weeks for ANA production, compared to several months for most spontaneous models [Citation103]. Furthermore, unlike spontaneous models that develop disease at different time points, the disease onset and progression in this model is highly synchronised. This permits the use of appropriately sized cohorts to test novel therapeutics during different stages of SLE/LN.
3.4. Humanised models of lupus
Though mouse models provide insights on SLE/LN pathogenesis, they do not fully recapitulate the complexities of human SLE. There are major differences between the mouse and human immune system [Citation104,Citation105] and genetics [Citation106,Citation107] pose challenges in understanding the complexities of SLE and evaluation of therapeutic protocols [Citation105]. There have been some attempts to bridge the differences between the murine and human immune systems by using humanised mice as models of SLE [Citation108]. Immune cell-mediated lupus-like disease can be initiated by engrafting immunodeficient mice with either peripheral blood leukocytes from SLE patients or healthy haematopoietic stem cells, followed by lupus induction with pristane [Citation108]. Proteinuria, a marker of kidney injury in LN, was observed using both approaches [Citation109,Citation110]. The first approach was used to evaluate the therapeutic potential of targeting the Kv1.3 channel on T cells with nanoparticles [Citation109]. However, the injury induced by human immune cells to murine renal parenchyma may not provide a reliable model for in-depth mechanistic studies or hold clinical relevance.
4. Conclusions
Novel pathogenic mechanisms of human SLE and LN are continuously identified. Progress has been made in the understanding of genetic risks to LN. For example, polymorphisms in the human major histocompatibility complex region increase the risk of disease development regardless of ethnicity [Citation111,Citation112]. In contrast, the apolipoprotein L1 gene (APOL1) risk alleles G1/G2 are more prevalent in African Americans than European Americans [Citation113]. Even though this knowledge is more than two decades old, invasive kidney biopsy is still the gold standard for diagnosing and guiding the treatment of LN. Clinical research has severe limitations when evaluating the causal role of individual genes, environmental factors, and novel interventions to tease out fine mechanisms contributing to disease outcomes.
Herein lies the strength of spontaneous and inducible animal models of SLE/LN. As such, no singularly perfect model recapitulates all aspects of human SLE and LN. An astute selection of animal models will continue to help elucidate the intricacies of the genetics, immune, and parenchymal cell contributions to the pathogenesis of human disease. In summary, animal models of SLE/LN provide an understanding of the dominant and fine mechanisms contributing to disease. As such, animal models offer the opportunity to develop novel strategies for interventions in SLE/LN, which translates into more timely and effective therapy.
Disclosure statement
The authors have declared that no conflict of interest exists.
Additional information
Funding
References
- Brunner HI, Gladman DD, Ibañez D, et al. Difference in disease features between childhood-onset and adult-onset systemic lupus erythematosus. Arthritis Rheum. 2008;58(2):1–8.
- Liu Z, Davidson A. Taming lupus-a new understanding of pathogenesis is leading to clinical advances. Nat Med. 2012;18(6):871–882.
- Tsokos GC. Systemic lupus erythematosus. N Engl J Med. 2011;365(22):2110–2121.
- Rullo OJ, Tsao BP. Recent insights into the genetic basis of systemic lupus erythematosus. Ann Rheum Dis. 2013;72 Suppl 2(0 2):ii56–61.
- Almaani S, Meara A, Rovin BH. Update on lupus nephritis. Clin J Am Soc Nephrol. 2017;12(5):825–835.
- Madaio MP. The role of autoantibodies in the pathogenesis of lupus nephritis. Semin Nephrol. 1999;19(1):48–56.
- Fogo AB, Lusco MA, Najafian B, et al. AJKD atlas of renal pathology: focal and diffuse lupus nephritis (ISN/RPS class III and IV). Am J Kidney Dis. 2017;70(2):e9–e11.
- Londoño Jimenez A, Mowrey WB, Putterman C, et al. Brief report: tubulointerstitial damage in lupus nephritis: a comparison of the factors associated with tubulointerstitial inflammation and renal scarring. Arthritis Rheumatol. 2018;70(11):1801–1806.
- Rijnink EC, Teng YKO, Wilhelmus S, et al. Clinical and histopathologic characteristics associated with renal outcomes in lupus nephritis. Clin J Am Soc Nephrol. 2017;12(5):734–743.
