Abstract
Caspases are cysteine aspartases acting either as initiators (caspases 8, 9, and 10) or executioners (caspases 3, 6, and 7) to induce programmed cell death by apoptosis. Parasite infections by certain intracellular protozoans increase host cell life span by targeting caspase activation. Conversely, caspase activation, followed by apoptosis of lymphocytes and other cells, prevents effective immune responses to chronic parasite infection. Here we discuss how pharmacological inhibition of caspases might affect the immunity to protozoan infections, by either blocking or delaying apoptosis.
Introduction
Apoptosis ensues as a death program triggered by detection of an extra or intracellular unfavorable condition, i.e., deprivation of growth factors, DNA damage, or infection, leading to increased caspase and endonuclease activities, cleavage of target substrates, nuclear condensation, DNA fragmentation, and cell shrinkage. The detection of a pathogen within the host cell may result in cell self-destruction or in the elimination of infected cell by the immune system. In co-evolution with their hosts, many pathogens developed strategies to counteract host cell apoptosis and sustain infection. On the other hand, the host immune system displays several cytotoxic pathways to destroy infected cells, some of which can also cause immunopathology. Apoptosis of lymphocytes and removal of apoptotic cells by macrophages terminate harmful immune responses, but might contribute to the establishment of chronic infections ().
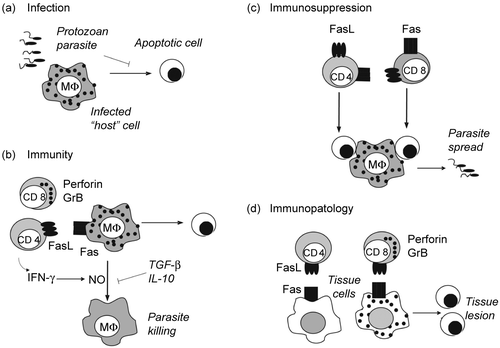
Figure 1. Apoptosis in protozoan infection. (a) Parasite infection. Protozoan parasites invade and multiply within host cells. Inhibition of host cell apoptosis by parasites contributes to the establishment of infection. (b) Cell-mediated immunity. CD8 T cells releasing perforin- and GrB-containing granules, and CD4 T cells expressing FasL induce apoptosis of infected cells. In addition, both CD4 and CD8 T cells secrete IFN-γ and activate macrophages to produce NO, which kills parasites. Cytokines TGF-β and IL-10 antagonize IFN-γ effects on macrophages and favor infection. (c) T-cell apoptosis. Activated CD4 and CD8 T cells express Fas and FasL and undergo activation-induced cell death. Either the disappearance of T cells or the effect of apoptotic cells on macrophages result in defective immune responses. Apoptotic cells induce macrophages to produce TGF-β, which increases parasite replication within infected macrophages. (d) Tissue lesion. CD8 T cells and CD4 T cells infiltrate tissues and kill both infected and non infected cells, causing tissue dysfunction.
Therefore, pharmacological manipulation of caspases and apoptosis may affect parasite-host interplay in several ways. Although we wish to give a broad view of the development in the field, our focus is on the studies designed to envisage the outcomes of blocking or delaying apoptosis in protozoan infections. In addition, by discussing recent advances, we will learn the protozoan strategies used to inhibit caspases and block host cell death.
Mechanisms of caspase activation and inhibition
CASPASES are Cysteinyl Aspartate-Specific Proteases belonging to three major groups: I, the inflammatory caspases 1, 4, 5, 11–14; II, the initiator caspases 2, 8, 9, and 10; III, the effector caspases 3, 6, and 7. Each caspase is expressed as a zymogen, which is cleaved and assembles into an active tetramer protease structure. Inflammatory caspases, which are activated by pathogens through the nuclear oligomerization domain (NOD) signaling pathway, usually induce secretion of pro-inflammatory cytokines, such as interleukin-1 (IL-1) and IL-18 upon proteolysis of cytokine precursors.(Citation1,Citation2) By contrast, the initiator caspases 8 and 9 are recruited to adapter proteins through the death effector domain (DED) or the caspase recruitment domain (CARD) to become proteolytically active and cleave executioner caspases. Executioner or effector caspases 3, 6 or 7 act on cell substrates, such as protein kinase Cδ (PKCδ), actin and laminA, to induce apoptosis. All caspases express a conserved cysteine in the active site and interact with aspartate containing tetrapeptides in their substrates. Peptide inhibitors were designed to fit better the active site of individual caspases and allow some specificity for inhibition, to facilitate entrance into the cells, and for reversible or irreversible inhibitory activity. Please refer to cited reviews for details on caspase activation, caspase structure, and interactions with inhibitory peptides or proteins.(Citation3–6)
There are three major mechanisms that can independently lead to the activation of caspase cascade and apoptosis; the extrinsic or death receptor pathway,(Citation5,Citation7) the intrinsic or mitochondrial pathway,(Citation4,Citation5) and the cytotoxic mechanism induced by perforin and granzymes(Citation8–10) (; please find abbreviations in the Table). The ligation of the trimeric ligands Fas ligand (FasL), tumor necrosis factor-α (TNF-α) or TNF-related apoptosis-inducing ligand (TRAIL) to their receptors assembles the death-inducing signaling complex (DISC) on the cytoplasmic membrane. The DISC is composed by the death domain-containing death receptors Fas, TNF-α receptor I (TNF-RI) or death receptor 5 (DR5), the adapter proteins Fas-associated death domain (FADD) and/or TNF receptor-associated death domain (TRADD), as well as the initiator caspase-8 or caspase-10.
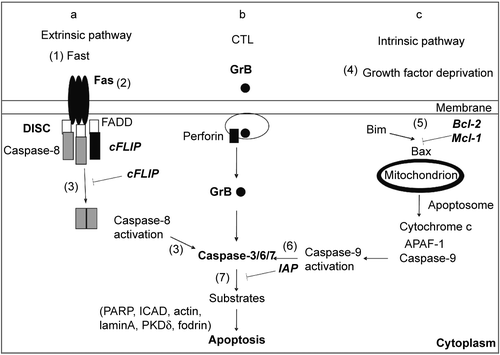
Figure 2. Molecular targets for inhibition of cell death signaling. (a) Extrinsic pathway. FasL induces apoptosis upon interaction with death receptor Fas, followed by recruitment of the adapter protein FADD and caspase-8 to the death inducing signaling complex (DISC). Caspase-8 activation depends on caspase-8 dimerization and is inhibited by heterodimer formation with cFLIP. (b) Cytotoxic pathway. Cytotoxic T cells (CTL) induce apoptosis by releasing granules containing Granzyme B (GrB) and perforin. Perforin promotes release of GrB to the cytoplasm, where GrB activates caspases. (c) Intrinsic pathway. Growth factor deprivation changes the ratio between pro and anti- apoptotic members of Bcl-2 family. Bim activates Bax, promoting the release of cytochrome c from mitochondria. Growth factors induce the expression of antiapoptotic Bcl-2 family members (Bcl-2 and Mcl-1), which sequester Bim and prevent cytochrome c release and apoptosis. Cytochrome c, APAF-1, and caspase-9 form the apoptosome in the cytoplasm, where caspase-9 becomes active. (a–c) The initiator caspases 8 and 9 or GrB cleave and activate effector caspases 3, 6 and 7. Apoptosis is induced by the action of effector caspases on cell substrates. Inhibitor of apoptosis proteins (IAPs) inhibit both caspase-9 and caspase-3. Potential targets for inhibition of apoptosis and caspase signaling were identified by numbers within parentheses (1–7).
In the intrinsic pathway, deprivation of growth factors leads to an imbalance of pro and antiapoptotic protein expression, increased mitochondrial membrane permeability, and cytochrome c release to the cytoplasm. Proapoptotic members of Bcl-2 family (Bim and Bid) activate Bax, promoting mitochondrial outer membrane permeabilization. On the other hand, antiapoptotic proteins (Bcl-2, Bcl-xL, and Mcl-1) sequester and neutralize Bim and Bid, preventing the release of cytochrome c into the cytoplasm. Cytochrome c, the adapter protein apoptotic protease-activating factor-1 (APAF-1), and caspase-9 assemble as the apoptosome, resulting in caspase-9 activation and apoptosis. Both initiator caspase-8 and caspase-9 propagate the apoptotic signaling by cleavage and activation of effector caspases, which act on substrates, such as poly(ADP ribose)polymerase (PARP), fodrin, and inhibitor of caspase-activated DNAse (ICAD). The cleavage of ICAD leads to DNAse activation and DNA fragmentation. Cell death signaling can be blocked by the caspase-8 inhibitor FLICE-like inhibitory protein (cFLIP) and by the Inhibitor of Apoptosis Proteins (IAPs), which inhibit caspase-9 and effector caspase-3.(Citation4,Citation5,Citation7)
Expression of some apoptotic regulatory proteins depends on the activation of the Nuclear Factor-κB (NF-κB) signaling pathway.(Citation11–13) Both apoptotic and antiapoptotic mechanisms are potential targets for pathogens or for therapy ( and Table). We will focus on the mechanisms that either induce or block caspase activation and discuss how they might affect the interactions between protozoan parasites and the host immune system.
Table 1. Pro- and antiapoptotic mechanism.
Induction and inhibition of apoptosis in protozoan host cells
Pathogens express diverse strategies to block or subvert host cell death and perpetuate infection.(Citation14–19) Recent reports show that protozoan parasites target caspases to inhibit apoptosis induced through either extrinsic or intrinsic pathways. Whereas virus genomes codify their own caspase inhibitory proteins,(Citation14,Citation20) such as viral-FLIPs (vFLIPs), protozoan parasites, instead, defray host cell apoptotic machinery(Citation21) or exploit mammalian inhibitory proteins(Citation22) to prevent cell death ().
Theileria parva infection transforms bovine T and B lymphocytes and causes the East Coast Fever. Productive infection in host cells relies on the inhibition of apoptosis, rendering parasitized cells independent of exogenous growth factors to proliferate and survive.(Citation23) Theileria infection protects cells from intrinsic and extrinsic apoptotic pathways. The NF-κB signaling pathway induced by Theileria infection is required to inhibit apoptosis in infected cells.(Citation23) In addition, c-Myc regulates the expression of antiapoptotic Mcl-1 protein in lymphocytes to prevent caspase-9 activation and apoptosis.(Citation24) Signaling pathways involving kinase cascades have also been implicated in the blockade of caspase activation and apoptosis by Theileria infection.(Citation25,Citation26)
Following elimination of the parasite by specific drugs, host T and B cells undergo apoptosis due to activation of caspase-9 and caspase-3, but apoptosis can be inhibited by the general and irreversible caspase inhibitor zVAD.fmk (zVAD).(Citation27) Theileria-infected T cells are also protected from the FasL and caspase-8 signaling, owing to the expression of caspase inhibitory proteins, such as cFLIP and IAPs, which are rapidly lost upon parasite death, rendering host cells susceptible to Fas-induced apoptosis.(Citation28)
Similarly, Trypanosoma cruzi, the agent of Chagas’ disease, reduces activation of caspase-8 and caspase-3, inhibiting apoptosis of host cells triggered by Fas and TNF-α.(Citation22,Citation29) Infection with T. cruzi induces expression of cFLIP and prevents Fas-caspase-8-induced death of host cells.(Citation22) Small interfering RNAs (siRNAs) used to block cFLIP expression restored apoptosis induction in parasite infected cells.(Citation22) Infected cardiomyocytes may also be protected from apoptosis by increased cFLIP expression(Citation22) or by a mechanism that involves activation of NF-κB, Bcl-2 expression, and inhibition of caspase-3 activity.(Citation30) In addition, cruzipain and transialidase, major parasite proteins, prevent cell death by a Bcl-2-dependent mechanisms(Citation31–33) and host heat shock protein 65 (HSP65) antagonizes apoptosis during infection with T. cruzi(Citation34) or Toxoplasma gondii.(Citation35)
Infection with T. gondii, the agent of toxoplasmosis in immunocompromised patients, interferes with apoptosis of host cells inducible by multiple ways,(Citation36,Citation37) suggesting a common downstream target in the signaling cascade. During T. gondii infection, growth factors secreted by infected cells inhibit neutrophil apoptosis, by inducing Mcl-1 expression.(Citation38) In addition, upregulation of the antiapoptotic protein A1 occurs in peritoneal neutrophils and macrophages during T. gondii infection.(Citation39) Delayed neutrophil apoptosis may result in a robust proinflammatory response.(Citation38)
Besides the indirect effects on neutrophils, infection with T. gondii directly inhibits host cell apoptosis propagated through the mitochondrial pathway.(Citation40,Citation41) The balance between pro and antiapoptotic Bcl-2 family members in host cells changes upon T. gondii infection, with downmodulation of Bad and Bax proapoptotic proteins,(Citation40) and upregulation of Mcl-1 protein, which inhibits cytochrome c release, caspase-9 and caspase-3 activation.(Citation41)
The NF-κB pathway, possibly by regulation of pro and antiapoptotic genes, including bcl-2, card, and iap family members, is required for inhibition of caspase activation and apoptosis by T. gondii.(Citation42,Citation43) However, viable T. gondii parasites or parasite lysates directly inhibit caspase-9 activation induced by treating cell extracts with cytochrome c, whereas apoptosis mediated by granzyme B, a component of the cytotoxic mechanism follows unaffected.(Citation44) Therefore, inhibition of apoptosis by parasite products also occurs downstream to the cytochrome release and independent of the expression of Bcl-2 family members.
