
Abstract
Immune-mediated demyelinating polyneuropathies (IMDPs) are rare disorders in which dysregulated adaptive immune responses cause peripheral nerve demyelinating inflammation and axonal injury in susceptible individuals. Despite significant advances in understanding IMDP pathogenesis guided by patient data and representative mammalian models, specific therapies are lacking. Significant knowledge gaps in IMDP pathogenesis still exist, e.g. precise antigen(s) and mechanisms that initially trigger immune system activation and identification of large population disease susceptibility factors. The initial directional cues for antigen-specific effector or autoreactive leukocyte trafficking into peripheral nerves are also unknown. An overview of current animal models, with emphasis on the experimental autoimmune neuritis and spontaneous autoimmune peripheral polyneuropathy models, is provided. Insights on the initial directional cues for peripheral nerve tissue specific autoimmunity using a novel Major Histocompatibility Complex class II conditional knockout mouse strain are also discussed, suggesting an essential research tool to study cell- and time-dependent adaptive immunity in autoimmune diseases.
Keywords:
Introduction
Autoimmune disorders affect ∼3–5% of the world’s population [Citation1]. Incidence and prevalence continue to rise [Citation1,Citation2]. Annual direct costs in the United States are conservatively estimated at over $100 billion [Citation2]. Despite significant advances in the pathogenesis, diagnosis and treatment of autoimmune disorders, major knowledge gaps exist. A variety of environmental factors can initiate autoimmunity combined with altered self-tolerance; however the precise contributions of these factors remains unknown. Furthermore, there still remains significant morbidity and mortality associated with autoimmune disorders despite current treatments. There is an urgent need to better understand the pathogenesis of autoimmunity, identify diagnostic, treatment efficacy and prognostic biomarkers, and discover more effective therapies with better patient outcomes [Citation2].
Guillain-Barré syndrome (GBS) is a severe tissue-specific autoimmune disorder that causes varying degrees of muscle weakness, sensory loss, autonomic dysfunction and cardiopulmonary compromise [Citation3]. GBS affects adults in a ∼1.3:1 male-to-female ratio irrespective of race, ethnicity and socioeconomic status, and there is increased prevalence with advancing age [Citation4]. Consistent with other autoimmune disorders, GBS prevalence is increasing worldwide [Citation4]. GBS results in death in 3–5% of patients. Significant morbidity (e.g. residual deficits, chronic neuropathic pain, loss of prior functional capabilities, employment loss), disability, and reduced overall productivity occur in survivors [Citation4]. GBS was estimated to cost the United States $1.7 billion annually in 2004 ($0.2 billion in direct costs and $1.5 billion in indirect costs due to loss of productivity) [Citation5]. This is equivalent to $2.75 billion in 2023 for an estimated GBS US prevalence of 4.2 per 100,000 population [Citation4] (∼$193,000 per patient).
Molecular mimicry is considered essential to GBS pathogenesis, with pathogenic antibodies generated against peripheral axon complex gangliosides or peripheral nerve myelin proteins or lipids [Citation6–8]. Definitive pathogenic antibodies responsible for the most common GBS variant worldwide, acute inflammatory demyelinating polyradiculoneuropathy (AIDP), remain elusive. AIDP is a CD4+ T-helper 1 (Th1)-mediated disorder based on cytokine profiling [Citation9,Citation10]. Major Histocompatibility Complex (MHC) or Human Leukocyte Antigen (HLA) class II susceptibility risk genes have not been definitively identified in GBS [Citation11–14]. Histopathological evaluation of AIDP-variant GBS patient biopsies indicates haematogenous leukocyte trafficking, with monocyte/macrophage, CD4+ and CD8+ T-cell and B-cell/plasma cell infiltration associated with demyelination and axonal degeneration [Citation15–20]. Importantly, electron dense intercellular contacts between endoneurial endothelial cells, indicative of tight junctions, are present during pathogenic leukocyte trafficking in AIDP, supporting the notion that leukocyte trafficking across microvessels is a coordinated, sequential process (i.e. multistep paradigm of leukocyte trafficking) dependent on dynamic leukocyte-endothelial cell interactions in vivo as seen in vitro [Citation19,Citation21–25], rather than passively due to a “leaky” blood-nerve barrier (BNB).
Monocytes/macrophages are the most commonly detected leukocyte subpopulation in AIDP patient peripheral nerves, with ultrastructural examination demonstrating invasion of myelin sheaths of structurally intact axons, hypothesised to be a response to specific antibody-antigen complexes on the myelin surface, on Schwann cells (the supportive glial cells of the peripheral nervous system) or at the node of Ranvier between myelin segments [Citation26–29]. Activated monocytes and tissue-resident macrophages may further enhance the pathogenic immune response, worsen local inflammation and induce axonal injury via cytokine and other pro-inflammatory molecule release. T-cells are the next most prevalent leukocyte subpopulation in peripheral nerve biopsies in AIDP, with CD4+ T-helper cells (polarized to Th1 > Th17 cytokine production) implicated in pathogenesis via endogenous chemokine and other pro-inflammatory cytokine production in peripheral nerves and nerve roots that enhance inflammation and activate macrophages. Definitive evidence of antigen-specific T-cell proliferation and cytokine secretion to myelin proteins/peptides in vitro is lacking [Citation30]. In addition, alterations in the number, but not the function, of circulating CD4+ CD25+ FoxP3+ regulatory T cells have been shown during the acute phase of GBS [Citation31,Citation32].
B-cells are also detected within peripheral nerve endoneurium in AIDP patient nerve biopsies [Citation20,Citation33] implying endogenous secretion of pathogenic polyclonal antibodies or other pro-inflammatory molecules, or local antigen presentation that may directly contribute to demyelination. Most commentaries suggest circulating pathogenic autoantibodies generated by activated B-cells gain access into peripheral nerves and nerve roots and cause demyelination or Schwann cell injury by antibody-dependent cellular cytotoxicity mediated by macrophages or complement fixation and activation resulting in membrane attack complex (C5b-9) [Citation34,Citation35]. In support of this, membrane bound complement components (e.g. complement activation marker, C3d and C5b-9) and immunoglobulin deposition have been described in peripheral nerve biopsies of AIDP patients [Citation16,Citation17,Citation36]. Since demyelination occurs in a multifocal pattern throughout the length of multiple peripheral nerves and nerve roots, the assumption is that these pathogenic autoantibodies readily cross the BNB in affected AIDP patients.
Despite the partial efficacy of antibody-modulating therapies (intravenous immunoglobulins and plasmapheresis), circulating antibodies against myelin glycoproteins, such as myelin protein zero (P0, the most prevalent peripheral nerve myelin protein) and peripheral nerve myelin 22 (PMP22) or the basic peripheral nerve protein P2 are absent or uncommonly detected in AIDP patients [Citation37–39]. Taking into account the variety of different infections associated with GBS and lack of definite evidence of specific autoantibodies against myelin components, an AIDP study hypothesised that primary peripheral nerve/nerve root damage occurs as a consequence of innate immunity-associated local inflammation following neurotropic virus egress, with polyclonal autoantibody production occurring as optional complementary secondary process [Citation40]. Despite advancements in the development of efficacious molecular-based targeted biologic therapies for immune-mediated disorders and cancers, there have been no major advances in GBS treatment for over 30 years [Citation3]. Unfortunately, the therapeutic targets or molecular therapies proposed in animal models have either not been developed or tested in large enough patient cohorts, or failed to translate into effective treatments for AIDP and other GBS variants [Citation41,Citation42].
Chronic inflammatory demyelinating polyradiculoneuropathy (CIDP) is a clinically heterogeneous immune-mediated, inflammatory disorder restricted to peripheral nerves and nerve roots with maximum severity attained >8 weeks following symptom onset. CIDP incidence ranges from 0.15–10.6 cases per 100,000 person-years and prevalence from 0.67–10.3 cases per 100,000 population. CIDP affects men and women in a 1.6:1 ratio, with increasing incidence and prevalence with age [Citation43–47]. CIDP is often under-recognized and may account for about 14% of chronic neurologic disability in adults above the age of 65 [Citation48]. CIDP consists of several clinical variants, including distal predominant and asymmetric forms, and may demonstrate a relapsing-remitting, steady or step-wise progressive course that results in chronic neuropathic pain and long-term disability (related to the degree of associated axonal injury) [Citation45,Citation49–54].
Several diagnostic criteria exist for clinical and research purposes. Consensus statements have been published by consortia of experts to aid clinicians diagnose patients early and institute therapy, as CIDP is a treatable disorder [Citation34,Citation45,Citation54,Citation55]. The criteria used most often in current clinical trials and medical practice were developed by the European Federation of Neurological Societies and the Peripheral Nerve Society (EFNS/PNS) [Citation45,Citation46,Citation56]. Although other diagnostic criteria are highly specific, they lack sensitivity, which may lead to CIDP under-diagnosis. In a study of 151 CIDP patients and 162 control patients, the EFNS/PNS criteria had sensitivity of 81.3% and specificity of 96.2% for definite or probable CIDP. Sensitivity of other diagnostic criteria ranged from 45.7% to 79.5% [Citation46]. Recent European Academy of Neurology (EAN)/PNS diagnostic criteria for CIDP have been published to increase diagnostic accuracy and differentiate CIDP from disorders with primary immune attack against the node of Ranvier (known as paranodopathies and nodopathies), necessary for salutatory conduction of nerve impulses in myelinated axons, and chronic immune sensory polyneuropathies [Citation57]. It is important to recognise that demyelinating features in the electrodiagnostic study criteria are not always present in patients with clinically suspected CIDP (especially with suspected proximal conduction block). Ancillary imaging studies such as ultrasound and magnetic resonance imaging may show nerve and nerve root enlargement to support diagnosing a chronic immune-mediated disorder [Citation45].