- Hong S, Healy H, Kassianos AJ. The emerging role of renal tubular epithelial cells in the immunological pathophysiology of lupus nephritis. Front Immunol. 2020;11:578952.
- Wilson PC, Kashgarian M, Moeckel G. Interstitial inflammation and interstitial fibrosis and tubular atrophy predict renal survival in lupus nephritis. Clin Kidney J. 2018;11(2):207–218.
- Chung SA, Brown EE, Williams AH, et al. Lupus nephritis susceptibility loci in women with systemic lupus erythematosus. J Am Soc Nephrol. 2014;25(12):2859–2870.
- Caza T, Wijewardena C, Al-Rabadi L, et al. Cell type-specific mechanistic target of rapamycin-dependent distortion of autophagy pathways in lupus nephritis. Transl Res. 2022;245:55–81.
- Wang A, Zhao J, Qin Y, et al. Alterations of the gut microbiota in the lupus nephritis: a systematic review. Ren Fail. 2023;45(2):2285877.
- Wang W, Lin Z, Feng J, et al. Identification of ferroptosis-related molecular markers in glomeruli and tubulointerstitium of lupus nephritis. Lupus. 2022;31(8):985–997.
- Vinuesa CG, Shen N, Ware T. Genetics of SLE: mechanistic insights from monogenic disease and disease-associated variants. Nat Rev Nephrol. 2023;19(9):558–572.
- Morel L. Genetics of SLE: evidence from mouse models. Nat Rev Rheumatol. 2010;6(6):348–357.
- Barnes BJ. Genetic versus non-genetic drivers of SLE: implications of IRF5 dysregulation in both roads leading to SLE. Curr Rheumatol Rep. 2019;21(1):2.
- Sang A, Yin Y, Zheng YY, et al. Animal models of molecular pathology systemic lupus erythematosus. Prog Mol Biol Transl Sci. 2012;105:321–370.
- Toumi E, Mezouar S, Plauzolles A, et al. Gut microbiota in SLE: from animal models to clinical evidence and pharmacological perspectives. Lupus Sci Med. 2023;10(1):e000776.
- Ma L, Roach T, Morel L. Immunometabolic alterations in lupus: where do they come from and where do we go from there? Curr Opin Immunol. 2022;78:102245.
- Wlazlo E, Mehrad B, Morel L, et al. Iron metabolism: an under investigated driver of renal pathology in lupus nephritis. Front Med (Lausanne). 2021;8:643686.
- Scindia Y, Wlazlo E, Ghias E, et al. Modulation of iron homeostasis with hepcidin ameliorates spontaneous murine lupus nephritis. Kidney Int. 2020;98(1):100–115.
- Rauch J, Murphy E, Roths JB, et al. A high frequency idiotypic marker of anti-DNA autoantibodies in MRL-Ipr/ipr mice. J Immunol. 1982;129(1):236–241.
- Theofilopoulos AN, Dixon FJ. Murine models of systemic lupus erythematosus. Adv Immunol. 1985;37:269–390.
- Crispín JC, Oukka M, Bayliss G, et al. Expanded double negative T cells in patients with systemic lupus erythematosus produce IL-17 and infiltrate the kidneys. J Immunol. 2008;181(12):8761–8766.
- Rodríguez-Rodríguez N, Apostolidis SA, Fitzgerald L, et al. Pro-inflammatory self-reactive T cells are found within murine TCR-alphabeta(+) CD4(-) CD8(-) PD-1(+) cells. Eur J Immunol. 2016;46(6):1383–1391.
- Cohen PL, Eisenberg RA. Lpr and gld: single gene models of systemic autoimmunity and lymphoproliferative disease. Annu Rev Immunol. 1991;9(1):243–269.
- Andrews BS, Eisenberg RA, Theofilopoulos AN, et al. Spontaneous murine lupus-like syndromes. Clinical and immunopathological manifestations in several strains. J Exp Med. 1978;148(5):1198–1215.
- Christensen SR, Shupe J, Nickerson K, et al. Toll-like receptor 7 and TLR9 dictate autoantibody specificity and have opposing inflammatory and regulatory roles in a murine model of lupus. Immunity. 2006;25(3):417–428.