Whereas T. gondii-infected cells can be destroyed by granule-mediated cytotoxicity,(Citation45) T. gondii renders host cells resistant to the extrinsic apoptotic pathway induced by Fas ligation and caspase-8 activity.(Citation21) Infection with T. gondii induces abnormal proteolysis of caspase-8 by mechanisms involving uncharacterized parasite proteins.(Citation21) Person et al. have recently shown that, rather than blocking FasL-mediated apoptosis of host cells, T. gondii infection induces necrosis upon a sequence of events including Fas ligation, caspase activation, intracellular calcium mobilization, and parasite egress from the host cell.(Citation46) Moreover, the general caspase inhibitor zVAD and caspase-8 inhibitor prevent parasite egress induced by anti-Fas or FasL. It is noteworthy that T. gondii takes advantage of host cell death by perforin or FasL to infect new cells, including the effector T lymphocytes.(Citation46) It remains to be investigated how pharmacological caspase inhibition in vivo will affect this mechanism of parasite escape and propagation in T. gondii infection.
Both Leishmania donovani(Citation47) and Leishmania major,(Citation48) respectively agents of visceral and cutaneous leishmaniasis, inhibit apoptosis of infected macrophages. Macrophages infected with L. donovani or treated with LPG (lipophosphoglycan from Leishmania) do not undergo apoptosis upon growth factor deprivation.(Citation47) L. donovani infection induces secretion of TNF-α, a factor that either inhibits(Citation47) or induces apoptosis. L. major infection also prevents apoptosis in macrophages deprived of growth factors, by blocking cytochrome c-induced caspase activation, but in a manner independent of TNF-α or NF-κB signaling.(Citation48)
By contrast, the extrinsic apoptotic pathway occurs in Leishmania-infected cells. In the presence of interferon-γ (IFN-γ), Fas expression increases in L. major-infected macrophages, which became susceptible to cytotoxic CD4 T cells expressing FasL.(Citation49) IFN-γ and FasL also synergize to enhance L. major killing by macrophages.(Citation50) Likewise, FasL-expressing CD4 T cells play a role in the control of L. donovani infection in the liver.(Citation51)
In human patients with visceral leishmaniasis, an increase in Fas-FasL expression was observed in the spleen, another site of parasite replication, and infection with L. donovani in vitro upregulates Fas expression on macrophages.(Citation52) Altogether, these studies suggest that infected macrophages bearing Fas are potential targets for FasL-expressing CD4 T cells, and that this mechanism contributes for the elimination of parasitized cells. These conclusions are supported by a number of studies in Fas-deficient lpr and FasL deficient gld mice.(Citation49–53) Both gld and lpr mice turn susceptible in the course of infections with L. major(Citation49,Citation50,Citation53) and L. donovani,(Citation51) despite a B6-resistant genetic background(Citation49–51) and pronounced Th1 responses.
Apoptosis of neutrophils might also affect immunity to Leishmania parasites. L. major infection reduces caspase-3 activity and delays spontaneous apoptosis of human neutrophils,(Citation54) which may serve as “trojan horses” to infect macrophages phagocytosing apoptotic cells.(Citation55) Macrophages that have interacted with either infected or uninfected neutrophils from humans or BALB/c mice become a suitable niche for parasite replication.(Citation55–57)
The role of Fas and FasL on neutrophil apoptosis was studied in BALB.gld mice. Interestingly, FasL plays a role in the recruitment of neutrophils upon L. major infection.(Citation58) In addition, FasL induces neutrophil apoptosis in susceptible BALB/c mice. In BALB.gld mice, the absence of FasL postpones neutrophil apoptosis, allowing that macrophage microbicidal activity ensues before apoptotic neutrophils are uptaken by macrophages.(Citation58) Indeed, BALB.gld mice are more resistant than BALB/c mice in the early phases of L. major infection.(Citation58)
The interactions between macrophages and neutrophils lead to different outcomes in susceptible BALB/c versus resistant B6 mice.(Citation56,Citation59) Whereas apoptotic neutrophils from BALB/c mice help parasite replication within alternatively activated macrophages, phagocytosis of neutrophils from B6 mice increases L. major killing by an elastase and TNF-α dependent mechanism.(Citation56,Citation59) These results suggest the existence of background modifiers affecting the clearance of neutrophils and susceptibility to infection in different mouse strains. For more comprehensive reviews, we suggest a complementary literature on Leishmania infection.(Citation60,Citation61)
Apoptosis and immunosuppression
Acute infections with pathogenic virus, bacteria and protozoans drive lymphocytes to proliferate and differentiate into effector immune cells that kill the pathogen or infected cells and undergo apoptosis (). Protozoan parasites, however, cannot be completely eliminated by the immune system, causing chronic infections, and diverse degrees of pathogenesis. High rates of apoptosis may overcome the expansion and differentiation of lymphocytes, causing defective immune responses that allow parasite persistence ().(Citation16,Citation19,Citation62) T cells from hosts infected with T. cruzi,(Citation63–65) T. gondii,(Citation66–68) Plasmodium,(Citation69–71) and Leishmania(Citation72–74) show defective proliferation, cytokine or immune responses, associated with increased T cells apoptosis.
Upon T cell receptor (TCR) stimulation, T cells express Fas ligand and undergo activation-induced cell death (AICD) through the Fas receptor pathway.(Citation75) T cells from T. cruzi-infected mice express Fas and FasL(Citation76–78) and undergo apoptosis upon TCR:CD3-induced activation.(Citation64,Citation78,Citation79) Immunity mediated by antigen-specific B cells is also negatively affected by apoptosis induced by Fas and FasL interactions.(Citation80,Citation81) IFN-γ, by inducing nitric oxide (NO) production and/or by regulating FasL-Fas expression, may be involved in apoptosis related immunosuppression.(Citation65,Citation82) In addition, B cells from T. cruzi-infected mice secrete Galectin-1, which induces apoptosis and reduces IFN-γ production by T cells.(Citation83)
There is a correlation between lymphocyte apoptosis in the host and parasite burden in T. gondii infection, despite a strong proinflammatory IFN-γ or IL-18 response.(Citation68,Citation84,Citation85) IFN-γ- and Fas-dependent T cell apoptosis occurs in the Peyer’s patches in the course of infection with T. gondii.(Citation86) During infection, CD4 and CD8 T cells, B cells, neutrophils, and NK cells express caspase activation, including caspases 8 and 9.(Citation87) Reduced apoptosis in FasL or TNF-RI-deficient mice implicates death receptors in cell loss during T. gondii infection.(Citation87) Apoptosis also increases in CD4 and CD8 T cells during chronic T. gondii infection in NF-κB2 (p100/p52) deficient mice, reducing IFN-γ production.(Citation88) Elimination of T cells expressing higher levels of Fas might account for increased toxoplasmic encephalitis and death in NF-κB2 KO mice.(Citation88)
As discussed below, T cell apoptosis occurs in both human and experimental infections with Leishmania parasites. In human cutaneous leishmaniasis, apoptosis of CD4 and CD8 T cells occurs in active lesions during infection with Leishmania braziliensis.(Citation73) In experimental L. major infection, T cell apoptosis progressively increases in lymphoid organs from susceptible BALB/c mice, whereas an early and transient apoptosis occurs in resistant CBA mice.(Citation89) Apoptosis has been implicated in T cell anergy to Leishmania amazonensis antigens.(Citation74) In BALB/c mice with progressive visceral leishmaniasis due to L. donovani infection, anergy and apoptosis correlate with defective IL-2 production and a reduced Bcl-2/Bax ratio in T cells.(Citation90) In addition, CD4 T cells from BALB/c, but not resistant B6, mice infected with L. donovani undergo apoptosis upon activation.(Citation72) It has been suggested that increased apoptosis account for reduced IL-2 and IFN-γ production by CD4 T cells and defective proliferative responses observed in susceptible BALB/c mice(Citation72) and human patients with acute visceral leishmaniasis.(Citation91) As increased Th1 responses occur in MLR.lpr or B6. gld mice during infection with L. major and L. donovani, it is likely that Fas-FasL-mediated death negatively affects IFN-γ-producing CD4 T cells.(Citation51,Citation53)
Acute human infection with Plasmodium falciparum(Citation69,Citation71) and severe primate infection with Plasmodium coatneyi(Citation70) induce T lymphocytopenia associated with increased T cell apoptosis. In mice, apoptosis occurs in CD4 T cells, CD8 T cells, B cells and macrophages during infection with Plasmodium chabaudi,(Citation92,Citation93) associated with increased expression of Fas and FasL in the spleen.(Citation92) Fas, FasL and perforin might be involved in macrophage apoptosis induced by cytotoxic CD8 T cells.(Citation94) Nonetheless, CD8 T cells, but not Fas, granzyme B or perforin, are required for immunity induced by Plasmodium berguei or Plasmodium yoelii vaccines, suggesting that protection is independent of cytotoxic mechanisms mediated by T cells.(Citation95,Citation96)
Notably, infection with P. berghei deletes parasite specific, but not ova-specific, CD4 T cells,(Citation97–99) indicating that apoptosis during malaria infection compromises the establishment of immunity through vaccination or natural infection. In this model, CD4 T cells produce Th1 cytokines and confer passive immunity independent on T cell help for antibody production.(Citation99) Xu et al. (2002) show that deletion of parasite-specific CD4 T cells depends on IFN-γ, but not Fas or TNF-RI.(Citation99) In agreement with these results, the course of infection is similar in lpr, gld or wild type (B6) mice.(Citation95,Citation100)
Finally, the removal of apoptotic lymphocytes by phagocytes () will further compromise the immune responses, exacerbating infection.(Citation101,Citation102) Interactions between macrophages infected with T. cruzi and apoptotic cells enhance parasite growth.(Citation101,Citation103) The proposed mechanism is the production of tumor growth factor-β (TGF-β), which turns macrophage arginine metabolism to polyamine production instead of parasite killing.(Citation101) TGF-β may also induce regulatory T (Treg) cells and inhibit effector T cells, contributing for immunosuppression.