Pathologically, CIDP is characterised by infiltration of predominantly monocytes and less commonly CD4+ and CD8+ T- and B-cells into peripheral nerve and nerve root endoneurium, associated with macrophage-mediated demyelination [Citation20,Citation34,Citation54]. This implies an important pathogenic role for activated haematogenous leukocyte infiltration into peripheral nerves and nerve roots across the BNB, although the precise trigger(s) for autoimmune attack and loss of self-tolerance are undetermined [Citation49,Citation58,Citation59]. There is an increase in IFN-γ-producing CD4+ Th1 and Th17, cytotoxic CD8+ and natural killer T-cells in CIDP patient blood and cerebrospinal fluid, suggesting crosstalk between the adaptive and innate immune systems in this disorder [Citation60–63]. B-cells have been implicated in CIDP pathogenesis, either via pathogenic antibody production from multiple clones of activated B-cells or via potent antigen presentation to CD4+ T-cells in secondary lymphoid organs [Citation34,Citation45,Citation54].
Current treatments for CIDP include corticosteroids, immunoglobulins (intravenous and subcutaneous) and plasma exchange based on randomised controlled trials, and immunosuppressant drugs such as Azathioprine, Mycophenolate, Cyclosporine, Cyclophosphamide and Rituximab based on small case series, case reports or specialist physician experience [Citation45–47,Citation64–66]. There is recent evidence to support the efficacy of subcutaneous Efgartigimod-alfa, a human IgG1 fragment that binds to the neonatal Fc receptor (FcRn also known as Fc gamma receptor and transporter) to inhibit the recycling of circulating IgG antibodies by endothelial cells into circulation, promoting their endogenous degradation, combined with recombinant human Hyaluronidase-PH20 to optimise biologic drug uptake, in a large international CIDP patient cohort vs. placebo (https://www.globenewswire.com/news-release/2023/07/17/2705309/0/en/argenx-Reports-Positive-Topline-Data-from-ADHERE-Study-of-VYVGART-Hytrulo-in-Patients-with-Chronic-Inflammatory-Demyelinating-Polyneuropathy.html).
Overall, CIDP prognosis is variable, with rare patients undergoing long term remission without need for immune-modulatory therapy or uncommon patients with unrelenting clinical progression despite currently available therapies [Citation46]. Among the first cohort of 53 CIDP patients reported in 1975, approximately 60% remained ambulatory, 25% became confined to a wheelchair or bed, and 10% died from their disease over a mean follow-up of 7.5 years [Citation67]. A cohort of 38 Japanese CIDP patients who had received immune-modulating therapy demonstrated 26% complete remission lasting at least 2 years without treatment, 61% with partial remission and 13% with severe disability and were unable to walk after 5 years follow-up [Citation68]. In a larger cohort of 106 CIDP patients followed for a mean of 6.4 years, 11% of patients were in remission without treatment for at least 5 years, 44% had active disease that was stabilised with treatment, 7% were improving after recent treatment initiation, and 18% were treatment naïve or treatment refractory with unstable active disease [Citation69]. In a survey of 41 CIDP patients followed for a mean duration of more than 7 years, almost 40% reported need for assistance with activities of daily living [Citation46,Citation70].
Healthcare resource utilisation and costs are substantial for CIDP patients. A 2011 study that analysed insurance claims data for 73 CIDP patients among 6.5 million covered lives in 9 United States commercial health plans showed an annual health plan cost per patient of ∼$57,000 [Citation71]. Pharmacy claims were the primary cost driver, accounting for 57% of health plan costs. Intravenous immunoglobulin accounted for 90% of drug costs, with a mean cost of $108,016 (± $18,437) per patient [Citation46,Citation71]. Although pain is usually considered a secondary CIDP symptom and is commonly not included as a clinical trial outcome measure, 13–17% of CIDP patients report severe neuropathic pain [Citation72], with direct health costs that run into several billion U.S. dollars per annum, excluding losses from reduced economic productivity [Citation73].
Despite significant advances in deducing the molecular pathogenesis of immune-mediated disorders and the development of disease-specific therapies for several autoimmune disorders, specific disease-modifying therapies for CIDP do not currently exist. It is plausible that preventing early leukocyte infiltration could reduce the extent of demyelination and axonal injury and severity of chronic neuropathic pain, resulting in improved patient outcomes in CIDP [Citation20,Citation41,Citation74]. Inflammatory leukocyte migration across microvessels is a sequential, coordinated process involving specific selectins, chemokines and adhesion molecules on endothelial cells, and selectin counterligands, chemokine receptors, integrins and matrix metalloproteases expressed by leukocytes [Citation20,Citation22,Citation24,Citation75,Citation76]. Specific chemokines activate selected integrins for firm leukocyte adhesion. Pathogenic IMDP patient trafficking at the human BNB via the paracellular route has been demonstrated in vitro and in situ [Citation19,Citation77].
Observational studies evaluating CIDP patient nerve biopsies, cerebrospinal fluid, plasma and sera have shown increased expression of specific pro-inflammatory cytokines, chemokines and chemokine receptors, adhesion molecules, matrix metalloproteinases and other inflammatory mediators, such as antibodies against peripheral nerve components [Citation20,Citation43,Citation45,Citation54,Citation78]. A major challenge in comprehensively deducing CIDP pathogenesis and developing specific therapies lies in its clinical heterogeneity and the complexities of the chronic adaptive immune response with possible antigenic tolerance and shift. It is feasible that subsets of CIDP patients with more uniform pathogenic features may response to specific therapies, ushering a personalised, disease-targeted therapy era for CIDP.
However, the initial directional cues that facilitate recently activated haematogenous leukocyte engagement with the human BNB during the early or initiating phases of IMDPs are unknown. Why is autoimmune attack on peripheral nerves and nerve roots a very rare event compared to more common exposure to bacteria, viruses, stress, surgery and trauma that would result in systemic immune activation and possible molecular mimicry? Based on human in vitro microvascular endothelial cell-leukocyte trafficking assays from other tissues, including the blood-brain barrier, it is proposed that recently activated antigen-specific effector memory CD4+ T-cells generated in secondary lymphoid organs are released into circulation to interact with vascular endothelial cell MHC class II molecules that present tissue-specific antigens [Citation79–85].
These vascular endothelial cells express specific co-stimulatory molecules under basal condition that support their roles as semi-professional antigen presenting cells. Endothelial cell MHC class II-presented antigen recognition by activated effector memory CD4+ T-cells would result in a chemokine-dependent or independent transmigration into tissues with subsequent local pro-inflammatory cytokine release that further activates endothelial cells to express additional chemokines and other adhesion molecules, driving further specific haematogenous leukocyte subpopulation transmigration into the tissue [Citation83]. Despite the limitations of in vitro endothelial cell-leukocyte transmigration assays, the dearth of leukocytes from patients with autoimmune neuropathies at disease onset, and the challenges in generating physiologic tissue antigen-specific CD4+ T-cells ex vivo, further evaluation using animal models of IMDPs could provide additional insights on the role of endothelial cells during disease initiation, in addition to providing preclinical models to test therapeutic drug efficacy guided by biologically relevant molecular targets hypothesised from well-characterised patient studies.
Animal models of acute inflammatory demyelinating polyradiculoneuropathy
Experimental autoimmune neuritis (EAN)
The first AIDP animal model was induced by injecting peripheral nervous tissue homogenates emulsified with complete Freund adjuvant (CFA) to susceptible rabbits ∼60 years ago [Citation86]. Subsequent models inject purified peripheral nerve whole myelin or purified myelin proteins or peptides (e.g. P0, P2, PMP22) in susceptible rats, mice, rabbits, guinea pigs and monkeys [Citation74]. Peripheral nervous system myelin significantly differs from central nervous system in protein (glycoproteins vs basic proteins) and lipid composition [Citation87]. The typical course of active immunisation EAN is monophasic, with onset about 7–14 days after disease induction and peak weakness 1–2 weeks later, followed by a plateau phase that lasts <1 week. Increased nociception (mechanical allodynia and thermal hyperalgesia) occurs in this model, associated with peak weakness [Citation88,Citation89], as described in GBS patients. Partial or complete recovery may occur, dependent in animal strain, antigen and disease severity [Citation33,Citation74,Citation90].
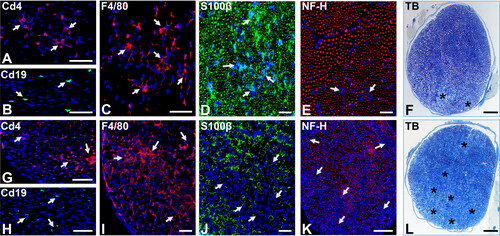
The Lewis rat EAN model is generally considered the most reliable EAN model, but limitations of available reagents compared to the mouse, and known similarities in murine and human neural-immune pathways, has limited its more generalised use to identify and validate AIDP therapeutic targets and test drug efficacy. In the Lewis rat, most CD4 + T-cell-dependent autoimmune responses associated with autoimmune diseases are MHC class II RT1.BL (RT1-Ba)-restricted, homologous to murine H2-Aa and human HLA-DQA1 [Citation91]. Active immunisation EAN onset is associated by perivascular or endoneurial CD4+ T-cell infiltration that precedes the earliest signs of hind limb weakness by 2–3 days [Citation92]. Subsequent haematogenous monocytes, CD4+ and CD8+ T-cells and B-cells infiltration occurs, with associated demyelination and axonal degeneration, as seen in biopsies of severely affected untreated AIDP patients [Citation20,Citation74,Citation93]. Guided by observational data from human AIDP patient biopsies, we demonstrated endogenous expression of presumed pathogenic chemokine ligand-receptor pairs, and ascertained an important role for CCR2 and CD11b (αM-integrin) during the effector phases of acute demyelinating neuritis in the severe murine (sm)-EAN model [Citation19,Citation94–96]. Representative photomicrographs demonstrating early phase and peak severity leukocyte infiltration and demyelination in sm-EAN-affected adult female SJL mice are shown in .