- Teichmann LL, Schenten D, Medzhitov R, et al. Signals via the adaptor MyD88 in B cells and DCs make distinct and synergistic contributions to immune activation and tissue damage in lupus. Immunity. 2013;38(3):528–540.
- Ols ML, Cullen JL, Turqueti-Neves A, et al. Dendritic cells regulate extrafollicular autoreactive B cells via T cells expressing fas and fas ligand. Immunity. 2016;45(5):1052–1065.
- Tilstra JS, Avery L, Menk AV, et al. Kidney-infiltrating T cells in murine lupus nephritis are metabolically and functionally exhausted. J Clin Invest. 2018;128(11):4884–4897.
- Bao L, Haas M, Quigg RJ. Complement factor H deficiency accelerates development of lupus nephritis. J Am Soc Nephrol. 2011;22(2):285–295.
- Watanabe H, Garnier G, Circolo A, et al. Modulation of renal disease in MRL/lpr mice genetically deficient in the alternative complement pathway factor B. J Immunol. 2000;164(2):786–794.
- Matsumoto K, Watanabe N, Akikusa B, et al. Fc receptor-independent development of autoimmune glomerulonephritis in lupus-prone MRL/lpr mice. Arthritis Rheum. 2003;48(2):486–494.
- Vidal S, Kono DH, Theofilopoulos AN. Loci predisposing to autoimmunity in MRL-Fas lpr and C57BL/6-Faslpr mice. J Clin Invest. 1998;101(3):696–702.
- Kyttaris VC, Zhang Z, Kuchroo VK, et al. Cutting edge: IL-23 receptor deficiency prevents the development of lupus nephritis in C57BL/6-lpr/lpr mice. J Immunol. 2010;184(9):4605–4609.
- Ramani K, Biswas PS. Interleukin 17 signaling drives type I interferon induced proliferative crescentic glomerulonephritis in lupus-prone mice. Clin Immunol. 2016;162:31–36.
- Li P, Jiang M, Li K, et al. Glutathione peroxidase 4-regulated neutrophil ferroptosis induces systemic autoimmunity. Nat Immunol. 2021;22(9):1107–1117.
- Marinov AD, Wang H, Bastacky SI, et al. The type II anti-CD20 antibody obinutuzumab (GA101) is more effective than rituximab at depleting B cells and treating disease in a murine lupus model. Arthritis Rheumatol. 2021;73(5):826–836.
- Blanco LP, Pedersen HL, Wang X, et al. Improved mitochondrial metabolism and reduced inflammation following attenuation of murine lupus with coenzyme Q10 analog idebenone. Arthritis Rheumatol. 2020;72(3):454–464.
- Maibaum MA, Haywood ME, Walport MJ, et al. Lupus susceptibility loci map within regions of BXSB derived from the SB/Le parental strain. Immunogenetics. 2000;51(4–5):370–372.
- Murphy ED, Roths JB. A Y chromosome associated factor in strain BXSB producing accelerated autoimmunity and lymphoproliferation. Arthritis Rheum. 1979;22(11):1188–1194.
- Hudgins CC, Steinberg RT, Klinman DM, et al. Studies of consomic mice bearing the Y chromosome of the BXSB mouse. J Immunol. 1985;134(6):3849–3854.
- Pisitkun P, Deane JA, Difilippantonio MJ, et al. Autoreactive B cell responses to RNA-related antigens due to TLR7 gene duplication. Science. 2006;312(5780):1669–1672.
- Subramanian S, Tus K, Li QZ, et al. A Tlr7 translocation accelerates systemic autoimmunity in murine lupus. Proc Natl Acad Sci U S A. 2006;103(26):9970–9975.
- Han S, Zhuang H, Shumyak S, et al. Mechanisms of autoantibody production in systemic lupus erythematosus. Front Immunol. 2015;6:228.
- McGaha TL, Sorrentino B, Ravetch JV. Restoration of tolerance in lupus by targeted inhibitory receptor expression. Science. 2005;307(5709):590–593.
- Theofilopoulos AN, Dixon FJ. Etiopathogenesis of murine SLE. Immunol Rev. 1981;55(1):179–216.
- Yoshida H, Fujiwara H, Fujiwara T, et al. Quantitative analysis of myocardial infarction in (NZW x BXSB)F1 hybrid mice with systemic lupus erythematosus and small coronary artery disease. Am J Pathol. 1987;129(3):477–485.