Positive or negative effects of apoptotic cells on antigen presenting cells (APC) such as dendritic cells (DCs) should also be considered.(Citation102) Whereas P. berguei inhibits caspase activation and apoptosis of hepatocytes,(Citation104)attenuated parasites, which allow hepatocyte apoptosis, enhance immunity, perhaps by facilitating antigen delivery to DCs.(Citation105,Citation106) By contrast, P. falciparum-infected erythrocytes, which mimic apoptotic cells, induce DCs to produce IL-10 and affect DC ability to stimulate T cells.(Citation107) Parasite infection might also directly affect APCs. DCs infected with T. gondii promote T cell apoptosis or unresponsiveness.(Citation108)
General caspase inhibition in vivo
Inhibition of caspases may work as a strategy to stimulate the immune system and treat emerging infectious diseases, where more specific therapies or vaccines are yet to be developed.(Citation109) In a broader sense, the concept might also be applied to counteract apoptosis-related immunosuppression in older infectious diseases, including protozoan infections. It is noteworthy that current treatments for some parasitic diseases do not work in immunosuppressed hosts.(Citation110,Citation111) Moreover, treatment with a currently used drug is followed by the generation of T cell memory in experimental models(Citation112,Citation113) and it is likely that inhibition of apoptosis matters.(Citation112)
Caspases are potential targets for therapeutic intervention in human sepsis, an often fatal bacterial infection.(Citation114) Therefore, experimental sepsis is a prototype model for studying the effects of caspase inhibitors and other strategies to block or delay lymphocyte apoptosis, helping the immune system to fight bacterial infection.(Citation115) In cecum ligation and puncture-induced sepsis, treatments with general caspase inhibitors or transgenic expression of Bcl-2 in lymphocytes improve immunity to bacteria and animal survival.(Citation116) Conversely, injection of apoptotic cells increases bacteremia and mortality, and negatively affects immune responses, such as IFN-γ production.(Citation117)
Similarly, injection of apoptotic cells increases parasitemia in experimental infection with T. cruzi.(Citation101) The experiments with in vivo injection of dead lymphocytes lead to the conclusion that apoptotic cells can directly modulate immune responses, as demonstrated in vitro for T. cruzi infection.(Citation101) T cells from T. cruzi-infected mice show increased levels of active caspase-3 in vitro.(Citation118) Treatment of cells with zVAD prior to culture abrogates the expression of active caspase-3 and both spontaneous and activation-induced cell death. Remarkably, infected mice treated with the general caspase inhibitor zVAD developed higher IFN-γ responses to parasite antigens and reduced parasitemia.(Citation118)
There are several possible explanations for the effects of caspase inhibitors attenuating the immunosuppression induced by apoptosis. First, caspase inhibitor may reduce or delay lymphocyte death. In agreement with this idea, both B and T cells, including effector memory CD8 T cells, accumulate in the spleen and peritoneum of T. cruzi-infected mice upon a 3 wk therapy with zVAD, owing to apoptosis inhibition.(Citation118) In this case, a heightened T lymphocyte response might account for the activation of macrophages, which produce inflammatory cytokines, nitric oxide and kill intracellular parasites.(Citation118) Second, reduction or delay of lymphocyte apoptosis prevents the suppressive effects of apoptotic cells on infected macrophages. This hypothesis is also compatible with the higher activation state of macrophages upon zVAD therapy in infected mice.(Citation118) Third, under the effect of caspase inhibitors, some cells alternatively die by necrosis(Citation119) or autophagic cell death.(Citation120) Necrotic cells may directly activate macrophages and DCs(Citation57,Citation121,Citation122) and potentiate the immune response to infection. Although the general idea of treating immunosuppression by caspase inhibition is a valid concept for both acute and chronic infections, the mechanisms of apoptosis may differ and, therefore, the proposal of specific therapies will require an advance in our current knowledge in each infection model.
Neutralizing the death receptor pathway
The most effective way to prevent apoptosis seems to be the use of reagents that interfere with the interactions between death receptors and their ligands. These apoptosis inhibitors, usually neutralizing antibodies or soluble decoy receptors, bind to the ligands and block the death receptor signaling before it is initiated. Other strategies used to block death receptor signaling in vivo include the use of a soluble Fas fusion protein(Citation123) and caspase-8 or Fas siRNAs,(Citation124) which prevent apoptosis and improve survival in experimental sepsis. In contrast to the studies of death receptor or ligand deficient mice, in vivo treatment works as a temporary inhibition of cell death, avoiding the interference of chronic deficiency phenotypes, which may lead to distorted conclusions. The combination of both strategies, however, is a powerful tool to demonstrate the mechanisms of apoptosis involved during infection.
As an example, infection with the intracellular bacteria Listeria monocytogenes leads to acute elimination of splenic lymphocytes in the first 48 h.(Citation125) Early apoptosis of lymphocytes and other leucocytes decreased upon treatment with soluble DR5 (an antagonist TRAIL ligand) and in TRAIL-deficient mice, which are more resistant to L. monocytogenes infection, with reduced bacterial burden, tissue injure, and mortality.(Citation126) By contrast, Fas plays a role in the control of bacterial burden and late apoptosis in the liver.(Citation127,Citation128)
In vitro experiments lead to the conclusion that both CD4 and CD8 T cells from T. cruzi infection undergo activation-induced cell death mediated by FasL, but not TRAIL or TNF-α.(Citation76,Citation78) In vivo, lymphocyte death follows parasitemia curve and peaks 3 weeks upon T. cruzi infection. Chronic stimulation of T cells occurs in the course of parasite infection and leads to increased susceptibility to Fas-induced apoptosis.(Citation78) Infected mice treated with anti-FasL are more resistant to T. cruzi infection, due to inhibition of apoptosis and increased immune responses.(Citation78)
As an immunomodulatory therapy the most interesting aspect of the inhibition of FasL-Fas interaction is the blockade of AICD followed by proliferation of both CD4(Citation76) and CD8(Citation78) T cells. Like in humans, Fas or FasL deficiency in lpr or gld mice, leads to accumulation of T and B cells in the lymphoid organs.(Citation7) In agreement with this idea, neutralization of FasL increased T(Citation78) and B(Citation80) cell numbers in the spleen and lymph nodes of mice infected with T. cruzi. In addition, T cells accumulated in the peritoneum, where activated macrophages controlled better infection with an increase in nitric oxide production.(Citation78)
A disputable issue is whether Fas-death pathway controls both T helper 1 (Th1) and Th2 cells or preferentially targets CD4 T cells producing Th1 cytokines.(Citation129) In T. cruzi infection, CD8 T cells secrete IFN-γ, whereas CD4 T cells produce both Th1 and Th2 cytokines. In vivo blockade of FasL increased early Th1 and late Th2 cytokine responses to T. cruzi antigens, whereas in vitro experiments using anti-CD3 stimulation show that secretion of Th2 cytokines increases by treating CD4 T cells with anti-FasL.(Citation78) Similarly, both lpr(Citation130) and gld(Citation76) mice express biased Th2 responses and increased susceptibility to T. cruzi infection. Some discrepancies between experiments in gld mice and treatment with anti-FasL might be related to differential kinetics of Th2 responses, as previously discussed.(Citation129)
The inhibition of caspase-8-mediated signaling
Experimental attempts to block caspase-8-induced apoptosis brought a new light on the role of caspases in the immune responses and may have deep implications for the use of therapies based on caspase inhibitors. T-cell conditional caspase-8 knock out(Citation131) or transgenic mice expressing caspase 8 inhibitor vFLIP(Citation132,Citation133) or cFLIP(Citation134) have defective T-cell mediated immunity, similar to caspase-8-deficient human patients.(Citation135) The critical and unexpected finding of these studies is that, in the absence of caspase-8 signaling, T cells are more susceptible to die upon T cell receptor (TCR)-stimulation, affecting IL-2 production and proliferation. A link between the triggering of TCR, caspase-8 activation, and induction of NF-κB signaling pathway was found,(Citation136) but the putative substrate for caspase-8 activity has been missing.(Citation7)
Although CD8 T cells are particularly affected,(Citation132) CD4 T cell-mediated immunity is also compromised.(Citation133,Citation134) CD4 and CD8 T cells from T. cruzi-infected mice express twice as more caspase activity, including caspase-8, compared with T cells from normal mice.(Citation133) Caspase activity increases in stimulated T cell cultures, leading to AICD, which is blocked by the caspase-8 inhibitor zIETD.(Citation133) We found that caspase-8 inhibition differentially affects T cells from normal and infected mice, reducing or increasing the recovery of activated T cells, respectively. IL-2-producing naïve T cells are negatively affected due to reduced NF-κB activation.(Citation133) By contrast, chronic stimulated T cells from T. cruzi-infected mice, mainly those producing IL-4 and IL-10, are rescued from AICD.(Citation133) Therefore, the development of immune responses upon infection might be negatively affected by caspase-8 inhibition. Indeed, transgenic T cells expressing vFLIP do not expand upon infection with T. cruzi, but develop higher late Th2 responses, leading to increased susceptibility to parasite infection.(Citation133) Similar results were found in mice treated with the caspase-8, but not caspase-9, inhibitor, which were more susceptible to T. cruzi infection.(Citation133) Caspase-8 inhibition also affects CD4 and CD8 T cells from mice infected with L. major, reducing IFN-γ production.(Citation137) These results suggest that caspase-8, being at the cross road of multiple signaling pathways,(Citation138) is not a reliable target when the goal is to improve immune responses to parasite infection.
Inhibition of caspase-9 activity
Distinctly of activation-induced cell death that controls ongoing immune responses at high antigen concentration, apoptosis of lymphocytes by lack of co-stimulation or deprivation of growth factors is a regulatory mechanism that contributes either to prevent or to the terminate immune responses. Apoptosis occurs through the intrinsic pathway and requires caspase-9 activation.(Citation12,Citation139) Simultaneous activation of many T cell clones belonging to a TCRVβ family by superantigens also promotes apoptosis through the intrinsic pathway induced by Bim,(Citation140) perhaps due to the high consumption of growth factors by activated T cells. Interestingly, activated T cells undergo Bim-mediated apoptosis in the presence of CD25 (IL-2Rα)-expressing Treg cells, which avidly consume IL-2 through high affinity receptors.(Citation141) However, in the absence of cytokine signaling, Treg cells also undergo apoptosis through the intrinsic death pathway.(Citation142) Reckling et al. investigated how Bim deficiency would affect immunity to L. major in an experimental model where effector T cells are controlled by Treg cells.(Citation143) The authors found improved memory T-cell responses to L. major infection in Bim-deficient mice.(Citation143) These results indicate that Bim-induced apoptosis downmodulates effector, but not regulatory, T cells in L. major infection. Therefore, inhibition of caspase-9 might counteract the suppression by Treg cells and enhance immunity in some parasite infections.
In T. cruzi infection, T cells undergo apoptosis in vitro in the absence of growth factors, but are protected in the presence of IL-2 or IL-4.(Citation64,Citation144) In addition, cell recovery and cytokine production increase in T cell cultures from mesenteric lymph nodes (MLN) treated with the caspase-9 inhibitor zLEHD.(Citation145) Acute atrophy of MLN contrasts with the expansion of other peripheral lymph nodes and the splenomegaly observed during T. cruzi infection.(Citation77) Treatments with either zVAD or the caspase-9 inhibitor prevent the atrophy of MLN during T. cruzi infection.(Citation145) Perhaps the presence of Treg cells contributes to cytokine withdrawn and T cell apoptosis by factor deprivation in MLN during infection.
Caspase activation and the pathogenesis of parasite infection
Several studies have shown caspase activation in tissues affected by protozoan infections. Therefore, therapies based on caspase inhibitors might be useful to dampen the pathogenic effects of apoptosis in parasitic diseases. It should be considered, however, whether inhibition of lymphocyte apoptosis could increase immune responses and damage in target tissues ().
Although in vitro experiments indicate that T. cruzi infection reduces apoptosis in cardiomyocytes, apoptotic leukocytes and myocytes have been observed in the hearts from animals and humans infected with T. cruzi.(Citation146–148) Proteomic and western blot analyses show increased expression and processing of caspase-8 in the hearts of patients with chronic Chagas cardiomyopathy.(Citation149) Caspase-8 might be expressed by myocytes undergoing death or by apoptotic lymphocytes within inflammatory infiltrates, as TCR expression was also detected in the hearts.(Citation149) An issue that remains unclear is how apoptosis or caspase inhibition will affect inflammation and damage in the hearts upon T. cruzi infection. In the experimental model of Chagas’ disease, treatment with a general caspase inhibitor did not affect the extension of inflammatory infiltrates in the hearts, but the death of cardiomyocytes was not assessed.(Citation118)
Interestingly, patients with heart failure have more apoptotic cells within inflammatory infiltrates in the hearts, despite similar inflammation, compared to asymptomatic patients.(Citation150) Moreover, parasite antigen-induced apoptosis, and increased Fas/FasL expression were observed in lymphocytes from patients with heart failure, but not in those from asymptomatic patients.(Citation150) Therefore, T cell apoptosis seems to correlate with parasite escape, leading to chronic infection and heart pathology. These results suggest that the blockade of FasL would increase immune response and reduce both infection and pathology in the hearts.