Figure 1. Severe murine experimental autoimmune neuritis in adult female SJL mice. Representative digital photomicrographs of axial cryostat 10 µm and glutaraldehyde-fixed, osmium tetroxide post-fixed, epoxy resin-embedded, toluidine blue-stained 1 µm sciatic nerve sections from sm-EAN-affected female SJL mice 2–3 days after disease onset (A-F) and at peak severity (G-L) are shown. Foci of Cd4+ T-cells (A, white arrows) associated with rare Cd19+ B-cells (B, white arrows) and more diffuse foci of F4/80+ macrophages (C, white arrows) are seen during the early stages of sm-EAN. Infiltrating leukocytes are associated with Schwann cell membranes (D, white arrows) with some focal reduction in axon density (E, white arrows). Foci of demyelination with axonal degeneration occur in association with endoneurial leukocyte infiltration (F, asterisk).
At peak severity, there is further leukocyte infiltration with more diffuse foci of Cd4+ T-cells (G, white arrows), persistent Cd19+ B-cell infiltration (H, white arrows) and increased foci or near confluent infiltration of F4/80+ monocytes/macrophages (I, white arrows). Leukocyte infiltration is associated with foci or near confluent loss of Schwann cell membranes (J, white arrows), consistent with demyelination, as well as reduction in axonal density (K, white arrows), consistent with axonal loss. Multiple foci or near confluent regional demyelination and axonal degeneration (L, asterisk) occurs with endoneurial leukocyte infiltration at peak severity. S100β: marker of myelinating Schwann cell membranes; NF-H: Neurofilament-heavy chain, marker of axon intermediate filaments, TB: toluidine blue. Scale bars: A, B, F, G, H, K, L = 100 µm and C, D, E, I, J = 50 µm.
Adoptive transfer EAN is induced in the Lewis rat following exogenous stimulated T-cells with P0 or P2 protein or peptides, and myelin associated glycoprotein, and in Balb/C mice following stimulation with myelin basic protein, with disease onset typically 3–7 days after injection, peak severity 2–3 days later, followed by a 1–2 day plateau phase and gradual recovery with residual weakness from 18–21 days post-injection [Citation97,Citation98]. A Lewis rat adoptive transfer EAN model using antigen-specific neuritogenic T-cells (P2 peptides 55–78) demonstrated neuritogenic T-cell-selective BNB transmigration and endoneurial entry from day 5 after transfer, with an inability of in vitro stimulated non-antigen-specific T-cells to cross the BNB despite rapid accumulation around peripheral nerves and in peripheral nerve epineurium [Citation98]. This study supports the importance of antigen-specific T-cell trafficking into peripheral nerves as a prerequisite for acute demyelinated neuritis, as implied by a classic active immunisation EAN study [Citation92]. Similar to active immunisation EAN, monocytes/macrophages are the most predominant endoneurial leukocyte, followed by T-cells with a cytokine/chemokine profile consistent with a CD4+ Th1-dependent disorder, as seen in AIDP [Citation74].
129/Sv-C57BL/6 mixed strain transgenic mice encoding the H2-Abb chain covalently bound to Eα chain–derived peptide (Eα52-68) bred into mice lacking H2-Ab, invariant chain, and β2-microglobulin (known as TKO mice) spontaneously developed a peripheral nerve tissue-specific IMDP with histopathological features consistent with EAN, with the earliest onset being 10 weeks of age, with 66% of mice (71% male and 58% female) affected by 20 weeks of age. Disease development was hypothesised to occur secondary to CD4+ T cells autoreactive to the H2-Abb-Eα52-68 complex, owing to its low expression on thymic dendritic cells responsible for tolerance induction. Infiltration of CD4+ αβ T-cell receptor (TCR)+ T-cells and monocytes/macrophages into sciatic nerve endoneurium were observed a week after symptom onset, with leukocyte infiltration independent of increased H2-Abb-Eα52-68 complex expression on BNB-forming endoneurial microvessels [Citation99]. However, it has been established that the murine α-chain (H2-Aa) is responsible for MHC class II antigen-specific binding, with β-chain (H2-Ab) redundancy occurring in order to generate functional MHC class II molecules [Citation100].
BNB-forming endoneurial microvessels actively participate in peripheral nerve immune surveillance by monocytes, B- and T-cells, required to maintain and restore homeostasis in normal physiological states or after inflammation/injury respectively [Citation63,Citation93,Citation101–104]. Increased MHC Class II expression occurs on multiple cell types in IMDP-patient nerve biopsies. These include monocytes/macrophages, Schwann cells, infiltrating T-cells, perineurial cells and BNB-forming endoneurial endothelial cells [Citation105–110]. Human and mouse BNB-forming microvascular endothelial cells possess the molecular machinery to serve as secondary or semi-professional antigen presenting cells during adaptive immune responses [Citation111,Citation112].
In order to determine the role(s) of BNB-forming microvascular endothelial cell MHC class II in peripheral nerve tissue-specific autoimmunity, we developed a Tamoxifen-inducible microvascular endothelial cell-specific conditional MHC class II knockout SJL mouse (generated from heterozygous MHC Class II antigen A-α chain (H2-Aa) floxed [H2-Aaflox/+] C57BL/6 mice [formally designated as C57BL/6-H2-Aatm1c(KOMP)Wtsi/UbeeMmmh] that were backcrossed and intercrossed to the SJL background to generate homozygous H2-Aaflox/flox SJL mice [formally designated as SJL.B6-H2-Aatm1c(KOMP)Wtsi/UbeeMmmh] and bred with hemizygous Tamoxifen-inducible von Willebrand factor [vWF] Cre recombinase (vWF-iCre/+) SJL mice [formally designated as SJL.Cg-Tg(VWF-Cre/ERT2)C1014/UbeeMmmh] to generate H2-Aaflox/flox; vWF-iCre/+ SJL mice in which the MHC class II gene is selectively deleted only in microvascular endothelial cells in a time-dependent manner [Citation113].
Our initial work demonstrated that Tamoxifen-treated H2-Aaflox/flox; vWF-iCre/+ adult female SJL mice with Cre-mediated excision of the H2-Aa gene prior to disease induction were resistant to sm-EAN based on neurobehavioral, electrophysiological and histological parameters despite extensive MHC class II expression in lymphoid and non-lymphoid tissues. Importantly, low basal sciatic nerve endoneurial endothelial cell MHC class II (H2-A) expression was observed in healthy control adult female SJL mice with upregulation observed in sm-EAN affected mice at disease onset prior to diffuse leukocyte infiltration [Citation113], as shown in . These observations imply that BNB-forming microvascular endothelial cell MHC Class II expression of endogenous antigens could be the necessary pre-requisite for antigen-specific peripheral nerve-restricted autoimmunity via interactions with recently activated effector memory CD4+ T-cells generated in response to the same antigen after exposure and systemic immune activation. shows the hypothetical initial effector memory CD4+ Th1 T-cell trafficking at the BNB that may facilitate tissue-specific autoimmunity in AIDP.
Figure 2. Severe murine experimental autoimmune neuritis in adult female H2-Aaflox/flox; vWF-iCre/+ SJL mice. Representative photomicrographs of axial cryostat 10 µm sciatic nerve sections from sm-EAN-affected adult female wildtype SJL mouse 3 days after disease-onset demonstrates increased MHC class II (H2-A) expression in peripheral nerve endoneurium (A) with some co-localization with Cd31+ endothelium (B and C, white arrows). Higher magnification images of an endoneurial microvessel from the same mouse show mononuclear cell diapedesis associated with H2-A membrane expression (D-G). Diffusely increased MHC class II (H2-A) expression is observed in the endoneurium at sm-EAN peak severity in an adult female Tamoxifen-treated H2-Aaflox/flox; +/+ SJL mouse, associated with Cd45+ leukocyte infiltration (H-J, white arrows). In contrast, an adult female Tamoxifen-treated H2-Aaflox/flox; vWF-iCre/+ SJL mice with microvascular endothelial cell specific MHC class II deletion after Cre-mediated recombination does not develop sm-EAN, with rare Cd45+ leukocytes observed outside the endoneurium (K, white arrows). Increased MHC class II expression is observed in the perineurium (p), as well as lymphoid and non-lymphoid organs (not shown), supporting the notion that microvascular endothelial cell MHC class II expression is necessary for tissue- or organ-specific autoimmunity.
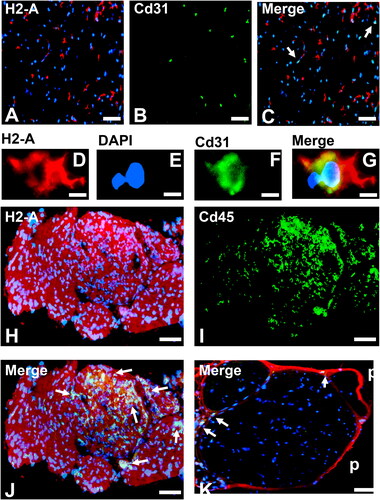
Figure 3. Hypothetical initial antigen-specific effector memory CD4+ Th1 T-cell trafficking at the blood-nerve barrier. Guided by published data from human in vitro leukocyte trafficking assays across microvascular endothelial cells from other organs, and unpublished data from untreated adult AIDP patient single cell transcriptomics compared to healthy age- and sex-matched controls, recently activated antigen-specific effector memory CD4+ Th1 T-cells roll on the endoneurial endothelium luminal surface via interactions between P-Selectin Glycoprotein-1 interacting with P-Selectin, followed haptotaxis driven by chemokines constitutively expressed by endoneurial endothelial cells (e.g. CX3CL1) bound to specific glycosaminoglycan that attract leukocytes expressing the corresponding chemokine receptor (e.g. CX3CR1). Effector memory CD4+ T-cells then engage with endothelial cells expressing endogenous peripheral nerve-derived peptides on the MHC class II α-chain via its TCRαβ complex.