- Hashimoto Y, Kawamura M, Ichikawa K, et al. Anticardiolipin antibodies in NZW x BXSB F1 mice. A model of antiphospholipid syndrome. J Immunol. 1992;149(3):1063–1068.
- Akkerman A, Huang W, Wang X, et al. CTLA4Ig prevents initiation but not evolution of anti-phospholipid syndrome in NZW/BXSB mice. Autoimmunity. 2004;37(6–7):445–451.
- Levine JS, Branch DW, Rauch J. The antiphospholipid syndrome. N Engl J Med. 2002; 346(10):752–763.
- Helyer BJ, Howie JB. Renal disease associated with positive lupus erythematosus tests in a cross-bred strain of mice. Nature. 1963;197(4863):197–197.
- Rudofsky UH, Evans BD, Balaban SL, et al. Differences in expression of lupus nephritis in New Zealand mixed H-2z homozygous inbred strains of mice derived from New Zealand black and New Zealand white mice. Lab Invest. 1993;68(4):419–426.
- Papoian R, Pillarisetty R, Talal N. Immunological regulation of spontaneous antibodies to DNA and RNA. II. Sequential switch from IgM to IgG in NZB/NZW F1 mice. Immunology. 1977;32(1):75–79.
- Ramanujam M, Wang X, Huang W, et al. Similarities and differences between selective and nonselective BAFF blockade in murine SLE. J Clin Invest. 2006;116(3):724–734.
- Liu Z, Bethunaickan R, Huang W, et al. IFN-alpha confers resistance of systemic lupus erythematosus nephritis to therapy in NZB/W F1 mice. J Immunol. 2011;187(3):1506–1513.
- Berthier CC, Kretzler M, Davidson A. From the large scale expression analysis of lupus nephritis to targeted molecular medicine. J Data Mining Genomics Proteomics. 2012; 3(3):1000123.
- Cornaby C, Elshikha AS, Teng X, et al. Efficacy of the combination of metformin and CTLA4Ig in the (NZB x NZW)F1 mouse model of lupus nephritis. Immunohorizons. 2020;4(6):319–331.
- Morel L, Rudofsky UH, Longmate JA, et al. Polygenic control of susceptibility to murine systemic lupus erythematosus. Immunity. 1994;1(3):219–229.
- Morel L. Mapping lupus susceptibility genes in the NZM2410 mouse model. Adv Immunol. 2012;115:113–139.
- Morel L, Croker BP, Blenman KR, et al. Genetic reconstitution of systemic lupus erythematosus immunopathology with polycongenic murine strains. Proc Natl Acad Sci U S A. 2000;97(12):6670–6675.
- Teng X, Brown J, Choi SC, et al. Metabolic determinants of lupus pathogenesis. Immunol Rev. 2020;295(1):167–186.
- Yin Y, Choi SC, Xu Z, et al. Normalization of CD4+ T cell metabolism reverses lupus. Sci Transl Med. 2015;7(274):274ra18.
- Yin Y, Choi SC, Xu Z, et al. Glucose oxidation is critical for CD4+ T cell activation in a mouse model of systemic lupus erythematosus. J Immunol. 2016;196(1):80–90.
- Sisirak V, Ganguly D, Lewis KL, et al. Genetic evidence for the role of plasmacytoid dendritic cells in systemic lupus erythematosus. J Exp Med. 2014;211(10):1969–1976.
- Sobel ES, Mohan C, Morel L, et al. Genetic dissection of SLE pathogenesis: adoptive transfer of Sle1 mediates the loss of tolerance by bone marrow-derived B cells. J Immunol. 1999;162(4):2415–2421.
- Rudofsky UH, Lawrence DA. New Zealand mixed mice: a genetic systemic lupus erythematosus model for assessing environmental effects. Environ Health Perspect. 1999; 107 Suppl 5(Suppl 5):713–721.
- Waters ST, Fu SM, Gaskin F, et al. NZM2328: a new mouse model of systemic lupus erythematosus with unique genetic susceptibility loci. Clin Immunol. 2001;100(3):372–383.
- Ge Y, Brown MG, Wang H, et al. Genetic approach to study lupus glomerulonephritis. Methods Mol Biol. 2012;900:271–290.