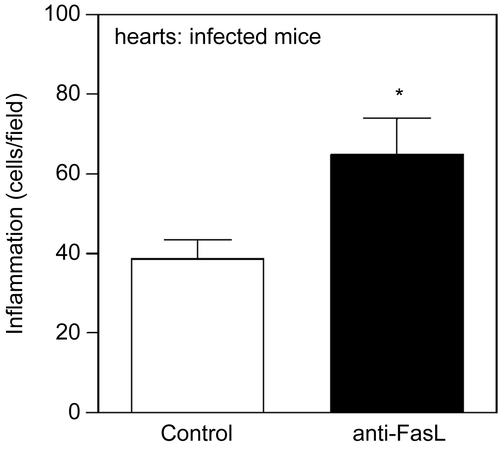
To investigate the role of FasL-induced death in the pathogenesis of Chagas’ disease, we analyzed the hearts of mice infected with T. cruzi and treated with anti-FasL, where we found increased inflammatory infiltrates at the end of acute infection (). Other groups performed experiments in lpr or gld mice.(Citation151,Citation152) Whereas CD8 T cells predominate in the hearts of BALB/c mice, IL-10-producing CD4 and double negative T cells were collected from the hearts of BALB.gld mice infected with T. cruzi.(Citation152) On the other hand, more CD8 T cells were detected in the hearts of perforin deficient mice.(Citation152,Citation153) Therefore, FasL and perforin differentially control the presence of CD4 and CD8 T cells in the hearts during acute infection with T. cruzi,(Citation152) whereas both mechanisms are necessary to prevent animal death during infection with a high virulent T. cruzi strain.(Citation151)
Figure 3. Treatment with anti-FasL increases inflammation in the hearts of T. cruzi- infected mice. BALB/c mice were infected with T. cruzi and received two injection of anti-FasL (0.15 mg/mouse). Control group received either saline or a control hamster IgG antibody.(Citation78) Hearts were collected 25 days post infection and analyzed for inflammatory infiltrates in slides stained with Hematoxilin and eosin. Results were expressed as number of cells/microscopic field. The number of cells/field in the normal mouse heart (6.4) was deducted from the results for each infected mouse. Significant difference is indicated (*) for p< 0.05, n = 4 mice/group.
Fas-FasL regulation also plays a role in the experimental model of eye pathology induced by intraocular Toxoplasma injection. Increased inflammation was observed in both lpr and gld mice.(Citation154) The authors suggest that FasL-induced apoptosis eliminates Fas-expressing T cells infiltrating the eyes, but may also damage ocular tissues.
For L. major infection, TRAIL and FasL expressed on macrophages and T lymphocytes may induce apoptosis in human keratinocytes.(Citation155,Citation156) Neutralization of the ligands TRAIL and FasL might prevent the death of keratinocytes and ameliorate lesions.(Citation155,Citation156) By contrast, injection of soluble FasL helped FasL-deficient B6.gld mice to heal their lesions and control L. major infection, possibly by killing infected macrophages.(Citation49) In visceral leishmaniasis, FasL controls parasite burden in the liver and spleen, and prevents tissue necrosis associated with uncontrolled Th1 response.(Citation51) Therefore, the blockade of Fas-FasL interactions could be deleterious for Leishmania infection. The death receptor TNF-RI might also play a role in the resolution of inflammatory lesions in L. major(Citation157) and T. cruzi(Citation158) infections.
The opportunist parasite Cryptosporidium parvum, which causes diarrhea in humans and animals, induces apoptosis of infected epithelial cells.(Citation159,Citation160) Apoptosis might contribute to the pathogenesis of cholangitis in patients with acquired immunodeficiency syndrome AIDS).(Citation161,Citation162) Both infected and uninfected cells undergo apoptosis induced by soluble or membrane FasL.(Citation161) Neutralization with anti-FasL, as well as the use of general and caspase-8 inhibitors block apoptosis upon C. parvum infection.(Citation159,Citation161,Citation162) Furthermore, in the absence of Fas or FasL, lpr and gld mice are protected from weight loss during infection with Cryptosporidium.(Citation163) Therefore, death receptors and caspase activation are potential therapeutic targets to prevent pathogenic apoptosis in cryptosporidiosis. A caveat of this treatment, however, may be the increase in the number of Cryptosporidium-infected cells in the presence of caspase inhibitor.(Citation159) Moreover, C. parvum infection inhibit apoptosis triggered by strong stimuli,(Citation160) possibly as a mechanism to preserve host cells and perpetuate infection, similar to other intracellular protozoans.(Citation16,Citation164)
Perhaps the best correlation between caspase activation and pathogenesis induced by protozoan infection was demonstrated in experimental malaria. Caspase activation occurs in several tissues upon infection with P. berghei and may be involved in the pathogenesis of cerebral malaria. In the brain, neurons, microglia, and astrocytes express active caspase-3.(Citation100,Citation165) In addition, endothelial cells express caspases early during infection.(Citation165) Endothelial cell apoptosis might cause blood-brain barrier dysfunction.
The interactions between red cells infected with P. falciparum and endothelial cells induce the expression of several proapoptotic genes, such as Fas, FasL, Bad, caspase-3, and inducible NO synthase (iNOS). Both caspase-8 and -9 induce endothelial cell apoptosis, which can be inhibited by zVAD in this model.(Citation166) Fas-FasL-deficient lpr and gld mice recover from cerebral malaria, implicating Fas-induced death in the pathogenesis and lethality of Plasmodium infection.(Citation100,Citation167) The authors suggest that Fas induces caspase-3 activation and apoptosis in astrocytes, contributing to pathogenesis in cerebral malaria.(Citation100)
Fatal outcome of malaria infection can be caused by thrombocytopenia: platelets express caspases 1, 3, 6, 8, and 9, and are deleted upon infection with P. berghei.(Citation168) Treatment with the general caspase inhibitor zVAD improved animal survival, possibly due to reduced thrombocytopenia(Citation168) or to other beneficial effects of blocking caspase activation. Caspase-3 activity also increases in hepatocytes during infection with P. yoelli. Reduced Bcl-2/Bax ratio in the liver and cytochrome c release might be associated with hepatic dysfunction induced by malaria infection.(Citation169) These results suggest that the inhibition of caspases is a promising target for therapy in some, but not all, parasitic diseases, either by improving immune responses or by defraying pathogenic apoptosis. In some cases, however, apoptosis is required for effective immunity (elimination of infected cells) or for the control of deleterious inflammation.
Concluding remarks and perspectives
The therapeutic potential of caspase inhibitors is promising, with the discovery of new drugs, some being already tested by clinical trials.(Citation3–5,Citation114) There are, however, several issues that will require further investigation, including experiments with caspase inhibition in vivo.
A considerable challenge is to analyze the experimental data in order to predict the clinical outcomes of caspase inhibition. The blockade of lymphocyte cell death might improve immune responses, but also increase inflammatory infiltrates in affected tissues. Inhibition of caspases and apoptosis in DCs would promote autoimmunity.(Citation170) In addition, the use of caspase inhibitors could prevent apoptosis of infected cells and favor infection. There are examples of bacteria and fungi infections, where caspase inhibition actually increased microbial burden.(Citation171,Citation172) Special attention should be given to how caspase inhibitors affect the production of cytokines(Citation1,Citation133,Citation171–173) and the mechanisms of cytotoxicity that eliminate infected cells and parasites.(Citation46) The use of cell-directed therapy, with antibody associated to caspase inhibitor, is a possibility to target the desired cell population, i.e., a T cell subset.
Some parasites express features similar to apoptotic mechanisms,(Citation18,Citation174,Citation175) yet, the expression of caspases has not been demonstrated. Nonetheless, caspase inhibitors might directly affect the unknown functions of caspase-related molecules, such as metacaspases, expressed by parasites.(Citation176) Finally, drugs already in clinical use for other goals might also be useful for blocking apoptosis in vivo, such as Benznidazole in experimental Chagas’ disease(Citation112) and protease inhibitors used in human immunodeficiency virus (HIV) infection, which inhibit apoptosis in vitro and in a model for stroke.(Citation177) This will reduce the high costs of new drug development for treatment of neglected diseases caused by protozoan infections.
Acknowledgments
We are grateful to all co-authors that contributed to our discoveries on the role of apoptosis in Chagas’ disease, in special to George A. DosReis. This investigation received financial support from the Rio de Janeiro State Science Foundation (Fundação Carlos Chagas Filho de Amparo à Pesquisa do Estado do Rio de Janeiro, FAPERJ), UNICEF/UNDP/World Bank/WHO Special Program for Research and Training in Tropical Diseases (TDR), and the Brazilian National Research Council (Conselho Nacional de Desenvolvimento Científico e Tecnológico, CNPq). LVCG has a CNPq post doc fellowship and MFL is a CNPq research fellow.
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Petrilli, V.; Dostert, C.; Muruve, D. A.; Tschopp, J. The inflammasome: A danger sensing complex triggering innate immunity. Curr Opin Immunol. 2007, 19 (6), 615–622.
- Muruve, D. A.; Petrilli, V.; Zaiss, A. K.; White, L. R.; Clark, S. A.; Ross, P. J.; Parks, R. J.; Tschopp, J. The inflammasome recognizes cytosolic microbial and host DNA and triggers an innate immune response. Nature. 2008, 452 (7183), 103–107.
- Concha, N. O.; Abdel-Meguid, S. S. Controlling apoptosis by inhibition of caspases. Curr Med Chem. 2002, 9 (6), 713–726.
- Green, D. R.; Kroemer, G. Pharmacological manipulation of cell death: Clinical applications in sight? J Clin Invest. 2005, 115 (10), 2610–2617.
- Lavrik, I. N.; Golks, A.; Krammer, P. H. Caspases:Pharmacological manipulation of cell death. J Clin Invest. 2005, 115 (10), 2665–2672.
- Riedl, S. J.; Shi, Y. Molecular mechanisms of caspase regulation during apoptosis. Nat Rev Mol Cell Biol. 2004, 5 (11), 897–907.
- Bidere, N.; Su, H. C.; Lenardo, M. J. Genetic disorders of programmed cell death in the immune system. Annu Rev Immunol. 2006, 24, 321–352.
- Atkinson, E. A.; Barry, M.; Darmon, A. J.; Shostak, I.; Turner, P. C.; Moyer, R. W.; Bleackley, R. C. Cytotoxic T lymphocyte-assisted suicide. Caspase 3 activation is primarily the result of the direct action of granzyme B. J Biol Chem. 1998, 273 (33), 21261–21266.
- Voskoboinik, I.; Smyth, M. J.; Trapani, J. A. Perforin-mediated target-cell death and immune homeostasis. Nat Rev Immunol. 2006, 6 (12), 940–952.
- Pipkin, M. E.; Lieberman, J. Delivering the kiss of death: Progress on understanding how perforin works. Curr Opin Immunol. 2007, 19 (3), 301–308.
- Wang, C. Y.; Mayo, M. W.; Korneluk, R. G.; Goeddel, D. V.; Baldwin, A. S., Jr. NF-kappaB antiapoptosis: Induction of TRAF1 and TRAF2 and c-IAP1 and c-IAP2 to suppress caspase-8 activation. Science 1998, 281 (5383), 1680–1683.
- Khoshnan, A.; Tindell, C.; Laux, I.; Bae, D.; Bennett, B.; Nel, A. E. The NF-kappa B cascade is important in Bcl-xL expression and for the anti-apoptotic effects of the CD28 receptor in primary human CD4+ lymphocytes. J Immunol. 2000, 165 (4), 1743–1754.
- Micheau, O.; Lens, S.; Gaide, O.; Alevizopoulos, K.; Tschopp, J. NF-kappaB signals induce the expression of c-FLIP. Mol Cell Biol. 2001, 21 (16), 5299–5305.
- Roulston, A.; Marcellus, R. C.; Branton, P. E. Viruses and apoptosis. Annu Rev Microbiol. 1999, 53 577–628.
- Gao, L. Y.; Kwaik, Y. A. The modulation of host cell apoptosis by intracellular bacterial pathogens. Trends Microbiol. 2000, 8 (7), 306–313.
- Luder, C. G.; Gross, U.; Lopes, M. F. Intracellular protozoan parasites and apoptosis: Diverse strategies to modulate parasite-host interactions. Trends Parasitol. 2001, 17 (10), 480–486.