If there is a mismatch, the effector memory CD4+ T-cell disengages from the peripheral nerve microvascular endothelium surface back into circulation. If there is a match, co-stimulation (e.g. via CD40L-CD40 or ICOS-ICOSL interactions) activates the antigen-specific effector CD4+ T-cell with consequential conformational change in leukocyte integrins (e.g. CD11b, CD49d) from inactive to active forms that facilitate firm adhesion to the endothelium by via binding to selective cell adhesion molecules (e.g. ICAM-1, VCAM-1). Effector memory CD4+ T-cells then develop protrusions and crawl to inter-endothelial cell junctions, and undergo diapedesis through homophilic interactions (e.g. CD99) with the endoneurial endothelial cell junctional membrane. Matrix metalloproteinases (e.g. MMP2, MMP9 [not shown]) are secreted from the leading protrusion to break down the endothelium basement membrane to permit effector memory CD4+ T-cell entry into the endoneurium.
![Figure 3. Hypothetical initial antigen-specific effector memory CD4+ Th1 T-cell trafficking at the blood-nerve barrier. Guided by published data from human in vitro leukocyte trafficking assays across microvascular endothelial cells from other organs, and unpublished data from untreated adult AIDP patient single cell transcriptomics compared to healthy age- and sex-matched controls, recently activated antigen-specific effector memory CD4+ Th1 T-cells roll on the endoneurial endothelium luminal surface via interactions between P-Selectin Glycoprotein-1 interacting with P-Selectin, followed haptotaxis driven by chemokines constitutively expressed by endoneurial endothelial cells (e.g. CX3CL1) bound to specific glycosaminoglycan that attract leukocytes expressing the corresponding chemokine receptor (e.g. CX3CR1). Effector memory CD4+ T-cells then engage with endothelial cells expressing endogenous peripheral nerve-derived peptides on the MHC class II α-chain via its TCRαβ complex.If there is a mismatch, the effector memory CD4+ T-cell disengages from the peripheral nerve microvascular endothelium surface back into circulation. If there is a match, co-stimulation (e.g. via CD40L-CD40 or ICOS-ICOSL interactions) activates the antigen-specific effector CD4+ T-cell with consequential conformational change in leukocyte integrins (e.g. CD11b, CD49d) from inactive to active forms that facilitate firm adhesion to the endothelium by via binding to selective cell adhesion molecules (e.g. ICAM-1, VCAM-1). Effector memory CD4+ T-cells then develop protrusions and crawl to inter-endothelial cell junctions, and undergo diapedesis through homophilic interactions (e.g. CD99) with the endoneurial endothelial cell junctional membrane. Matrix metalloproteinases (e.g. MMP2, MMP9 [not shown]) are secreted from the leading protrusion to break down the endothelium basement membrane to permit effector memory CD4+ T-cell entry into the endoneurium.](/cms/asset/70b4a2f9-35fc-4aaa-9ed3-31253948e844/iaut_a_2361745_f0003_c.jpg)
Although EAN recapitulates the onset and kinetics of the monophasic clinical course, electrophysiological abnormalities and histopathologic features of AIDP, a cellular immune response or autoantibody production against a peripheral myelin protein/peptide or lipid component is present in only a minority of patients. Furthermore, aggressive induction protocols that induce systemic immune activation (which uncommonly occurs in patients) are required to induce acute demyelinating neuritis in these models, implying an inherent resistance to tissue-specific autoimmunity in murine peripheral nerves despite putative antigenic exposure or T-cell activation. The translational failure from EAN models to AIDP is well-known, but not unique when compared to other animal models of human disease. It would be important to determine whether biologically relevant pathogenic molecules and pathways deduced from well-characterised AIDP patient nerve specimens also occur in representative EAN models, and test whether therapeutic intervention after disease onset during the early, mid- or late effector phases, or at peak severity alter outcomes to suggest potential suitability for affected patients.
Spatial transcriptomics and proteomics provide an exciting avenue to more comprehensively characterise AIDP and ascertain common pathogenic mechanisms and pathways in affected nerve biopsies. It is also important to determine whether the therapeutic drug acts systemically (e.g. blocks haematogenous pathogenic leukocyte trafficking) or in situ (e.g. inhibits Schwann cell pro-inflammatory cytokine production) as drug design will have to consider BNB permeability and endoneurial retention characteristics.
Animal models of chronic inflammatory demyelinating polyradiculoneuropathy
Chronic relapsing Experimental autoimmune neuritis
Chronic relapsing EAN can be induced in the Dark Agouti rat by active immunisation using recurrent administrations of bovine peripheral nerve myelin emulsified in CFA, resulting in a CIDP-like disorder [Citation114]. Active immunisation of the Lewis rat with S-palmitoylated P0 peptide 180–199 emulsified in CFA induced a chronic progressive or relapsing EAN with accumulation of CD4+ Th17 T-cells and monocytes/macrophages within the sciatic nerve during the late disease phase [Citation115]. A less reliable chronic relapsing EAN model can be induced with peripheral nerve myelin immunisation in the Lewis rat treated with low dose Cyclosporine (hypothesised to modulate interleukin (Il)-2 and −17 homeostasis, with inhibition of regulatory T-cell proliferation, survival and activity). Adoptive transfer of P2-reactive T-cell lines can also induce chronic relapsing EAN in the Lewis rat [Citation74].
Unlike CIDP that is rarely associated with complete recovery, chronic relapsing EAN rats typically improve with less severe disease or return to normal over time, and in most cases, repetitive disease induction is required to maintain the disorder [Citation116]. As with EAN, a peripheral nerve myelin cellular immune response or autoantibody production is lacking in most CIDP patients and the trigger(s) for CIDP disease induction are currently elusive. Over the past 20 years, spontaneous CIDP mouse models have been developed and characterised, providing more reliable, reproducible and reagent-available tools to better understand potential mechanisms of disease induction, pathogenesis and therapeutic efficacy in chronic IMDPs.
Spontaneous autoimmune peripheral polyneuropathy
A naturally occurring inbred mouse strain, the non-obese diabetic (NOD) mouse with MHC Class II H2-Aa g7; H2-Ea null alloantigens (g7 haplotype; in contrast to b with C57BL/6 and s2 in SJL/J mice), has a high propensity towards spontaneous chronic tissue- and organ-specific autoimmunity. These mice were initially observed to develop Type I diabetes, sialadenitis, thyroiditis and gastritis without polyneuropathy [Citation117–119]. Germline knockout of co-stimulatory molecule Cd86 (B7-2) prevented diabetes; however 100% of female mice and ∼30% of male mice developed spontaneous autoimmune peripheral polyneuropathy (SAPP) [Citation117]. This is a tissue-restricted chronic progressive demyelinating neuritis with earliest onset of weakness at 20 weeks of age and peak severity reached between 32–35 weeks of age [Citation120]. SAPP recapitulates the neurobehavioral, electrophysiological and histopathological features of severe chronic progressive CIDP, with infiltration of monocytes/macrophages, T-cells, B-cells and dendritic cells described [Citation117,Citation120]. SAPP in Cd86-/- NOD mice is a CD4+ Th1 interferon (IFN)-γ expressing T cell- and autoantibody dependent disorder reactive to myelin P0, with P0 peptides1–25 and 180–199 identified as pathogenic in this model [Citation117,Citation118,Citation121,Citation122].
NOD mice, with a dominant loss-of-function G228W mutation in the autoimmune regulator (Aire) gene (NOD.AireGW/+) develop SAPP similar to Cd86-/- NOD mice, associated with thymic dysregulation against tissue-specific self-antigens, including P0, during development [Citation63,Citation123]. SAPP severity in this model was shown to worsen with Cd86 knockout or Cd80 and Cd86 blockade, supporting the notion that these co-stimulatory molecules are necessary to antigen-specific regulatory T-cell activation, proliferation and function in NOD mice [Citation124]. NOD.129S4(B6)-Icam1tm1Jcgr/J mice, which express alternative splice isoforms of the adhesion molecule intercellular adhesion molecule-1 (Icam-1) to render them deficient (NOD.Icam-1-/-), develop a CD4+ Th17 T-cell-dependent SAPP without alternation in regulatory T-cell numbers [Citation125]. ICAM-1 has been described as an endothelial cell adhesion molecule important for human CD4+ effector memory T-cell transmigration during tissue-specific autoimmunity (see ), as well as pathogenic human peripheral blood mononuclear leukocyte trafficking across the human in vitro BNB and during the effector phases of peripheral nerve autoimmune responses in animal models in vivo [Citation20,Citation77,Citation93]. However, altered thymic selection against P0 autoreactive T-cells was ascertained to cause SAPP in this mouse strain [Citation125].