- Bagavant H, Deshmukh US, Wang H, et al. Role for nephritogenic T cells in lupus glomerulonephritis: progression to renal failure is accompanied by T cell activation and expansion in regional lymph nodes. J Immunol. 2006;177(11):8258–8265.
- Bagavant H, Kalantarinia K, Scindia Y, et al. Novel therapeutic approaches to lupus glomerulonephritis: translating animal models to clinical practice. Am J Kidney Dis. 2011;57(3):498–507.
- Deshmukh US, Bagavant H, Fu SM. Role of anti-DNA antibodies in the pathogenesis of lupus nephritis. Autoimmun Rev. 2006;5(6):414–418.
- Satoh M, Reeves WH. Induction of lupus-associated autoantibodies in BALB/c mice by intraperitoneal injection of pristane. J Exp Med. 1994;180(6):2341–2346.
- Satoh M, Kumar A, Kanwar YS, et al. Anti-nuclear antibody production and immune-complex glomerulonephritis in BALB/c mice treated with pristane. Proc Natl Acad Sci U S A. 1995;92(24):10934–10938.
- Reeves WH, Lee PY, Weinstein JS, et al. Induction of autoimmunity by pristane and other naturally occurring hydrocarbons. Trends Immunol. 2009;30(9):455–464.
- Chowdhary VR, Grande JP, Luthra HS, et al. Characterization of haemorrhagic pulmonary capillaritis: another manifestation of pristane-induced lupus. Rheumatology (Oxford). 2007;46(9):1405–1410.
- Nguyen VA, Gotwald T, Prior C, et al. Acute pulmonary edema, capillaritis and alveolar hemorrhage: pulmonary manifestations coexistent in antiphospholipid syndrome and systemic lupus erythematosus? Lupus. 2005;14(7):557–560.
- Wooley PH, Seibold JR, Whalen JD, et al. Pristane-induced arthritis. The immunologic and genetic features of an experimental murine model of autoimmune disease. Arthritis Rheum. 1989;32(8):1022–1030.
- Smith DL, Dong X, Du S, et al. A female preponderance for chemically induced lupus in SJL/J mice. Clin Immunol. 2007;122(1):101–107.
- Zhuang H, Szeto C, Han S, et al. Animal models of interferon signature positive lupus. Front Immunol. 2015;6:291.
- Bossaller L, Christ A, Pelka K, et al. TLR9 deficiency leads to accelerated renal disease and myeloid lineage abnormalities in Pristane-Induced murine lupus. J Immunol. 2016;197(4):1044–1053.
- Lerner RA, Glassock RJ, Dixon FJ. The role of anti-glomerular basement membrane antibody in the pathogenesis of human glomerulonephritis. J Exp Med. 1967; 126(6):989–1004.
- Dammacco F, Battaglia S, Gesualdo L, et al. Goodpasture’s disease: a report of ten cases and a review of the literature. Autoimmun Rev. 2013;12(11):1101–1108.
- Groggel GC, Salant DJ, Darby C, et al. Role of terminal complement pathway in the heterologous phase of antiglomerular basement membrane nephritis. Kidney Int. 1985;27(4):643–651.
- Park SY, Ueda S, Ohno H, et al. Resistance of Fc receptor- deficient mice to fatal glomerulonephritis. J Clin Invest. 1998;102(6):1229–1238.
- Kaneko Y, Nimmerjahn F, Madaio MP, et al. Pathology and protection in nephrotoxic nephritis is determined by selective engagement of specific Fc receptors. J Exp Med. 2006;203(3):789–797.
- Sheerin NS, Springall T, Carroll MC, et al. Protection against anti-glomerular basement membrane (GBM)-mediated nephritis in C3- and C4-deficient mice. Clin Exp Immunol. 1997;110(3):403–409.
- Hébert MJ, Takano T, Papayianni A, et al. Acute nephrotoxic serum nephritis in complement knockout mice: relative roles of the classical and alternate pathways in neutrophil recruitment and proteinuria. Nephrol Dial Transplant. 1998;13(11):2799–2803.
- Schrijver G, Bogman MJ, Assmann KJ, et al. Anti-GBM nephritis in the mouse: role of granulocytes in the heterologous phase. Kidney Int. 1990;38(1):86–95.