- Heussler, V. T.; Kuenzi, P.; Rottenberg, S. Inhibition of apoptosis by intracellular protozoan parasites. Int J Parasitol. 2001, 31 (11), 1166–1176.
- James, E. R.; Green, D. R. Manipulation of apoptosis in the host-parasite interaction. Trends Parasitol. 2004, 20 (6), 280–287.
- Schaumburg, F.; Hippe, D.; Vutova, P.; Luder, C. G. Pro- and anti-apoptotic activities of protozoan parasites. Parasitology. 2006, 132 (Suppl), S69–85.
- Thome, M.; Schneider, P.; Hofmann, K.; Fickenscher, H.; Meinl, E.; Neipel, F.; Mattmann, C.; Burns, K.; Bodmer, J. L.; Schroter, M.; Scaffidi, C.; Krammer, P. H.; Peter, M. E.; Tschopp, J. Viral FLICE-inhibitory proteins (FLIPs) prevent apoptosis induced by death receptors. Nature. 1997, 386 (6624), 517–521.
- Vutova, P.; Wirth, M.; Hippe, D.; Gross, U.; Schulze-Osthoff, K.; Schmitz, I.; Luder, C. G. Toxoplasma gondii inhibits Fas/CD95-triggered cell death by inducing aberrant processing and degradation of caspase 8. Cell Microbiol. 2007, 9 (6), 1556–1570.
- Hashimoto, M.; Nakajima-Shimada, J.; Aoki, T. Trypanosoma cruzi posttranscriptionally up-regulates and exploits cellular FLIP for inhibition of death-inducing signal. Mol Biol Cell. 2005, 16 (8), 3521–3528.
- Heussler, V. T.; Machado, J., Jr.; Fernandez, P. C.; Botteron, C.; Chen, C. G.; Pearse, M. J.; Dobbelaere, D. A. The intracellular parasite Theileria parva protects infected T cells from apoptosis. Proc Natl Acad Sci U S A. 1999, 96 (13), 7312–7317.
- Dessauge, F.; Hilaly, S.; Baumgartner, M.; Blumen, B.; Werling, D.; Langsley, G. c-Myc activation by Theileria parasites promotes survival of infected B-lymphocytes. Oncogene. 2005, 24 (6), 1075–1083.
- Dessauge, F.; Lizundia, R.; Langsley, G. Constitutively activated CK2 potentially plays a pivotal role in Theileria-induced lymphocyte transformation. Parasitology. 2005, 130 (Suppl), S37–44.
- Lizundia, R.; Sengmanivong, L.; Guergnon, J.; Muller, T.; Schnelle, T.; Langsley, G.; Shorte, S. L. Use of micro-rotation imaging to study JNK-mediated cell survival in Theileria parva-infected B-lymphocytes. Parasitology. 2005, 130 (Pt 6), 629–635.
- Guergnon, J.; Dessauge, F.; Langsley, G.; Garcia, A. Apoptosis of Theileria-infected lymphocytes induced upon parasite death involves activation of caspases 9 and 3. Biochimie. 2003, 85 (8), 771–776.
- Kuenzi, P.; Schneider, P.; Dobbelaere, D. A. Theileria parva-transformed T cells show enhanced resistance to Fas/Fas ligand-induced apoptosis. J Immunol. 2003, 171 (3), 1224–1231.
- Nakajima-Shimada, J.; Zou, C.; Takagi, M.; Umeda, M.; Nara, T.; Aoki, T. Inhibition of Fas-mediated apoptosis by Trypanosoma cruzi infection. Biochim Biophys Acta. 2000, 1475 (2), 175–183.
- Petersen, C. A.; Krumholz, K. A.; Carmen, J.; Sinai, A. P.; Burleigh, B. A. Trypanosoma cruzi infection and nuclear factor kappa B activation prevent apoptosis in cardiac cells. Infect Immun. 2006, 74 (3), 1580–1587.
- Chuenkova, M. V.; Pereira, M. A. A trypanosomal protein synergizes with the cytokines ciliary neurotrophic factor and leukemia inhibitory factor to prevent apoptosis of neuronal cells. Mol Biol Cell. 2000, 11 (4), 1487–1498.
- Chuenkova, M. V.; Furnari, F. B.; Cavenee, W. K.; Pereira, M. A. Trypanosoma cruzi trans-sialidase: A potent and specific survival factor for human Schwann cells by means of phosphatidylinositol 3-kinase/Akt signaling. Proc Natl Acad Sci U S A. 2001, 98 (17), 9936–9941.
- Aoki Mdel, P.; Cano, R. C.; Pellegrini, A. V.; Tanos, T.; Guinazu, N. L.; Coso, O. A.; Gea, S. Different signaling pathways are involved in cardiomyocyte survival induced by a Trypanosoma cruzi glycoprotein. Microbes Infect. 2006, 8 (7), 1723–1731.
- Sakai, T.; Hisaeda, H.; Ishikawa, H.; Maekawa, Y.; Zhang, M.; Nakao, Y.; Takeuchi, T.; Matsumoto, K.; Good, R. A.; Himeno, K. Expression and role of heat-shock protein 65 (HSP65) in macrophages during Trypanosoma cruzi infection: involvement of HSP65 in prevention of apoptosis of macrophages. Microbes Infect. 1999, 1 (6), 419–427.
- Hisaeda, H.; Sakai, T.; Ishikawa, H.; Maekawa, Y.; Yasutomo, K.; Good, R. A.; Himeno, K. Heat shock protein 65 induced by gammadelta T cells prevents apoptosis of macrophages and contributes to host defense in mice infected with Toxoplasma gondii. J Immunol. 1997, 159 (5), 2375–2381.
- Nash, P. B.; Purner, M. B.; Leon, R. P.; Clarke, P.; Duke, R. C.; Curiel, T. J. Toxoplasma gondii-infected cells are resistant to multiple inducers of apoptosis. J Immunol. 1998, 160 (4), 1824–1830.
- Goebel, S.; Luder, C. G.; Gross, U. Invasion by Toxoplasma gondii protects human-derived HL-60 cells from actinomycin D-induced apoptosis. Med Microbiol Immunol. 1999, 187 (4), 221–226.
- Channon, J. Y.; Miselis, K. A.; Minns, L. A.; Dutta, C.; Kasper, L. H. Toxoplasma gondii induces granulocyte colony-stimulating factor and granulocyte-macrophage colony-stimulating factor secretion by human fibroblasts: Implications for neutrophil apoptosis. Infect Immun. 2002, 70 (11), 6048–6057.
- Orlofsky, A.; Somogyi, R. D.; Weiss, L. M.; Prystowsky, M. B. The murine antiapoptotic protein A1 is induced in inflammatory macrophages and constitutively expressed in neutrophils. J Immunol. 1999, 163 (1), 412–419.
- Carmen, J. C.; Hardi, L.; Sinai, A. P. Toxoplasma gondii inhibits ultraviolet light-induced apoptosis through multiple interactions with the mitochondrion-dependent programmed cell death pathway. Cell Microbiol. 2006, 8 (2), 301–315.
- Goebel, S.; Gross, U.; Luder, C. G. Inhibition of host cell apoptosis by Toxoplasma gondii is accompanied by reduced activation of the caspase cascade and alterations of poly(ADP-ribose) polymerase expression. J Cell Sci. 2001, 114 (Pt 19), 3495–3505.
- Payne, T. M.; Molestina, R. E.; Sinai, A. P. Inhibition of caspase activation and a requirement for NF-kappaB function in the Toxoplasma gondii- mediated blockade of host apoptosis. J Cell Sci. 2003, 116 (Pt 21), 4345–4358.
- Molestina, R. E.; Payne, T. M.; Coppens, I.; Sinai, A. P. Activation of NF-kappaB by Toxoplasma gondii correlates with increased expression of antiapoptotic genes and localization of phosphorylated IkappaB to the parasitophorous vacuole membrane. J Cell Sci. 2003, 116 (Pt 21), 4359–4371.
- Keller, P.; Schaumburg, F.; Fischer, S. F.; Hacker, G.; Gross, U.; Luder, C. G. Direct inhibition of cytochrome c-induced caspase activation in vitro by Toxoplasma gondii reveals novel mechanisms of interference with host cell apoptosis. FEMS Microbiol Lett. 2006, 258 (2), 312–319.
- Nakano, Y.; Hisaeda, H.; Sakai, T.; Zhang, M.; Maekawa, Y.; Zhang, T.; Nishitani, M.; Ishikawa, H.; Himeno, K. Granule-dependent killing of Toxoplasma gondii by CD8+ T cells. Immunology. 2001, 104 (3), 289–298.
- Persson, E. K.; Agnarson, A. M.; Lambert, H.; Hitziger, N.; Yagita, H.; Chambers, B. J.; Barragan, A.; Grandien, A. Death receptor ligation or exposure to perforin trigger rapid egress of the intracellular parasite Toxoplasma gondii. J Immunol. 2007, 179 (12), 8357–8365.
- Moore, K. J.; Matlashewski, G. Intracellular infection by Leishmania donovani inhibits macrophage apoptosis. J Immunol. 1994, 152 (6), 2930–2937.
- Akarid, K.; Arnoult, D.; Micic-Polianski, J.; Sif, J.; Estaquier, J.; Ameisen, J. C. Leishmania major-mediated prevention of programmed cell death induction in infected macrophages is associated with the repression of mitochondrial release of cytochrome c. J Leukoc Biol. 2004, 76 (1), 95–103.
- Conceicao-Silva, F.; Hahne, M.; Schroter, M.; Louis, J.; Tschopp, J. The resolution of lesions induced by Leishmania major in mice requires a functional Fas (APO-1, CD95) pathway of cytotoxicity. Eur J Immunol. 1998, 28 (1), 237–245.
- Chakour, R.; Guler, R.; Bugnon, M.; Allenbach, C.; Garcia, I.; Mauel, J.; Louis, J.; Tacchini-Cottier, F. Both the Fas ligand and inducible nitric oxide synthase are needed for control of parasite replication within lesions in mice infected with Leishmania major whereas the contribution of tumor necrosis factor is minimal. Infect Immun. 2003, 71 (9), 5287–5295.
- Alexander, C. E.; Kaye, P. M.; Engwerda, C. R. CD95 is required for the early control of parasite burden in the liver of Leishmania donovani-infected mice. Eur J Immunol. 2001, 31 (4), 1199–1210.
- Eidsmo, L.; Wolday, D.; Berhe, N.; Sabri, F.; Satti, I.; El Hassan, A. M.; Sundar, S.; Chiodi, F.; Akuffo, H. Alteration of Fas and Fas ligand expression during human visceral leishmaniasis. Clin Exp Immunol. 2002, 130 (2), 307–313.
- Huang, F. P.; Xu, D.; Esfandiari, E. O.; Sands, W.; Wei, X. Q.; Liew, F. Y. Mice defective in Fas are highly susceptible to Leishmania major infection despite elevated IL-12 synthesis, strong Th1 responses, and enhanced nitric oxide production. J Immunol. 1998, 160 (9), 4143–4147.
- Aga, E.; Katschinski, D. M.; van Zandbergen, G.; Laufs, H.; Hansen, B.; Muller, K.; Solbach, W.; Laskay, T. Inhibition of the spontaneous apoptosis of neutrophil granulocytes by the intracellular parasite Leishmania major. J Immunol. 2002, 169 (2), 898–905.
- van Zandbergen, G.; Klinger, M.; Mueller, A.; Dannenberg, S.; Gebert, A.; Solbach, W.; Laskay, T. Cutting edge: neutrophil granulocyte serves as a vector for Leishmania entry into macrophages. J Immunol. 2004, 173 (11), 6521–6525.
- Ribeiro-Gomes, F. L.; Otero, A. C.; Gomes, N. A.; Moniz-De-Souza, M. C.; Cysne-Finkelstein, L.; Arnholdt, A. C.; Calich, V. L.; Coutinho, S. G.; Lopes, M. F.; DosReis, G. A. Macrophage interactions with neutrophils regulate Leishmania major infection. J Immunol. 2004, 172 (7), 4454–4462.
- Afonso, L.; Borges, V. M.; Cruz, H.; Ribeiro-Gomes, F. L.; Dosreis, G. A.; Dutra, A. N.; Clarencio, J.; de Oliveira, C. I.; Barral, A.; Barral-Netto, M.; Brodskyn, C. I. Interactions with apoptotic but not with necrotic neutrophils increase parasite burden in human macrophages infected with Leishmania amazonensis. J Leukoc Biol. 2008, 84 (2), –.