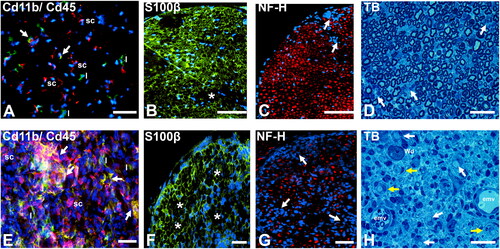
Antibody-depleted Il-2 NOD mice develop SAPP dependent on altered regulatory T-cell homeostasis, exacerbated by further antibody-mediated regulatory T-cell depletion [Citation126]. NOD mice genetically altered at the H2-Aa locus from g7 to b do not develop spontaneous autoimmune diabetes mellitus, but develop SAPP in co-stimulatory molecule PD-1 germline knockouts (NOD.H-2b.PD-1-/-), as well as sialadenitis with gastritis, associated with upregulation of co-stimulatory molecules Ctla-4 and Cd86 in the mouse sciatic nerves [Citation127]. NOD mice deficient in inducible costimulatory molecule Icos and its ligand, Icos-l also spontaneously develop peripheral nervous system autoimmunity, including SAPP and myositis [Citation128]. Guided by observational data from human CIDP patient biopsies, we demonstrated an important role for the alternatively spliced fibronectin variant, fibronectin connecting segment-1 (FNCS-1, a known counterligand for Cd49d, also known as α4-integrin) during the effector phases of chronic demyelinating neuritis in the SAPP model, suggesting that BNB-forming endoneurial endothelial cell FN CS-1 or VCAM-1 interactions with leukocyte CD49d are pathogenic in severe CIDP, supported by in vitro studies [Citation59]. Representative photomicrographs demonstrating early phase and peak severity leukocyte infiltration and demyelination in SAPP-affected Cd86-/- female NOD mice are shown in .
Figure 4. Spontaneous autoimmune peripheral polyneuropathy in adult female Cd86-/- NOD mice. Representative photomicrographs of axial cryostat 10 µm and glutaraldehyde-fixed, osmium tetroxide post-fixed, epoxy resin-embedded, toluidine blue-stained 1 µm sciatic nerve sections from SAPP-affected adult female Cd86-/- NOD mice at 22 weeks (early phase, 2 weeks after symptomatic onset; A-D) and 40 weeks (maximum severity plateau phase, 18 weeks after symptomatic onset, E-H) of age are shown. At 22 weeks of age, there is early infiltration of Cd11b+ (red) Cd45+ (green) leukocytes, consistent with monocytes (white arrows), as well as Cd11b- Cd45+ lymphocytes (l) into the endoneurium, as shown in A. Cd11b + Cd45- cells, consistent with Schwann cells (sc) also observed. Foci of demyelination (asterisk, B) and reduction in axonal density associated mononuclear cell infiltration (white arrows, C) are seen during the early phases of SAPP, supported by epoxy resin-embedded sections showing mononuclear cellular infiltrates (white arrows, D) with normal appearing axons.
At 40 weeks of age, there is more diffuse multi-focal infiltration of Cd11b + CD45+ monocytes/macrophages (white arrows) associated with Schwann cell (sc) proliferation and increased numbers of lymphocytes (l) in the endoneurium, as shown in E . This is associated with multi-focal demyelination (asterisk, F) and reduction in axonal density (white arrows, G). Mononuclear cell infiltration, associated with completely demyelinated axons (white arrows) and demyelinated axons surrounded by concentric layers of Schwann cell membranes (i.e. onion bulbs, yellow arrows), indicative of repeated demyelination-remyelination, is shown in H. Endoneurial microvessels (emv) with perivascular mononuclear cells and an axon undergoing Wallerian degeneration (Wd) are also shown. S100β: marker of myelinating Schwann cell membranes; NF-H: Neurofilament-heavy chain, marker of axon intermediate filaments, TB: toluidine blue. Scale bars: A-C = 100 µm and D-H = 50 µm.
C57BL/6 mice overexpressing Il-10 under a human vitelliform macular dystrophy 2 gene promoter (mutated in macular degeneration) spontaneously developed a FasL-dependent, macrophage-mediated demyelinating polyneuropathy similar to CIDP within 10 to 20 weeks of age, associated with increased Il-10 expression in the eyes, sciatic nerves, spinal cords and brains of symptomatic mice [Citation129]. The relative peripheral nerve susceptibility to autoimmune injury compared to other tissues was undetermined. Sciatic nerve sections from symptomatic mice showed increased expression of Il-12, Ifn-γ, Fasl, and Icam-1. Il-10-mediated Icam-1 upregulation at the BNB was hypothesised to induce haematogenous monocyte transmigration in this model [Citation74,Citation129]; recognising that Il-12 and Ifn-γ would polarise an adaptive immune response towards a CD4+ Th1 phenotype, as seen in sm-EAN [Citation130]. Readers are referred to a published table summarising autoimmune polyneuropathy animal models, including axonal forms not discussed in this review [Citation74].
A common theme with these NOD mouse SAPP models is polyneuropathy associated with germline knockout of co-stimulatory molecules or immune tolerance genes in an autoimmune disease-prone strain. Immune-related adverse events occur in 30–60% of patients treated with immune checkpoint inhibitors for cancers such as melanoma, adenocarcinomas, non-small cell lung cancer and lymphomas. 1–3% of these adverse events are restricted to the nervous system with a predilection for the peripheral nervous system (muscle, neuromuscular junction and peripheral nerves). Fulminant polyneuropathies presenting as AIDP or acute-presentation CIDP, associated with lymphocytic pleocytosis and elevated cerebrospinal fluid protein, may occur [Citation63,Citation131]. Factors that increase differential susceptibility to peripheral nervous system tissue- or organ-specific autoimmunity in the setting of malignancy and modulation of specific co-stimulatory molecules necessary for CD4+ TCR-antigen presenting cell MHC class II activation are currently unknown. It is provocative to speculate that specific HLA-D haplotypes (e.g. HLA-DQA1) may increase risk based on animal model observations.
In CIDP, definitive triggers or putative antigens responsible for inducing peripheral nerve and nerve root autoimmune attack are lacking. Cell- and antibody-mediated responses to myelin proteins such as P0 in CIDP occur in <20% of patients [Citation38,Citation132,Citation133]. Similarly, there is no definitive evidence for deficiencies in molecules required for thymic tolerance of self-antigens or co-stimulation necessary for physiological CD4+ TCR-MHC class II-mediated adaptive immune responses in CIDP patients to date. SAPP is akin to severe forms of progressive CIDP, and not the more common relapsing-remitting disorder with varying degrees of severity. This could preclude effective identification and validation of effective translatable therapeutic targets for CIDP patients. As discussed in AIDP, spatial transcriptomics and proteomics provide an avenue to more comprehensively characterise CIDP and ascertain common pathogenic mechanisms and pathways in affected nerve biopsies to identify “druggable” molecular targets to modulate in SAPP following systemic administration dependent on its cellular location and/or requirement for BNB permeability and drug retention within peripheral nerve and nerve root endoneurium.
A recent single cell transcriptome paper in the NOD.Icam-1-/- mouse provides further insights into SAPP immunopathogenesis [Citation134], recognising that cellular dissociation from tissues may induce transcripts not expressed in situ [Citation111], and relative loss of rare cells (e.g. vascular endothelial cells) may occur. Single cell transcriptomics of untreated, remitted, medically refractory, or chronic progressive CIDP patients could provide information on the molecular changes that may occur during different phases of the disease and aid with clinically useful biomarker identification of disease progression or treatment outcomes.
Conclusions
IMDP animal models have provided significant insights into AIDP and CIDP pathogenesis with respect to regulation of thymic selection, regulation of immune tolerance, immune system activation, generation of pathogenic autoreactive effector leukocytes and autoantibodies, activation, proliferation and function of regulatory leukocytes (particularly T-cells) and immune system deactivation. Early studies using a novel conditional knockout mouse strain suggest that microvascular endothelial cell MHC II expression is the directional cue for recently activated antigen-specific effector memory CD4+ T-cells to engage the microvascular endothelium and subsequently undergo transmigration to facilitate tissue- and organ-specific autoimmune responses. The Tamoxifen-inducible H2-Aaflox/flox; vWF-iCre/+ SJL mouse can undergo backcross to other strains to study antigen presentation by different cells during tissue- or organ-specific adaptive immune responses in a time-dependent manner. This is an advantage over germline knockouts in which adaptive or compensatory gene translation or signalling mechanisms may occur during development. IMDP animal models, despite their inherent limitations, provide tools to test the efficacy of therapeutics which should be designed and guided by human observational data from AIDP and CIDP patient biopsies. Transcriptomics and proteomics provide avenues to discover potentially pathogenic, biologically relevant molecules and signalling pathways in IMDP patients that should further expand the pipeline of disease-specific, molecular targeted drugs with high translational potential in IMDPs, with the ultimate goal of biomarker-driven personalised molecular therapies for these clinically important disabling autoimmune disorders.
Acknowledgements
Special thanks to current and past members and collaborators of the Neuromuscular Immunopathology Research Laboratory (NIRL), University of Alabama at Birmingham (UAB) for insightful discussions and experimental work. The content is solely the responsibility of the author and does not necessarily represent the official views of UAB or NIH.
