
Abstract
Background
Doxorubicin (DOX) resistance remains a major challenge for adriamycin-based treatment of breast cancer (BC). Transforming growth factor β (TGF-β) has been reported to contribute to drug resistance. Although the role of long noncoding RNAs (LncRNAs) in cancer progression has been widely studied, its effect on TGF-β-induced resistance remains limited. This study aimed to investigate the role of LncRNA on the regulation of TGF-β-induced drug resistance.
Methods
Cell counting kit-8 (CCK-8) and an EdU assay were used to evaluate cell viability and proliferation. The level of LncRNA mRNA expression in BC tissues and cells was examined by quantitative real-time PCR. Changes in epithelial-mesenchymal transition (EMT) and cell apoptosis were quantified by Western blot and immunofluorescence.
Results
TGF-β induced EMT and promoted DOX resistance. LncRNA urothelial carcinoma-associated 1(lncRNA UCA1) associated with TGF-β was upregulated in BC cells and tissues. LncRNA UCA1 silencing enhanced sensitivity to DOX decreased cellular proliferation and increased apoptosis in BC cells. The effect of TGF-β on EMT and DOX resistance disappeared following a lncRNA UCA1 knockdown.
Conclusions
These findings suggest that lncRNA-UCA1, a mediator of TGF-β signaling, could predispose BC patients to EMT and DOX resistance.
Introduction
Breast cancer (BC) is the most frequent malignant tumor that threatens the health of women throughout the world [Citation1]. Doxorubicin (DOX) is one of the main chemotherapy drugs used as first-line therapy for women with early and metastatic BC [Citation2,Citation3]. However, approximately one-third of patients develop drug resistance with a poor prognosis for a period of time following DOX treatment [Citation4]. Both inherent and acquired drug resistance affects the efficacy of DOX-based therapies [Citation5]. Therefore, a greater understanding of the mechanism of DOX resistance contributes to improving the prognosis of BC patients.
The epithelial-mesenchymal transition (EMT) is known to play a pivotal role in cancer progression, dissemination, and drug resistance [Citation6,Citation7]. Transforming growth factors β (TGF-β) acts as a potent inducer of the EMT and plays a predominant role in tumor progression, migration, invasion, and metastasis [Citation8,Citation9]. Recently, an imbalance in TGF-β signaling has been reported to affect drug resistance [Citation10]. Therefore, understanding the mechanisms by which TGF-β affects drug resistance will be of benefit to BC patients.
Long noncoding RNAs (LncRNAs) represent a group of highly conservative RNA molecules longer than 200 nucleotides, which have little or no protein-coding capacity. A number of studies have reported that LncRNAs are aberrantly expressed in various human cancers and are involved in cancer carcinogenesis and metastasis as cancer-promoting or cancer-suppressing factors [Citation11,Citation12]. LncRNA urothelial carcinoma associated 1 (lncRNA-UCA1) is markedly elevated in numerous cancers (e.g. oral squamous cell carcinoma [Citation13], colorectal cancer [Citation14], and gastric cancer [Citation15]) and acts as a cancer-promoting factor. The study by Yi et al. reported that the level of UCA1 was positively associated with PTP1B expression in breast tumor tissues and UCA1-induced PTP1B expression facilitated cellular proliferation [Citation16]. The report by Tuo et al. showed that UCA1 affected BC cell growth and apoptosis through the negative regulation of miR-143 [Citation17]. In addition, Liu et al. demonstrated that UCA1 mediated Paclitaxel resistance in BC cells through the miR-613/CDK12 axis [Citation18]; however, the relationship between lncRNA-UCA1 and the development of TGF-β induced-chemo-resistance of BC has not been well established.
In our present study, we explored the role of LncRNA in TGF-β-induced DOX resistance. We found that lncRNA-UCA1 was significantly overexpressed in BC cells following TGF-β treatment. Moreover, UCA1 silencing mediated TGF-β-induced DOX chemo-resistance in BC cells by inhibiting EMT. Our study provides a greater understanding of the role of lncRNA-UCA1 in BC chemoresistance and the associated mechanism.
Materials and methods
Patient samples
Tumor specimens and paired noncancerous tissues were collected from 15 patients with BC who underwent resection between January 2015 and January 2016. The tissue samples were stored at −80 °C. Written informed consent for research was provided by all of the patients. This study was approved by the Ethics Committee of the First Affiliated Hospital of Zhejiang Chinese Medicine University.
Cell culture
Breast cell lines (MDA-MB-231, MDA-MB-468, and MCF-7 cells) were purchased from the American Type Culture Collection (ATCC). Cells were cultured at 37 °C in 5% CO2 with DMEM media supplemented with 10% fetal bovine serum (FBS).
Cell viability assay
Cell viability was measured with a cell counting kit-8 (CCK-8) assay. BC cells were seeded into 96-well plates and subsequently treated with different concentrations of DOX (0, 0.25, 0.5, 1, 2, and 4 μg/mL) for 48 h. Following three washes with PBS, the cells were incubated with 100 µL media containing 10 µL CCK-8 solution at 37 °C for 2 h, and the absorbance was measured at 450 nm using a microplate reader.