- RibeiroGomes, F. L.; Moniz-de-Souza, M. C.; Borges, V. M.; Nunes, M. P.; Mantuano-Barradas, M.; D’Avila, H.; Bozza, P. T.; Calich, V. L.; DosReis, G. A. Turnover of neutrophils mediated by Fas ligand drives Leishmania major infection. J Infect Dis. 2005, 192 (6), 1127–1134.
- Ribeiro-Gomes, F. L.; Moniz-de-Souza, M. C.; Alexandre-Moreira, M. S.; Dias, W. B.; Lopes, M. F.; Nunes, M. P.; Lungarella, G.; DosReis, G. A. Neutrophils activate macrophages for intracellular killing of Leishmania major through recruitment of TLR4 by neutrophil elastase. J Immunol. 2007, 179 (6), 3988–3994.
- Louis, J. A.; Conceicao-Silva, F.; Himmelrich, H.; Tacchini-Cottier, F.; Launois, P. Anti-leishmania effector functions of CD4+ Th1 cells and early events instructing Th2 cell development and susceptibility to Leishmania major in BALB/c mice. Adv Exp Med Biol. 1998, 452 53–60.
- Ribeiro-Gomes, F. L.; Silva, M. T.; Dosreis, G. A. Neutrophils, apoptosis and phagocytic clearance: An innate sequence of cellular responses regulating intramacrophagic parasite infections. Parasitology. 2006, 132 (Suppl), S61–68.
- Gavrilescu, L. C.; Denkers, E. Y. Apoptosis and the balance of homeostatic and pathologic responses to protozoan infection. Infect Immun. 2003, 71 (11), 6109–6115.
- Lopes, M. F.; dos Reis, G. A. Trypanosoma cruzi-induced immunosuppression: blockade of costimulatory T-cell responses in infected hosts due to defective T-cell receptor-CD3 functioning. Infect Immun. 1994, 62 (4), 1484–1488.
- Lopes, M. F.; da Veiga, V. F.; Santos, A. R.; Fonseca, M. E.; DosReis, G. A. Activation-induced CD4+ T cell death by apoptosis in experimental Chagas’ disease. J Immunol. 1995, 154 (2), 744–752.
- Martins, G. A.; Cardoso, M. A.; Aliberti, J. C.; Silva, J. S. Nitric oxide-induced apoptotic cell death in the acute phase of Trypanosoma cruzi infection in mice. Immunol Lett. 1998, 63 (2), 113–120.
- Khan, I. A.; Matsuura, T.; Kasper, L. H. Activation-mediated CD4+ T cell unresponsiveness during acute Toxoplasma gondii infection in mice. Int Immunol. 1996, 8 (6), 887–896.
- Luder, C. G.; Gross, U. Apoptosis and its modulation during infection with Toxoplasma gondii: molecular mechanisms and role in pathogenesis. Curr Top Microbiol Immunol. 2005, 289, 219–237.
- Lee, Y. H.; Channon, J. Y.; Matsuura, T.; Schwartzman, J. D.; Shin, D. W.; Kasper, L. H. Functional and quantitative analysis of splenic T cell immune responses following oral Toxoplasma gondii infection in mice. Exp Parasitol. 1999, 91 (3), 212–221.
- Balde, A. T.; Sarthou, J. L.; Roussilhon, C. Acute Plasmodium falciparum infection is associated with increased percentages of apoptotic cells. Immunol Lett. 1995, 46 (1–2), 59–62.
- Matsumoto, J.; Kawai, S.; Terao, K.; Kirinoki, M.; Yasutomi, Y.; Aikawa, M.; Matsuda, H. Malaria infection induces rapid elevation of the soluble Fas ligand level in serum and subsequent T lymphocytopenia: Possible factors responsible for the differences in susceptibility of two species of Macaca monkeys to Plasmodium coatneyi infection. Infect Immun. 2000, 68 (3), 1183–1188.
- Kemp, K.; Akanmori, B. D.; Adabayeri, V.; Goka, B. Q.; Kurtzhals, J. A.; Behr, C.; Hviid, L. Cytokine production and apoptosis among T cells from patients under treatment for Plasmodium falciparum malaria. Clin Exp Immunol. 2002, 127 (1), 151–157.
- Das, G.; Vohra, H.; Rao, K.; Saha, B.; Mishra, G. C. Leishmania donovani infection of a susceptible host results in CD4+ T-cell apoptosis and decreased Th1 cytokine production. Scand. J Immunol. 1999, 49 (3), 307–310.
- Bertho, A. L.; Santiago, M. A.; Da-Cruz, A. M.; Coutinho, S. G. Detection of early apoptosis and cell death in T CD4+ and CD8+ cells from lesions of patients with localized cutaneous leishmaniasis. Braz J Med Biol Res. 2000, 33 (3), 317–325.
- Pinheiro, R. O.; Pinto, E. F.; Benedito, A. B.; Lopes, U. G.; Rossi-Bergmann, B. The T-cell anergy induced by Leishmania amazonensis antigens is related with defective antigen presentation and apoptosis. An Acad Bras Cienc. 2004, 76 (3), 519–527.
- Budd, R. C. Activation-induced cell death. Curr. Opin. Immunol. 2001, 13 (3), 356–362.
- Lopes, M. F.; Nunes, M. P.; Henriques-Pons, A.; Giese, N.; Morse, H. C., 3rd; Davidson, W. F.; Araujo-Jorge, T. C.; DosReis, G. A. Increased susceptibility of Fas ligand-deficient gld mice to Trypanosoma cruzi infection due to a Th2-biased host immune response. Eur J Immunol. 1999, 29 (1), 81–89.
- de Meis, J.; Mendes-da-Cruz, D. A.; Farias-de-Oliveira, D. A.; Correa-de-Santana, E.; Pinto-Mariz, F.; Cotta-de-Almeida, V.; Bonomo, A.; Savino, W. Atrophy of mesenteric lymph nodes in experimental Chagas’ disease: Differential role of Fas/Fas-L and TNFRI/TNF pathways. Microbes Infect. 2006, 8 (1), 221–231.
- Guillermo, L. V.; Silva, E. M.; Ribeiro-Gomes, F. L.; De Meis, J.; Pereira, W. F.; Yagita, H.; DosReis, G. A.; Lopes, M. F. The Fas death pathway controls coordinated expansions of type 1 CD8 and type 2 CD4 T cells in Trypanosoma cruzi infection. J Leukoc Biol. 2007, 81 (4), 942–951.
- Lopes, M. F.; DosReis, G. A. Trypanosoma cruzi-induced immunosuppression: selective triggering of CD4+ T-cell death by the T-cell receptor-CD3 pathway and not by the CD69 or Ly-6 activation pathway. Infect Immun. 1996, 64 (5), 1559–1564.
- Zuniga, E.; Motran, C. C.; Montes, C. L.; Yagita, H.; Gruppi, A. Trypanosoma cruzi infection selectively renders parasite-specific IgG+ B lymphocytes susceptible to Fas/Fas ligand-mediated fratricide. J Immunol. 2002, 168 (8), 3965–3973.
- Acosta Rodriguez, E. V.; Zuniga, E. I.; Montes, C. L.; Merino, M. C.; Bermejo, D. A.; Amezcua Vesely, M. C.; Motran, C. C.; Gruppi, A. Trypanosoma cruzi infection beats the B-cell compartment favouring parasite establishment: can we strike first? Scand. J Immunol. 2007, 66 (2–3), 137–142.
- Martins, G. A.; Vieira, L. Q.; Cunha, F. Q.; Silva, J. S. Gamma interferon modulates CD95 (Fas) and CD95 ligand (Fas-L) expression and nitric oxide-induced apoptosis during the acute phase of Trypanosoma cruzi infection: A possible role in immune response control. Infect Immun. 1999, 67 (8), 3864–3871.
- Zuniga, E.; Rabinovich, G. A.; Iglesias, M. M.; Gruppi, A. Regulated expression of galectin-1 during B-cell activation and implications for T-cell apoptosis. J Leukoc Biol. 2001, 70 (1), 73–79.
- Mordue, D. G.; Monroy, F.; La Regina, M.; Dinarello, C. A.; Sibley, L. D. Acute toxoplasmosis leads to lethal overproduction of Th1 cytokines. J Immunol. 2001, 167 (8), 4574–4584.
- Gavrilescu, L. C.; Denkers, E. Y. IFN-gamma overproduction and high level apoptosis are associated with high but not low virulence Toxoplasma gondii infection. J Immunol. 2001, 167 (2), 902–909.
- Liesenfeld, O.; Kosek, J. C.; Suzuki, Y. Gamma interferon induces Fas-dependent apoptosis of Peyer’s patch T cells in mice following peroral infection with Toxoplasma gondii. Infect Immun. 1997, 65 (11), 4682–4689.
- Gavrilescu, L. C.; Denkers, E. Y. Interleukin-12 p40- and Fas ligand-dependent apoptotic pathways involving STAT-1 phosphorylation are triggered during infection with a virulent strain of Toxoplasma gondii. Infect Immun. 2003, 71 (5), 2577–2583.
- Caamano, J.; Tato, C.; Cai, G.; Villegas, E. N.; Speirs, K.; Craig, L.; Alexander, J.; Hunter, C. A. Identification of a role for NF-kappa B2 in the regulation of apoptosis and in maintenance of T cell-mediated immunity to Toxoplasma gondii. J Immunol. 2000, 165 (10), 5720–5728.
- Desbarats, J.; Stone, J. E.; Lin, L.; Zakeri, Z. F.; Davis, G. S.; Pfeiffer, L. M.; Titus, R. G.; Newell, M. K. Rapid early onset lymphocyte cell death in mice resistant, but not susceptible to Leishmania major infection. Apoptosis. 2000, 5 (2), 189–196.
- Mukherjee, P.; Sen, P. C.; Ghose, A. C. Lymph node cells from BALB/c mice with chronic visceral leishmaniasis exhibiting cellular anergy and apoptosis: Involvement of Ser/Thr phosphatase. Apoptosis. 2006, 11 (11), 2013–2029.
- Potestio, M.; D’Agostino, P.; Romano, G. C.; Milano, S.; Ferlazzo, V.; Aquino, A.; Di Bella, G.; Caruso, R.; Gambino, G.; Vitale, G.; Mansueto, S.; Cillari, E. CD4+ CCR5+ and CD4+ CCR3+ lymphocyte subset and monocyte apoptosis in patients with acute visceral leishmaniasis. Immunology. 2004, 113 (2), 260–268.
- Helmby, H.; Jonsson, G.; Troye-Blomberg, M. Cellular changes and apoptosis in the spleens and peripheral blood of mice infected with blood-stage Plasmodium chabaudi chabaudi AS. Infect Immun. 2000, 68 (3), 1485–1490.
- Sanchez-Torres, L.; Rodriguez-Ropon, A.; Aguilar-Medina, M.; Favila-Castillo, L. Mouse splenic CD4+ and CD8+ T cells undergo extensive apoptosis during a Plasmodium chabaudi chabaudi AS infection. Parasite Immunol. 2001, 23 (12), 617–626.
- Beattie, L.; Engwerda, C. R.; Wykes, M.; Good, M. F. CD8+ T lymphocyte-mediated loss of marginal metallophilic macrophages following infection with Plasmodium chabaudi chabaudi. AS J Immunol. 2006, 177 (4), 2518–2526.
- Renggli, J.; Hahne, M.; Matile, H.; Betschart, B.; Tschopp, J.; Corradin, G. Elimination of P. berghei liver stages is independent of Fas (CD95/Apo-I) or perforin-mediated cytotoxicity. Parasite Immunol. 1997, 19 (3), 145–148.
- Doolan, D. L.; Hoffman, S. L. The complexity of protective immunity against liver-stage malaria. J Immunol. 2000, 165 (3), 1453–1462.
- Hirunpetcharat, C.; Good, M. F. Deletion of Plasmodium berghei-specific CD4+ T cells adoptively transferred into recipient mice after challenge with homologous parasite. Proc Natl Acad Sci USA. 1998, 95 (4), 1715–1720.