Disclosure statement
E.E.U. received research support from the National Institute of Neurological Disorders and Stroke/NIH, CSL Behring, Sanofi-Genzyme and Argenx BBVA, and royalties from a non-exclusive commercial licence for a simian virus 40 large T-cell immortalised human endoneurial endothelial cell line (held by Baylor College of Medicine Ventures) and books published by Springer Science + Business Media. He has served as a paid consultant for UCB, Latigo Biotherapeutics, Seismic Therapeutics, and Argenx BBVA, and paid grant reviewer for the Peer Reviewed Medical Research Program and Paul G. Allen Frontiers Group over the past 5 years. C57BL/6 H2-Aa conditional knockout (C57BL/6-H2-Aatm1c(KOMP)Wist/UbeeMmmh; RRID:MMRRC_046053-MU), SJL H2-Aa conditional knockout (SJL.B6-H2-Aatm1c(KOMP)Wtsi/UbeeMmmh; RRID:MMRRC_071292-MU) and Tamoxifen-inducible von Willebrand factor (SJL.Cg-Tg(VWF-Cre/ERT2)C1014/UbeeMmmh; RRID:MMRRC_071293-MU) transgenic mouse strains are commercially available from the University of Missouri MMRRC based on Materials Transfer Agreements/commercial licencing contracts with the UAB Harbert Institute for Innovation and Entrepreneurship (HIEE).
Additional information
Funding
References
- Wang L, Wang FS, Gershwin ME. Human autoimmune diseases: a comprehensive update. J Intern Med. 2015;278(4):1–16.
- Miller FW. The increasing prevalence of autoimmunity and autoimmune diseases: an urgent call to action for improved understanding, diagnosis, treatment, and prevention. Curr Opin Immunol. 2023;80:102266.
- Liu S, Dong C, Ubogu EE. Immunotherapy of Guillain-Barre syndrome. Hum Vaccin Immunother. 2018;14(11):2568–2579.
- Bragazzi NL, Kolahi A-A, Nejadghaderi SA, et al. Global, regional, and national burden of Guillain-Barre syndrome and its underlying causes from 1990 to 2019. J Neuroinflammation. 2021;18(1):264.
- Frenzen PD. Economic cost of Guillain-Barre syndrome in the United States. Neurology. 2008;71(1):21–27.
- van den Berg B, Walgaard C, Drenthen J, et al. Guillain-Barre syndrome: pathogenesis, diagnosis, treatment and prognosis. Nat Rev Neurol. 2014;10(8):469–482.
- Willison HJ, Jacobs BC, van Doorn PA. Guillain-Barre syndrome. Lancet. 2016;388(10045):717–727.
- Hughes RAC, Cornblath DR, Willison HJ. Guillain-Barre syndrome in the 100 years since its description by guillain, barre and strohl. Brain. 2016;139(11):3041–3047.
- Zhang HL, Zheng XY, Zhu J. Th1/Th2/Th17/treg cytokines in Guillain-Barre syndrome and experimental autoimmune neuritis. Cytokine Growth Factor Rev. 2013;24(5):443–453.
- Li S, Jin T, Zhang H-L, et al. Circulating Th17, Th22, and Th1 cells are elevated in the Guillain-Barre syndrome and downregulated by IVIg treatments. Mediators Inflamm. 2014;2014:740947–740910.
- Magira EE, Papaioakim M, Nachamkin I, et al. Differential distribution of HLA-DQ beta/DR beta epitopes in the two forms of Guillain-Barre syndrome, acute motor axonal neuropathy and acute inflammatory demyelinating polyneuropathy (AIDP): identification of DQ beta epitopes associated with susceptibility to and protection from AIDP. J Immunol. 2003;170(6):3074–3080.
- Fekih-Mrissa N, Mrad M, Riahi A, et al. Association of HLA-DR/DQ polymorphisms with Guillain-Barre syndrome in tunisian patients. Clin Neurol Neurosurg. 2014;121:19–22.
- Jin P-P, Sun L-L, Ding B-J, et al. Human leukocyte antigen DQB1 (HLA-DQB1) polymorphisms and the risk for Guillain-Barre syndrome: a systematic review and Meta-Analysis. PLoS One. 2015;10(7):e0131374.
- Hayat S, Jahan I, Das A, et al. Human leukocyte antigen-DQB1 polymorphisms and haplotype patterns in Guillain-Barre syndrome. Ann Clin Transl Neurol. 2019;6(9):1849–1857.
- Hughes R, Atkinson P, Coates P, et al. Sural nerve biopsies in Guillain-Barre syndrome: axonal degeneration and macrophage-associated demyelination and absence of cytomegalovirus genome. Muscle Nerve. 1992;15(5):568–575.
- Nyland H, Matre R, Mørk S. Immunological characterization of sural nerve biopsies from patients with Guillain-Barre syndrome. Ann Neurol. 1981;9 Suppl(S1):80–86.
- Putzu GA, Figarella-Branger D, Bouvier-Labit C, et al. Immunohistochemical localization of cytokines, C5b-9 and ICAM-1 in peripheral nerve of Guillain-Barre syndrome. J Neurol Sci. 2000;174(1):16–21.
- Schmidt B, Toyka KV, Kiefer R, et al. Inflammatory infiltrates in sural nerve biopsies in Guillain-Barre syndrome and chronic inflammatory demyelinating neuropathy. Muscle Nerve. 1996;19(4):474–487.
- Dong C, Palladino SP, Helton ES, et al. The pathogenic relevance of alphaM-integrin in Guillain-Barre syndrome. Acta Neuropathol. 2016;132(5):739–752.
- Ubogu EE. Inflammatory neuropathies: pathology, molecular markers and targets for specific therapeutic intervention. Acta Neuropathol. 2015;130(4):445–468.
- Ley K, Laudanna C, Cybulsky MI, et al. Getting to the site of inflammation: the leukocyte adhesion Cascade updated. Nat Rev Immunol. 2007;7(9):678–689.
- Man S, Ubogu EE, Ransohoff RM. Inflammatory cell migration into the Central nervous system: a few new twists on an old tale. Brain Pathol. 2007;17(2):243–250.
- Muller WA. Leukocyte-endothelial-cell interactions in leukocyte transmigration and the inflammatory response. Trends Immunol. 2003;24(6):327–334.
- Muller WA. Mechanisms of leukocyte transendothelial migration. Annu Rev Pathol. 2011;6(1):323–344.
- Muller WA. How endothelial cells regulate transmigration of leukocytes in the inflammatory response. Am J Pathol. 2014;184(4):886–896.
- Prineas JW. Acute idiopathic polyneuritis. An electron microscope study. Lab Invest. 1972;26(2):133–147.
- Prineas JW. Pathology of the Guillain-Barre syndrome. Ann Neurol. 1981;9 Suppl(S1):6–19.
- Kieseier BC, Kiefer R, Gold R, et al. Advances in understanding and treatment of immune-mediated disorders of the peripheral nervous system. Muscle Nerve. 2004;30(2):131–156.
- Brechenmacher C, Vital C, Deminiere C, et al. Guillain-Barre syndrome: an ultrastructural study of peripheral nerve in 65 patients. Clinical Neuropathology. 1987;6(1):19–24.
- Csurhes PA, Sullivan AA, Green K, et al. T cell reactivity to P0, P2, PMP-22, and myelin basic protein in patients with Guillain-Barre syndrome and chronic inflammatory demyelinating polyradiculoneuropathy. J Neurol Neurosurg Psychiatry. 2005;76(10):1431–1439.
- Chi LJ, Wang HB, Zhang Y, et al. Abnormality of circulating CD4(+)CD25(+) regulatory T cell in patients with Guillain-Barre syndrome. J Neuroimmunol. 2007;192(1-2):206–214.
- Harness J, McCombe PA. Increased levels of activated T-cells and reduced levels of CD4/CD25+ cells in peripheral blood of Guillain-Barre syndrome patients compared to controls. J Clin Neurosci. 2008;15(9):1031–1035.
- Xia RH, Yosef N, Ubogu EE. Clinical, electrophysiological and pathologic correlations in a severe murine experimental autoimmune neuritis model of Guillain-Barre syndrome. J Neuroimmunol. 2010;219(1-2):54–63.
- Dalakas MC. Pathogenesis of immune-mediated neuropathies. Biochim Biophys Acta. 2015;1852(4):658–666.
- Rinaldi S. Update on Guillain-Barre syndrome. J Peripheral Nervous Sys. 2013;18(2):99–112.
- Hafer-Macko CE, Sheikh KA, Li CY, et al. Immune attack on the schwann cell surface in acute inflammatory demyelinating polyneuropathy. Ann Neurol. 1996;39(5):625–635.
- Zweiman B, Rostami A, Lisak RP, et al. Immune reactions to P2 protein in human inflammatory demyelinative neuropathies. Neurology. 1983;33(2):234–237.
- Inglis HR, Csurhes PA, McCombe PA. Antibody responses to peptides of peripheral nerve myelin proteins P0 and P2 in patients with inflammatory demyelinating neuropathy. J Neurol Neurosurg Psychiatry. 2007;78(4):419–422.
- Makowska A, Pritchard J, Sanvito L, et al. Immune responses to myelin proteins in Guillain-Barre syndrome. J Neurol Neurosurg Psychiatry. 2008;79(6):664–671.
- Ziganshin RH, Ivanova OM, Lomakin YA, et al. The pathogenesis of the demyelinating form of Guillain-Barre syndrome (GBS): proteo-peptidomic and immunological profiling of physiological fluids. Mol Cell Proteomics. 2016;15(7):2366–2378.
- Meyer zu Hörste G, Hartung H, Kieseier B. From bench to bedside–experimental rationale for immune-specific therapies in the inflamed peripheral nerve. Nat Clin Pract Neurol. 2007;3(4):198–211.
- Motamed-Gorji N, Matin N, Tabatabaie O, et al. Biological drugs in Guillain-Barre syndrome: an update. Curr Neuropharmacol. 2017;15(7):938–950.
- Dalakas MC, Medscape. Advances in the diagnosis, pathogenesis and treatment of CIDP. Nat Rev Neurol. 2011;7(9):507–517.
- Laughlin RS, Dyck PJ, Melton LJ, 3rd, et al. Incidence and prevalence of CIDP and the association of diabetes mellitus. Neurology. 2009;73(1):39–45.