- Hochheiser K, Engel DR, Hammerich L, et al. Kidney dendritic cells become pathogenic during crescentic glomerulonephritis with proteinuria. J Am Soc Nephrol. 2011;22(2):306–316.
- Tipping PG, Holdsworth SR. T cells in crescentic glomerulonephritis. J Am Soc Nephrol. 2006;17(5):1253–1263.
- Paust HJ, Turner JE, Steinmetz OM, et al. The IL-23/Th17 axis contributes to renal injury in experimental glomerulonephritis. J Am Soc Nephrol. 2009;20(5):969–979.
- Alli AA, Desai D, Elshika A, et al. Kidney tubular epithelial cell ferroptosis links glomerular injury to tubulointerstitial pathology in lupus nephritis. Clin Immunol. 2023;248:109213.
- Soloviova K, Puliaiev M, Foster A, et al. The parent-into-F1 murine model in the study of lupus-like autoimmunity and CD8 cytotoxic T lymphocyte function. Methods Mol Biol. 2012;900:253–270.
- Morris SC, Cohen PL, Eisenberg RA. Experimental induction of systemic lupus erythematosus by recognition of foreign Ia. Clin Immunol Immunopathol. 1990;57(2):263–273.
- Morris SC, Cheek RL, Cohen PL, et al. Allotype-specific immunoregulation of autoantibody production by host B cells in chronic graft-versus host disease. J Immunol. 1990;144(3):916–922.
- Morris SC, Cheek RL, Cohen PL, et al. Autoantibodies in chronic graft versus host result from cognate T-B interactions. J Exp Med. 1990;171(2):503–517.
- Chen F, Maldonado MA, Madaio M, et al. The role of host (endogenous) T cells in chronic graft-versus-host autoimmune disease. J Immunol. 1998;161(11):5880–5885.
- Choudhury A, Cohen PL, Eisenberg RA. B cells require "nurturing" by CD4 T cells during development in order to respond in chronic graft-versus-host model of systemic lupus erythematosus. Clin Immunol. 2010;136(1):105–115.
- Han X, Gu S, Hong SM, et al. Amelioration of autoimmunity in a lupus mouse model by modulation of T-Bet-Promoted energy metabolism in pathogenic age/autoimmune-associated B cells. Arthritis Rheumatol. 2023;75(7):1203–1215.
- Shay T, Jojic V, Zuk O, et al. Conservation and divergence in the transcriptional programs of the human and mouse immune systems. Proc Natl Acad Sci U S A. 2013;110(8):2946–2951.
- Koboziev I, Jones-Hall Y, Valentine JF, et al. Use of humanized mice to study the pathogenesis of autoimmune and inflammatory diseases. Inflamm Bowel Dis. 2015;21(7):1652–1673.
- Landgraf P, Rusu M, Sheridan R, et al. A mammalian microRNA expression atlas based on small RNA library sequencing. Cell. 2007;129(7):1401–1414.
- Noviello TMR, Di Liddo A, Ventola GM, et al. Detection of long non-coding RNA homology, a comparative study on alignment and alignment-free metrics. BMC Bioinf. 2018;19(1):407.
- Chen J, Liao S, Zhou H, et al. Humanized mouse models of systemic lupus erythematosus: opportunities and challenges. Front Immunol. 2021;12:816956.
- Khodoun M, Chimote AA, Ilyas FZ, et al. Targeted knockdown of Kv1.3 channels in T lymphocytes corrects the disease manifestations associated with systemic lupus erythematosus. Sci Adv. 2020;6(47):eabd1471.
- Gunawan M, Her Z, Liu M, et al. A novel human systemic lupus erythematosus model in humanised mice. Sci Rep. 2017;7(1):16642.
- Horton R, Wilming L, Rand V, et al. Gene map of the extended human MHC. Nat Rev Genet. 2004;5(12):889–899.
- Taylor KE, Chung SA, Graham RR, et al. Risk alleles for systemic lupus erythematosus in a large case-control collection and associations with clinical subphenotypes. PLoS Genet. 2011;7(2):e1001311.
- Freedman BI, Langefeld CD, Andringa KK, et al. End-stage renal disease in african americans with lupus nephritis is associated with APOL1. Arthritis Rheumatol. 2014;66(2):390–396.