- Wipasa, J.; Xu, H.; Stowers, A.; Good, M. F. Apoptotic deletion of Th cells specific for the 19-kDa carboxyl-terminal fragment of merozoite surface protein 1 during malaria infection. J Immunol. 2001, 167 (7), 3903–3909.
- Xu, H.; Wipasa, J.; Yan, H.; Zeng, M.; Makobongo, M. O.; Finkelman, F. D.; Kelso, A.; Good, M. F. The mechanism and significance of deletion of parasite-specific CD4(+) T cells in malaria infection. J Exp Med. 2002, 195 (7), 881–892.
- Potter, S. M.; Chan-Ling, T.; Rosinova, E.; Ball, H. J.; Mitchell, A. J.; Hunt, N. H. A role for Fas-Fas ligand interactions during the late-stage neuropathological processes of experimental cerebral malaria. J Neuroimmunol. 2006, 173 (1–2), 96–107.
- Freire-de-Lima, C. G.; Nascimento, D. O.; Soares, M. B.; Bozza, P. T.; Castro-Faria-Neto, H. C.; de Mello, F. G.; DosReis, G. A.; Lopes, M. F. Uptake of apoptotic cells drives the growth of a pathogenic trypanosome in macrophages. Nature. 2000, 403 (6766), 199–203.
- Lopes, M. F.; Freire-de-Lima, C. G.; DosReis, G. A. The macrophage haunted by cell ghosts:A pathogen grows. Immunol Today. 2000, 21 (10), 489–494.
- Nunes, M. P.; Andrade, R. M.; Lopes, M. F.; DosReis, G. A. Activation-induced T cell death exacerbates Trypanosoma cruzi replication in macrophages cocultured with CD4+ T lymphocytes from infected hosts. J Immunol. 1998, 160 (3), 1313–1319.
- van de Sand, C.; Horstmann, S.; Schmidt, A.; Sturm, A.; Bolte, S.; Krueger, A.; Lutgehetmann, M.; Pollok, J. M.; Libert, C.; Heussler, V. T. The liver stage of Plasmodium berghei inhibits host cell apoptosis. Mol Microbiol. 2005, 58 (3), 731–742.
- van Dijk, M. R.; Douradinha, B.; Franke-Fayard, B.; Heussler, V.; van Dooren, M. W.; van Schaijk, B.; van Gemert, G. J.; Sauerwein, R. W.; Mota, M. M.; Waters, A. P.; Janse, C. J. Genetically attenuated, P36p-deficient malarial sporozoites induce protective immunity and apoptosis of infected liver cells. Proc Natl Acad Sci USA. 2005, 102 (34), 12194–12199.
- Leiriao, P.; Mota, M. M.; Rodriguez, A. Apoptotic Plasmodium-infected hepatocytes provide antigens to liver dendritic cells. J Infect Dis. 2005, 191 (10), 1576–1581.
- Urban, B. C.; Willcox, N.; Roberts, D. J. A role for CD36 in the re gulation of dendritic cell function. Proc Natl Acad Sci USA. 2001, 98 (15), 8750–8755.
- Wei, S.; Marches, F.; Borvak, J.; Zou, W.; Channon, J.; White, M.; Radke, J.; Cesbron-Delauw, M. F.; Curiel, T. J. Toxoplasma gondii-infected human myeloid dendritic cells induce T-lymphocyte dysfunction and contact-dependent apoptosis. Infect Immun. 2002, 70 (4), 1750–1760.
- Parrino, J.; Hotchkiss, R. S.; Bray, M. Prevention of immune cell apoptosis as potential therapeutic strategy for severe infections. Emerg Infect Dis. 2007, 13 (2), 191–198.
- Molina, J.; Brener, Z.; Romanha, A. J.; Urbina, J. A. In vivo activity of the bis-triazole D0870 against drug-susceptible and drug-resistant strains of the protozoan parasite Trypanosoma cruzi. J Antimicrob Chemother. 2000, 46 (1), 137–140.
- Molina, J.; Martins-Filho, O.; Brener, Z.; Romanha, A. J.; Loebenberg, D.; Urbina, J. A. Activities of the triazole derivative SCH 56592 (posaconazole) against drug-resistant strains of the protozoan parasite Trypanosoma (Schizotrypanum) cruzi in immunocompetent and immunosuppressed murine hosts. Antimicrob Agents Chemother. 2000, 44 (1), 150–155.
- Olivieri, B. P.; Cotta-De-Almeida, V.; Araujo-Jorge, T. Benznidazole treatment following acute Trypanosoma cruzi infection triggers CD8+ T-cell expansion and promotes resistance to reinfection. Antimicrob. Agents Chemother. 2002, 46 (12), 3790–3796.
- Bustamante, J. M.; Bixby, L. M.; Tarleton, R. L. Drug-induced cure drives conversion to a stable and protective CD8+ T central memory response in chronic Chagas disease. Nat Med. 2008, 14 (5), 542–550.
- Wesche-Soldato, D. E.; Swan, R. Z.; Chung, C. S.; Ayala, A. The apoptotic pathway as a therapeutic target in sepsis. Curr Drug Targets. 2007, 8 (4), 493–500.
- Hotchkiss, R. S.; Nicholson, D. W. Apoptosis and caspases regulate death and inflammation in sepsis. Nat Rev Immunol. 2006, 6 (11), 813–822.
- Hotchkiss, R. S.; Tinsley, K. W.; Swanson, P. E.; Chang, K. C.; Cobb, J. P.; Buchman, T. G.; Korsmeyer, S. J.; Karl, I. E. Prevention of lymphocyte cell death in sepsis improves survival in mice. Proc Natl Acad Sci USA. 1999, 96 (25), 14541–14546.
- Hotchkiss, R. S.; Chang, K. C.; Grayson, M. H.; Tinsley, K. W.; Dunne, B. S.; Davis, C. G.; Osborne, D. F.; Karl, I. E. Adoptive transfer of apoptotic splenocytes worsens survival, whereas adoptive transfer of necrotic splenocytes improves survival in sepsis. Proc Natl Acad Sci USA. 2003, 100 (11), 6724–6729.
- Silva, E. M.; Guillermo, L. V.; Ribeiro-Gomes, F. L.; De Meis, J.; Nunes, M. P.; Senra, J. F.; Soares, M. B.; DosReis, G. A.; Lopes, M. F. Caspase inhibition reduces lymphocyte apoptosis and improves host immune responses to Trypanosoma cruzi infection. Eur J Immunol. 2007, 37 (3), 738–746.
- Scheller, C.; Knoferle, J.; Ullrich, A.; Prottengeier, J.; Racek, T.; Sopper, S.; Jassoy, C.; Rethwilm, A.; Koutsilieri, E. Caspase inhibition in apoptotic T cells triggers necrotic cell death depending on the cell type and the proapoptotic stimulus. J Cell Biochem. 2006, 97 (6), 1350–1361.
- Yu, L.; Alva, A.; Su, H.; Dutt, P.; Freundt, E.; Welsh, S.; Baehrecke, E. H.; Lenardo, M. J. Regulation of an ATG7-beclin 1 program of autophagic cell death by caspase–8. Science. 2004, 304 (5676), 1500–1502.
- Gallucci, S.; Lolkema, M.; Matzinger, P. Natural adjuvants: Endogenous activators of dendritic cells. Nat Med. 1999, 5 (11), 1249–1255.
- Sauter, B.; Albert, M. L.; Francisco, L.; Larsson, M.; Somersan, S.; Bhardwaj, N. Consequences of cell death: exposure to necrotic tumor cells, but not primary tissue cells or apoptotic cells, induces the maturation of immunostimulatory dendritic cells. J Exp Med. 2000, 191 (3), 423–434.
- Chung, C. S.; Yang, S.; Song, G. Y.; Lomas, J.; Wang, P.; Simms, H. H.; Chaudry, I. H.; Ayala, A. Inhibition of Fas signaling prevents hepatic injury and improves organ blood flow during sepsis. Surgery. 2001, 130 (2), 339–345.
- Wesche-Soldato, D. E.; Chung, C. S.; Lomas-Neira, J.; Doughty, L. A.; Gregory, S. H.; Ayala, A. In vivo delivery of caspase-8 or Fas siRNA improves the survival of septic mice. Blood. 2005, 106 (7), 2295–2301.
- Carrero, J. A.; Unanue, E. R. Lymphocyte apoptosis as an immune subversion strategy of microbial pathogens. Trends Immunol. 2006, 27 (11), 497–503.
- Zheng, S. J.; Jiang, J.; Shen, H.; Chen, Y. H. Reduced apoptosis and ameliorated listeriosis in TRAIL-null mice. J Immunol. 2004, 173 (9), 5652–5658.
- Fuse, Y.; Nishimura, H.; Maeda, K.; Yoshikai, Y. CD95 (Fas) may control the expansion of activated T cells after elimination of bacteria in murine listeriosis. Infect Immun. 1997, 65 (5), 1883–1891.
- Jensen, E. R.; Glass, A. A.; Clark, W. R.; Wing, E. J.; Miller, J. F.; Gregory, S. H. Fas (CD95)-dependent cell-mediated immunity to Listeria monocytogenes. Infect Immun. 1998, 66 (9), 4143–4150.
- Lopes, M. F.; Guillermo, L. V.; Silva, E. M. Decoding caspase signaling in host immunity to the protozoan Trypanosoma cruzi. Trends Immunol. 2007, 28 (8), 366–372.
- Martins, G. A.; Petkova, S. B.; MacHado, F. S.; Kitsis, R. N.; Weiss, L. M.; Wittner, M.; Tanowitz, H. B.; Silva, J. S. Fas-FasL interaction modulates nitric oxide production in Trypanosoma cruzi-infected mice. Immunology. 2001, 103 (1), 122–129.
- Salmena, L.; Lemmers, B.; Hakem, A.; Matysiak-Zablocki, E.; Murakami, K.; Au, P. Y.; Berry, D. M.; Tamblyn, L.; Shehabeldin, A.; Migon, E.; Wakeham, A.; Bouchard, D.; Yeh, W. C.; McGlade, J. C.; Ohashi, P. S.; Hakem, R. Essential role for caspase 8 in T-cell homeostasis and T-cell-mediated immunity. Genes Dev. 2003, 17 (7), 883–895.
- Wu, Z.; Roberts, M.; Porter, M.; Walker, F.; Wherry, E. J.; Kelly, J.; Gadina, M.; Silva, E. M.; DosReis, G. A.; Lopes, M. F.; O’shea, J.; Leonard, W. J.; Ahmed, R.; Siegel, R. M. Viral FLIP impairs survival of activated T cells and generation of CD8+ T cell memory. J Immunol. 2004, 172 (10), 6313–6323.
- Silva, E. M.; Guillermo, L. V.; Ribeiro-Gomes, F. L.; De Meis, J.; Pereira, R. M.; Wu, Z.; Calegari-Silva, T. C.; Seabra, S. H.; Lopes, U. G.; Siegel, R. M.; Dosreis, G. A.; Lopes, M. F. Caspase-8 activity prevents type 2 cytokine responses and is required for protective T cell-mediated immunity against Trypanosoma cruzi infection. J Immunol. 2005, 174 (10), 6314–6321.
- Hinshaw-Makepeace, J.; Huston, G.; Fortner, K. A.; Russell, J. Q.; Holoch, D.; Swain, S.; Budd, R. C. c-FLIP(S) reduces activation of caspase and NF-kappaB pathways and decreases T cell survival. Eur J Immunol. 2008, 38 (1), 54–63.
- Chun, H. J.; Zheng, L.; Ahmad, M.; Wang, J.; Speirs, C. K.; Siegel, R. M.; Dale, J. K.; Puck, J.; Davis, J.; Hall, C. G.; Skoda-Smith, S.; Atkinson, T. P.; Straus, S. E.; Lenardo, M. J. Pleiotropic defects in lymphocyte activation caused by caspase-8 mutations lead to human immunodeficiency. Nature. 2002, 419 (6905), 395–399.
- Su, H.; Bidere, N.; Zheng, L.; Cubre, A.; Sakai, K.; Dale, J.; Salmena, L.; Hakem, R.; Straus, S.; Lenardo, M. Requirement for caspase-8 in NF-kappaB activation by antigen receptor. Science. 2005, 307 (5714), 1465–1468.