- Bunschoten C, Jacobs BC, Van den Bergh PYK, et al. Progress in diagnosis and treatment of chronic inflammatory demyelinating polyradiculoneuropathy. Lancet Neurol. 2019;18(8):784–794.
- Ryan M, Ryan SJ. Chronic inflammatory demyelinating polyneuropathy: considerations for diagnosis, management, and population health. Am J Manag Care. 2018;24(17 Suppl):S371–S379.
- Dyck PJB, Tracy JA. History, diagnosis, and management of chronic inflammatory demyelinating polyradiculoneuropathy. Mayo Clin Proc. 2018;93(6):777–793.
- Chia L, Fernandez A, Lacroix C, et al. Contribution of nerve biopsy findings to the diagnosis of disabling neuropathy in the elderly. A retrospective review of 100 consecutive patients. Brain. 1996;119 (Pt 4)(Pt 4):1091–1098.
- Bouchard C, Lacroix C, Planté V, et al. Clinicopathologic findings and prognosis of chronic inflammatory demyelinating polyneuropathy. Neurology. 1999;52(3):498–503.
- Kieseier B, Dalakas M, Hartung H. Immune mechanisms in chronic inflammatory demyelinating neuropathy. Neurology. 2002;59(12 Suppl 6):S7–S12.
- Alabdali M, Abraham A, Alsulaiman A, et al. Clinical characteristics, and impairment and disability scale scores for different CIDP disease activity status classes. J Neurol Sci. 2017;372:223–227.
- Spina E, Topa A, Iodice R, et al. Early predictive factors of disability in CIDP. J Neurol. 2017;264(9):1939–1944.
- Bozovic I, Kacar A, Peric S, et al. Quality of life predictors in patients with chronic inflammatory demyelinating polyradiculoneuropathy. J Neurol. 2017;264(12):2481–2486.
- Mathey EK, Park SB, Hughes RAC, et al. Chronic inflammatory demyelinating polyradiculoneuropathy: from pathology to phenotype. J Neurol Neurosurg Psychiatry. 2015;86(9):973–985.
- EFNS/PNS. European federation of neurological societies/peripheral nerve society guideline on management of chronic inflammatory demyelinating polyradiculoneuropathy: report of a joint task force of the european federation of neurological societies and the peripheral nerve society–first revision. Journal of the Peripheral Nervous System: JPNS. 2010;15(1):1–9.
- Van den Bergh PYK, Hadden RDM, Bouche P, et al. European federation of neurological societies/peripheral nerve society guideline on management of chronic inflammatory demyelinating polyradiculoneuropathy: report of a joint task force of the european federation of neurological societies and the peripheral nerve society - first revision. Eur J Neurol. 2010;17(3):356–363.
- Van den Bergh PYK, van Doorn PA, Hadden RDM, et al. European academy of neurology/peripheral nerve society guideline on diagnosis and treatment of chronic inflammatory demyelinating polyradiculoneuropathy: report of a joint task Force-Second revision. J Peripher Nerv Syst. 2021;26(3):242–268.
- Rizzuto N, Morbin M, Cavallaro T, et al. Focal lesions area feature of chronic inflammatory demyelinating polyneuropathy (CIDP). Acta Neuropathol. 1998;96(6):603–609.
- Dong C, Greathouse KM, Beacham RL, et al. Fibronectin connecting segment-1 peptide inhibits pathogenic leukocyte trafficking and inflammatory demyelination in experimental models of chronic inflammatory demyelinating polyradiculoneuropathy. Exp Neurol. 2017;292:35–45.
- Chi LJ, Xu WH, Zhang ZW, et al. Distribution of Th17 cells and Th1 cells in peripheral blood and cerebrospinal fluid in chronic inflammatory demyelinating polyradiculoneuropathy. J Peripher Nerv Syst. 2010;15(4):345–356.
- Schneider-Hohendorf T, Schwab N, Uceyler N, et al. CD8+ T-cell immunity in chronic inflammatory demyelinating polyradiculoneuropathy. Neurology. 2012;78(6):402–408.
- Heming M, Schulte-Mecklenbeck A, Brix T, et al. Immune cell profiling of the cerebrospinal fluid provides pathogenetic insights into inflammatory neuropathies. Front Immunol. 2019;10:515.
- Wolbert J, Cheng MI, Meyer zu Horste G, et al. Deciphering immune mechanisms in chronic inflammatory demyelinating polyneuropathies. JCI Insight. 2020;5(3):e132411.
- Brannagan TH.3rd. Current treatments of chronic immune-mediated demyelinating polyneuropathies. Muscle Nerve. 2009;39(5):563–578.
- Nobile-Orazio E, Gallia F, Terenghi F, et al. Comparing treatment options for chronic inflammatory neuropathies and choosing the right treatment plan. Expert Rev Neurother. 2017;17(8):755–765.
- Oaklander AL, Lunn MP, Hughes RA, et al. Treatments for chronic inflammatory demyelinating polyradiculoneuropathy (CIDP): an overview of systematic reviews. Cochrane Database Syst Rev. 2017;1(1):CD010369.
- Dyck PJ, Lais AC, Ohta M, et al. Chronic inflammatory polyradiculoneuropathy. Mayo Clin Proc. 1975;50(11):621–637.
- Kuwabara S, Misawa S, Mori M, et al. Long term prognosis of chronic inflammatory demyelinating polyneuropathy: a five year follow up of 38 cases. J Neurol Neurosurg Psychiatry. 2006;77(1):66–70.
- Gorson KC, van Schaik IN, Merkies ISJ, et al. Chronic inflammatory demyelinating polyneuropathy disease activity status: recommendations for clinical research standards and use in clinical practice. J Peripher Nerv Syst. 2010;15(4):326–333.
- Santos PL, Almeida-Ribeiro GA, Silva DM, et al. Chronic inflammatory demyelinating polyneuropathy: quality of life, sociodemographic profile and physical complaints. Arq Neuropsiquiatr. 2014;72(3):179–183.
- Guptill JT, Bromberg MB, Zhu L, et al. Patient demographics and health plan paid costs in chronic inflammatory demyelinating polyneuropathy. Muscle Nerve. 2014;50(1):47–51.
- Merkies IS, Kieseier BC. Fatigue, pain, anxiety and depression in Guillain-Barre syndrome and chronic inflammatory demyelinating polyradiculoneuropathy. Eur Neurol. 2016;75(3-4):199–206.
- McCarberg BH, Billington R. Consequences of neuropathic pain: quality-of-life issues and associated costs. Am J Manag Care. 2006;12(9 Suppl):S263–S8. [pii].
- Schafflick D, Kieseier BC, Wiendl H, et al. Novel pathomechanisms in inflammatory neuropathies. J Neuroinflammation. 2017;14(1):232.
- Yonekawa K, Harlan JM. Targeting leukocyte integrins in human diseases. J Leukoc Biol. 2005;77(2):129–140.
- Simon SI, Green CE. Molecular mechanics and dynamics of leukocyte recruitment during inflammation. Annu Rev Biomed Eng. 2005;7(1):151–185.
- Yosef N, Ubogu EE. Alpha(M)beta(2)-integrin-intercellular adhesion molecule-1 interactions drive the flow-dependent trafficking of Guillain-Barre syndrome patient derived mononuclear leukocytes at the blood-nerve barrier in vitro. J Cell Physiol. 2012;227(12):3857–3875.
- Querol L, Devaux J, Rojas-Garcia R, et al. Autoantibodies in chronic inflammatory neuropathies: diagnostic and therapeutic implications. Nat Rev Neurol. 2017;13(9):533–547.
- Manes TD, Pober JS. Antigen presentation by human microvascular endothelial cells triggers ICAM-1-dependent transendothelial protrusion by, and fractalkine-dependent transendothelial migration of, effector memory CD4+ T cells. J Immunol. 2008;180(12):8386–8392.
- Manes TD, Pober JS. Identification of endothelial cell junctional proteins and lymphocyte receptors involved in transendothelial migration of human effector memory CD4+ T cells. J Immunol. 2011;186(3):1763–1768.
- Manes TD, Shiao SL, Dengler TJ, et al. TCR signaling antagonizes rapid IP-10-mediated transendothelial migration of effector memory CD4+ T cells. J Immunol. 2007;178(5):3237–3243.
- Manes TD, Wang V, Pober JS. Costimulators expressed on human endothelial cells modulate antigen-dependent recruitment of circulating T lymphocytes. Front Immunol. 2022;13:1016361.
- Pober JS, Merola J, Liu R, et al. Antigen presentation by vascular cells. Front Immunol. 2017;8:1907.
- Lopes Pinheiro MA, Kamermans A, Garcia-Vallejo JJ, et al. Internalization and presentation of myelin antigens by the brain endothelium guides antigen-specific T cell migration. Elife. 2016;5:e13149.
- Wheway J, Obeid S, Couraud PO, et al. The brain microvascular endothelium supports T cell proliferation and has potential for alloantigen presentation. PLoS One. 2013;8(1):e52586.
- Waksman BH, Adams RD. Allergic neuritis: an experimental disease of rabbits induced by the injection of peripheral nervous tissue and adjuvants. J Exp Med. 1955;102(2):213–236.
- Garbay B, Heape AM, Sargueil F, et al. Myelin synthesis in the peripheral nervous system. Prog Neurobiol. 2000;61(3):267–304.
- Taha O, Opitz T, Mueller M, et al. Neuropathic pain in experimental autoimmune neuritis is associated with altered electrophysiological properties of nociceptive DRG neurons. Exp Neurol. 2017;297:25–35.
- Renk P, Sgodzai M, Klimas R, et al. Small fibre integrity and axonal pathology in the rat model of experimental autoimmune neuritis. Brain Commun. 2024;6(2):fcae059.
- Koehler NK, Martin R, Wiethölter H. The antibody repertoire in experimental allergic neuritis: evidence for PMP-22 as a novel neuritogen. J Neuroimmunol. 1996;71(1-2):179–189.
- Wauben MH, van der Kraan M, Grosfeld-Stulemeyer MC, et al. Definition of an extended MHC class II-peptide binding motif for the autoimmune disease-associated lewis rat RT1.BL molecule. Int Immunol. 1997;9(2):281–290.
- Aström KE, Webster HD, Arnason BG. The initial lesion in experimental allergic neuritis. A phase and electron microscopic study. J Exp Med. 1968;128(3):469–495.
- Ubogu EE. Biology of the human blood-nerve barrier in health and disease. Exp Neurol. 2020;328:113272.
- Chiang S, Ubogu EE. The role of chemokines in Guillain-Barre syndrome. Muscle Nerve. 2013;48(3):320–330.
- Xia RH, Yosef N, Ubogu EE. Selective expression and cellular localization of pro-inflammatory chemokine ligand/receptor pairs in the sciatic nerves of a severe murine experimental autoimmune neuritis model of Guillain-Barre syndrome. Neuropathol Appl Neurobiol. 2010;36(5):388–398.
- Yuan F, Yosef N, Lakshmana Reddy C, et al. CCR2 gene deletion and pharmacologic blockade ameliorate a severe murine experimental autoimmune neuritis model of Guillain-Barre syndrome. PLoS One. 2014;9(3):e90463.
- Kiefer R, Funa K, Schweitzer T, et al. Transforming growth factor-beta 1 in experimental autoimmune neuritis. Cellular localization and time course. Am J Pathol. 1996;148(1):211–223.
- Mausberg AK, Szepanowski F, Odoardi F, et al. Trapped in the epineurium: early entry into the endoneurium is restricted to neuritogenic T cells in experimental autoimmune neuritis. J Neuroinflammation. 2018;15(1):217.
- Oono T, Fukui Y, Masuko S, et al. Organ-specific autoimmunity in mice whose T cell repertoire is shaped by a single antigenic peptide. J Clin Invest. 2001;108(11):1589–1596.
- Monzón-Casanova E, Rudolf R, Starick L, et al. The forgotten: identification and functional characterization of MHC class II molecules H2-Eb2 and RT1-Db2. J Immunol. 2016;196(3):988–999.
- Olsson Y. Microenvironment of the peripheral nervous system under normal and pathological conditions. Crit Rev Neurobiol. 1990;5(3):265–311.
- Rechthand E, Rapoport SI. Regulation of the microenvironment of peripheral nerve: role of the blood-nerve barrier. Prog Neurobiol. 1987;28(4):303–343.
- Gold R, Archelos JJ, Hartung HP. Mechanisms of immune regulation in the peripheral nervous system. Brain Pathol. 1999;9(2):343–360.
- Kieseier BC, Lehmann HC, Meyer Zu Hörste G. Autoimmune diseases of the peripheral nervous system. Autoimmun Rev. 2012;11(3):191–195.
- Van Rhijn I, Van den Berg LH, Bosboom WM, et al. Expression of accessory molecules for T-cell activation in peripheral nerve of patients with CIDP and vasculitic neuropathy. Brain. 2000;123 (Pt 10)(10):2020–2029.
- Matsumuro K, Izumo S, Umehara F, et al. Chronic inflammatory demyelinating polyneuropathy: histological and immunopathological studies on biopsied sural nerves. J Neurol Sci. 1994;127(2):170–178.
- Mitchell GW, Williams GS, Bosch EP, et al. Class II antigen expression in peripheral neuropathies. J Neurol Sci. 1991;102(2):170–176.
- Meyer Zu Horste G, Heidenreich H, Lehmann HC, et al. Expression of antigen processing and presenting molecules by schwann cells in inflammatory neuropathies. Glia. 2010;58(1):80–92.
- Pollard JD, Baverstock J, McLeod JG. Class II antigen expression and inflammatory cells in the Guillain-Barre syndrome. Ann Neurol. 1987;21(4):337–341.
- Pollard JD, McCombe PA, Baverstock J, et al. Class II antigen expression and T lymphocyte subsets in chronic inflammatory demyelinating polyneuropathy. J Neuroimmunol. 1986;13(2):123–134.
- Palladino SP, Helton ES, Jain P, et al. The human blood-nerve barrier transcriptome. Sci Rep. 2017;7(1):17477.
- Gerber D, Pereira JA, Gerber J, et al. Transcriptional profiling of mouse peripheral nerves to the single-cell level to build a sciatic nerve ATlas (SNAT). Elife. 2021;10:e58591.
- Ubogu EE, Conner JA, Wang Y, et al. Development of a major histocompatibility complex class II conditional knockout mouse to study cell-specific and time-dependent adaptive immune responses in peripheral nerves. bioRxiv. 2023.
- Jung S, Gaupp S, Korn T, et al. Biphasic form of experimental autoimmune neuritis in dark agouti rats and its oral therapy by antigen-specific tolerization. J Neurosci Res. 2004;75(4):524–535.
- Brun S, Beaino W, Kremer L, et al. Characterization of a new rat model for chronic inflammatory demyelinating polyneuropathies. J Neuroimmunol. 2015;278:1–10.
- Lassmann H, Fierz W, Neuchrist C, et al. Chronic relapsing experimental allergic neuritis induced by repeated transfer of P2-protein reactive T cell lines. Brain. 1991;114 (Pt 1B)(1):429–442.
- Salomon B, Rhee L, Bour-Jordan H, et al. Development of spontaneous autoimmune peripheral polyneuropathy in B7-2-deficient NOD mice. J Exp Med. 2001;194(5):677–684.
- Soliven B. Animal models of autoimmune neuropathy. Ilar J. 2014;54(3):282–290.
- Anderson MS, Bluestone JA. The NOD mouse: a model of immune dysregulation. Annu Rev Immunol. 2005;23(1):447–485.
- Ubogu EE, Yosef N, Xia RH, et al. Behavioral, electrophysiological, and histopathological characterization of a severe murine chronic demyelinating polyneuritis model. J Peripher Nerv Syst. 2012;17(1):53–61.
- Louvet C, Kabre BG, Davini DW, et al. A novel myelin P0-specific T cell receptor transgenic mouse develops a fulminant autoimmune peripheral neuropathy. J Exp Med. 2009;206(3):507–514.
- Kim HJ, Jung CG, Jensen MA, et al. Targeting of myelin protein zero in a spontaneous autoimmune polyneuropathy. J Immunol. 2008;181(12):8753–8760.
- Su MA, Davini D, Cheng P, et al. Defective autoimmune regulator-dependent Central tolerance to myelin protein zero is linked to autoimmune peripheral neuropathy. J Immunol. 2012;188(10):4906–4912.
- Zeng XL, Nagavalli A, Smith CJ, et al. Divergent effects of T cell costimulation and inflammatory cytokine production on autoimmune peripheral neuropathy provoked by aire deficiency. J Immunol. 2013;190(8):3895–3904.
- Meyer zu Horste G, Mausberg AK, Cordes S, et al. Thymic epithelium determines a spontaneous chronic neuritis in Icam1(tm1Jcgr)NOD mice. J Immunol. 2014;193(6):2678–2690.
- Setoguchi R, Hori S, Takahashi T, et al. Homeostatic maintenance of natural Foxp3(+) CD25(+) CD4(+) regulatory T cells by interleukin (IL)-2 and induction of autoimmune disease by IL-2 neutralization. J Exp Med. 2005;201(5):723–735.
- Yoshida T, Jiang F, Honjo T, et al. PD-1 deficiency reveals various tissue-specific autoimmunity by H-2b and dose-dependent requirement of H-2g7 for diabetes in NOD mice. Proc Natl Acad Sci U S A. 2008;105(9):3533–3538.
- Prevot N, Briet C, Lassmann H, et al. Abrogation of ICOS/ICOS ligand costimulation in NOD mice results in autoimmune deviation toward the neuromuscular system. Eur J Immunol. 2010;40(8):2267–2276.
- Dace DS, Khan AA, Stark JL, et al. Interleukin-10 overexpression promotes fas-ligand-dependent chronic macrophage-mediated demyelinating polyneuropathy. PLoS One. 2009;4(9):e7121.
- Calida DM, Kremlev SG, Fujioka T, et al. Experimental allergic neuritis in the SJL/J mouse: induction of severe and reproducible disease with bovine peripheral nerve myelin and pertussis toxin with or without interleukin-12. J Neuroimmunol. 2000;107(1):1–7.
- Farina A, Villagrán-García M, Vogrig A, et al. Neurological adverse events of immune checkpoint inhibitors and the development of paraneoplastic neurological syndromes. Lancet Neurol. 2024;23(1):81–94.
- Khalili-Shirazi A, Atkinson P, Gregson N, et al. Antibody responses to P0 and P2 myelin proteins in Guillain-Barre syndrome and chronic idiopathic demyelinating polyradiculoneuropathy. J Neuroimmunol. 1993;46(1-2):245–251.
- Yan WX, Archelos JJ, Hartung HP, et al. P0 protein is a target antigen in chronic inflammatory demyelinating polyradiculoneuropathy. Ann Neurol. 2001;50(3):286–292.
- Wolbert J, Li X, Heming M, et al. Redefining the heterogeneity of peripheral nerve cells in health and autoimmunity. Proc Natl Acad Sci USA. 2020;117(17):9466–9476.