- Pereira, W. F.; Guillermo, L. V.; Ribeiro-Gomes, F. L.; Lopes, M. F. Inhibition of caspase-8 activity reduces IFN-gamma expression by T cells from Leishmania major infection. An Acad Bras Cienc. 2008, 80 (1), 129–136.
- Siegel, R. M. Caspases at the crossroads of immune-cell life and death. Nat Rev Immunol. 2006, 6 (4), 308–317.
- Strasser, A.; Pellegrini, M. T-lymphocyte death during shutdown of an immune response. Trends Immunol. 2004, 25 (11), 610–615.
- Hildeman, D. A.; Zhu, Y.; Mitchell, T. C.; Bouillet, P.; Strasser, A.; Kappler, J.; Marrack, P. Activated T cell death in vivo mediated by proapoptotic bcl-2 family member bim. Immunity. 2002, 16 (6), 759–767.
- Pandiyan, P.; Zheng, L.; Ishihara, S.; Reed, J.; Lenardo, M. J. CD4+CD25+Foxp3+ regulatory T cells induce cytokine deprivation-mediated apoptosis of effector CD4+ T cells. Nat Immunol. 2007, 8 (12), 1353–1362.
- Pandiyan, P.; Lenardo, M. J. The control of CD4+CD25+Foxp3+ regulatory T cell survival. Biol Direct. 2008, 3 6.
- Reckling, S.; Divanovic, S.; Karp, C. L.; Wojciechowski, S.; Belkaid, Y.; Hildeman, D. Proapoptotic Bcl-2 family member Bim promotes persistent infection and limits protective immunity. Infect Immun. 2008, 76 (3), 1179–1185.
- Lopes, M. F.; DosReis, G. A. Apoptosis as a cause of T-cell unresponsiveness in experimental Chagas’ disease. Braz J Med Biol Res. 1995, 28 (8), 913–918.
- de Meis, J.; Ferreira, L. M.; Guillermo, L. V.; Silva, E. M.; Dosreis, G. A.; Lopes, M. F. Apoptosis differentially regulates mesenteric and subcutaneous lymph node immune responses to Trypanosoma cruzi. Eur J Immunol. 2008, 38 (1), 139–146.
- Zhang, J.; Andrade, Z. A.; Yu, Z. X.; Andrade, S. G.; Takeda, K.; Sadirgursky, M.; Ferrans, V. J. Apoptosis in a canine model of acute Chagasic myocarditis. J Mol Cell Cardiol. 1999, 31 (3), 581–596.
- Rossi, M. A.; Souza, A. C. Is apoptosis a mechanism of cell death of cardiomyocytes in chronic Chagasic myocarditis? Int J Cardiol. 1999, 68 (3), 325–331.
- Tostes, S., Jr.; Bertulucci Rocha-Rodrigues, D.; de Araujo Pereira, G.; Rodrigues, V., Jr. Myocardiocyte apoptosis in heart failure in chronic Chagas’ disease. Int J Cardiol. 2005, 99 (2), 233–237.
- Teixeira, P. C.; Iwai, L. K.; Kuramoto, A. C.; Honorato, R.; Fiorelli, A.; Stolf, N.; Kalil, J.; Cunha-Neto, E. Proteomic inventory of myocardial proteins from patients with chronic Chagas’ cardiomyopathy. Braz J Med Biol Res. 2006, 39 (12), 1549–1562.
- Rodrigues, V., Jr.; Agrelli, G. S.; Leon, S. C.; Silva Teixeira, D. N.; Tostes, S., Jr.; Rocha-Rodrigues, D. B. Fas/Fas-L expression, apoptosis and low proliferative response are associated with heart failure in patients with chronic Chagas’ disease. Microbes Infect. 2008, 10 (1), 29–37.
- Muller, U.; Sobek, V.; Balkow, S.; Holscher, C.; Mullbacher, A.; Museteanu, C.; Mossmann, H.; Simon, M. M. Concerted action of perforin and granzymes is critical for the elimination of Trypanosoma cruzi from mouse tissues, but prevention of early host death is in addition dependent on the FasL/Fas pathway. Eur J Immunol. 2003, 33 (1), 70–78.
- de Oliveira, G. M.; Diniz, R. L.; Batista, W.; Batista, M. M.; Bani Correa, C.; de Araujo-Jorge, T. C.; Henriques-Pons, A. Fas ligand-dependent inflammatory regulation in acute myocarditis induced by Trypanosoma cruzi infection. Am J Pathol. 2007, 171 (1), 79–86.
- Henriques-Pons, A.; Oliveira, G. M.; Paiva, M. M.; Correa, A. F.; Batista, M. M.; Bisaggio, R. C.; Liu, C. C.; Cotta-De-Almeida, V.; Coutinho, C. M.; Persechini, P. M.; Araujo-Jorge, T. C. Evidence for a perforin-mediated mechanism controlling cardiac inflammation in Trypanosoma cruzi infection. Int J Exp Pathol. 2002, 83 (2), 67–79.
- Hu, M. S.; Schwartzman, J. D.; Yeaman, G. R.; Collins, J.; Seguin, R.; Khan, I. A.; Kasper, L. H. Fas-FasL interaction involved in pathogenesis of ocular toxoplasmosis in mice. Infect Immun. 1999, 67 (2), 928–935.
- Eidsmo, L.; Nylen, S.; Khamesipour, A.; Hedblad, M. A.; Chiodi, F.; Akuffo, H. The contribution of the Fas/FasL apoptotic pathway in ulcer formation during Leishmania major-induced cutaneous leishmaniasis. Am J Pathol. 2005, 166 (4), 1099–1108.
- Eidsmo, L.; Fluur, C.; Rethi, B.; Eriksson Ygberg, S.; Ruffin, N.; De Milito, A.; Akuffo, H.; Chiodi, F. FasL and TRAIL induce epidermal apoptosis and skin ulceration upon exposure to Leishmania major. Am J Pathol. 2007, 170 (1), 227–239.
- Vieira, L. Q.; Goldschmidt, M.; Nashleanas, M.; Pfeffer, K.; Mak, T.; Scott, P. Mice lacking the TNF receptor p55 fail to resolve lesions caused by infection with Leishmania major, but control parasite replication. J Immunol. 1996, 157 (2), 827–835.
- Castanos-Velez, E.; Maerlan, S.; Osorio, L. M.; Aberg, F.; Biberfeld, P.; Orn, A.; Rottenberg, M. E. Trypanosoma cruzi infection in tumor necrosis factor receptor p55-deficient mice. Infect Immun. 1998, 66 (6), 2960–2968.
- Ojcius, D. M.; Perfettini, J. L.; Bonnin, A.; Laurent, F. Caspase-dependent apoptosis during infection with Cryptosporidium parvum. Microbes Infect. 1999, 1 (14), 1163–1168.
- McCole, D. F.; Eckmann, L.; Laurent, F.; Kagnoff, M. F. Intestinal epithelial cell apoptosis following Cryptosporidium parvum infection. Infect Immun. 2000, 68 (3), 1710–1713.
- Chen, X. M.; Gores, G. J.; Paya, C. V.; LaRusso, N. F. Cryptosporidium parvum induces apoptosis in biliary epithelia by a Fas/Fas ligand-dependent mechanism. Am J Physiol. 1999, 277 (3 Pt 1), G599–608.
- O’Hara, S. P.; Small, A. J.; Nelson, J. B.; Badley, A. D.; Chen, X. M.; Gores, G. J.; Larusso, N. F. The human immunodeficiency virus type 1 tat protein enhances Cryptosporidium parvum-induced apoptosis in cholangiocytes via a Fas ligand-dependent mechanism. Infect Immun. 2007, 75 (2), 684–696.
- Motta, I.; Gissot, M.; Kanellopoulos, J. M.; Ojcius, D. M. Absence of weight loss during Cryptosporidium infection in susceptible mice deficient in Fas-mediated apoptosis. Microbes Infect. 2002, 4 (8), 821–827.
- Leiriao, P.; Rodrigues, C. D.; Albuquerque, S. S.; Mota, M. M. Survival of protozoan intracellular parasites in host cells. EMBO Rep. 2004, 5 (12), 1142–1147.
- Lackner, P.; Burger, C.; Pfaller, K.; Heussler, V.; Helbok, R.; Morandell, M.; Broessner, G.; Tannich, E.; Schmutzhard, E.; Beer, R. Apoptosis in experimental cerebral malaria: Spatial profile of cleaved caspase-3 and ultrastructural alterations in different disease stages. Neuropathol Appl Neurobiol. 2007, 33 (5), 560–571.
- Pino, P.; Vouldoukis, I.; Kolb, J. P.; Mahmoudi, N.; Desportes-Livage, I.; Bricaire, F.; Danis, M.; Dugas, B.; Mazier, D. Plasmodium falciparum: Infected erythrocyte adhesion induces caspase activation and apoptosis in human endothelial cells. J Infect Dis. 2003, 187 (8), 1283–1290.
- Ohno, T.; Kobayashi, F.; Nishimura, M. Fas has a role in cerebral malaria, but not in proliferation or exclusion of the murine parasite in mice. Immunogenetics. 2005, 57 (3–4), 293–296.
- Piguet, P. F.; Kan, C. D.; Vesin, C. Thrombocytopenia in an animal model of malaria is associated with an increased caspase-mediated death of thrombocytes. Apoptosis. 2002, 7 (2), 91–98.
- Guha, M.; Kumar, S.; Choubey, V.; Maity, P.; Bandyopadhyay, U. Apoptosis in liver during malaria: Role of oxidative stress and implication of mitochondrial pathway. Faseb J. 2006, 20 (8), 1224–1226.
- Chen, M.; Wang, Y. H.; Wang, Y.; Huang, L.; Sandoval, H.; Liu, Y. J.; Wang, J. Dendritic cell apoptosis in the maintenance of immune tolerance. Science. 2006, 311 (5764), 1160–1164.
- Allen, H. L.; Deepe, G. S., Jr. Apoptosis modulates protective immunity to the pathogenic fungus Histoplasma capsulatum. J Clin Invest. 2005, 115 (10), 2875–2885.
- Popov, S. G.; Popova, T. G.; Grene, E.; Klotz, F.; Cardwell, J.; Bradburne, C.; Jama, Y.; Maland, M.; Wells, J.; Nalca, A.; Voss, T.; Bailey, C.; Alibek, K. Systemic cytokine response in murine anthrax. Cell Microbiol. 2004, 6 (3), 225–233.
- Sehra, S.; Patel, D.; Kusam, S.; Wang, Z. Y.; Chang, C. H.; Dent, A. L. A role for caspases in controlling IL-4 expression in T cells. J Immunol. 2005, 174 (6), 3440–3446.
- de Freitas Balanco, J. M.; Moreira, M. E.; Bonomo, A.; Bozza, P. T.; Amarante-Mendes, G.; Pirmez, C.; Barcinski, M. A. Apoptotic mimicry by an obligate intracellular parasite downregulates macrophage microbicidal activity. Curr Biol. 2001, 11 (23), 1870–1873.
- Wanderley, J. L.; Moreira, M. E.; Benjamin, A.; Bonomo, A. C.; Barcinski, M. A. Mimicry of apoptotic cells by exposing phosphatidylserine participates in the establishment of amastigotes of Leishmania (L) amazonensis in mammalian hosts. J Immunol. 2006, 176 (3), 1834–1839.
- Kosec, G.; Alvarez, V. E.; Aguero, F.; Sanchez, D.; Dolinar, M.; Turk, B.; Turk, V.; Cazzulo, J. J. Metacaspases of Trypanosoma cruzi: possible candidates for programmed cell death mediators. Mol Biochem Parasitol. 2006, 145 (1), 18–28.
- Weaver, J. G.; Tarze, A.; Moffat, T. C.; Lebras, M.; Deniaud, A.; Brenner, C.; Bren, G. D.; Morin, M. Y.; Phenix, B. N.; Dong, L.; Jiang, S. X.; Sim, V. L.; Zurakowski, B.; Lallier, J.; Hardin, H.; Wettstein, P.; van Heeswijk, R. P.; Douen, A.; Kroemer, R. T.; Hou, S. T.; Bennett, S. A.; Lynch, D. H.; Kroemer, G.; Badley, A. D. Inhibition of adenine nucleotide translocator pore function and protection against apoptosis in vivo by an HIV protease inhibitor. J Clin Invest. 2005, 115 (7), 1828–1838.