Quantitative real-time PCR (qRT-PCR) assay
A qRT-PCR assay was performed to quantify the level of lncRNA mRNA in the BC tissues and indicated BC cell lines. The total RNA was extracted from the tissues and cells using Trizol reagent (Invitrogen), and reverse transcribed into cDNA using Prime ScriptTM RT Master Mix (Takara Bio Inc., Dalian, China). qRT-PCR was performed using the 7500 Fast Real-time PCR System (Applied Biosystems) with TB Green® Premix Ex Taq™ II (Tli RNaseH Plus) (Takara). The sequences of primers were list in Supplementary file.
Western blot assay
Cell lysates were obtained using RIPA buffer (Beyotime, Shanghai, China), separated on a 10% SDS-polyacrylamide gel, and transferred to a polyvinylidene fluoride membrane. The membrane was incubated with respective primary antibodies for E-cadherin (1:1000, #3195, Cell Signaling Technology), vimentin (1:1000, #5741, Cell Signaling Technology), and GAPDH (1:1000, #2118S, Cell Signaling Technology) at 4 °C overnight. After washing three times, the membranes were incubated with HRP-conjugated secondary antibodies at 37 °C for 2 h. The bands were developed using enhanced chemiluminescence reagent (Beyotime, China) and the results were quantified with Image-Pro Plus 6.0 software.
EdU assay
Cell proliferation was detected using an EdU assay kit. Following transfection with the indicated constructs, BC cells in the logarithmic phase were seeded into 24-well plates and incubated overnight. The cells were subsequently treated with or without 10 ng/mL TGF-β. After 48 h, the cells were incubated with 10 mM EdU reagent (BeyoClick EdU-594, Beyotime, China) for 2 h at 37 °C. Nuclei were stained with Hoechst 33342 for 30 min. The number of positive cells and the total number of DAPI positive cells in three visual fields were randomly selected and calculated under a microscope.
Immunofluorescence
Cells on the slides were fixed in 4% paraformaldehyde for 10 min at room temperature. The slides were incubated with primary antibodies for E-cadherin and vimentin. After incubation with secondary antibodies for Alexa Fluor®488-labeled anti-Rabbit IgG (1:200, ZF-0514, Zhongshan Jinqiao), the slides were incubated with Hoechst (1:100) for nuclear staining and imaged with confocal microscopy.
Statistical analysis
Statistical analysis was performed using GraphPad Prism software 8. Data are represented as the mean ± SD. An unpaired Student’s t-test was used to evaluate the level of gene expression. All of the results are considered to be statistically significant at p < .05.
Results
TGF-β-induces EMT and promotes chemoresistance in BC cells
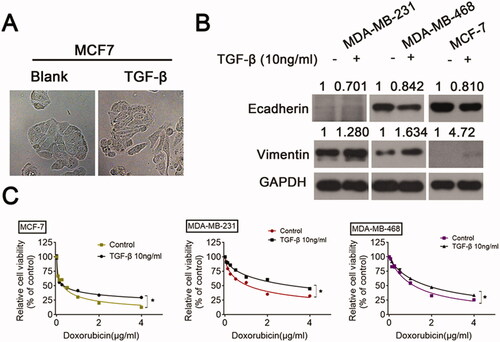
First, TGF-β (10 ng/mL) was used to treat MCF-7 cells. The morphological changes in MCF-7 cells with or without TGF-β were compared under an optical microscope. The MCF-7 cells were epithelioid and polygonal, whereas TGF-β-treated MCF-7 cells were fusiform and mesenchymal (). Next, we observed further changes in the level of protein expression. TGF-β stimulation led to the transition of the epithelium to mesenchyme involving a decrease in E-cadherin and an increase in vimentin (). Next, a CCK-8 assay was performed to explore the effect of TGF-β on the response to DOX (). The mean half-maximal inhibitory concentration (IC50) values for DOX in MDA-MB-231, MDA-MB-468, and MCF-7 cells were 1.66 ± 0.12 μM, 1.14 ± 0.09 μM, and 0.28 ± 0.03 μM, respectively. TGF-β treatment significantly elevated cell viability from the increase in the IC50 value. Following TGF-β treatment, IC50 values for DOX in MDA-MB-231, MDA-MB-468, and MCF-7 cells were 3.47 ± 0.27 μM, 1.92 ± 0.11 μM, and 0.41 ± 0.07 μM, respectively. These data indicate that TGF-β results in EMT and chemoresistance in BC cells.
Figure 1. TGF-β induces EMT and increases DOX chemoresistance in BC cells BC cells were incubated with either a vehicle or TGF-β (10 ng/mL) for 24 h. (A) The cellular morphology was observed under a light microscope. (B) The levels of E-cadherin and vimentin were examined by Western blot. (C) The responses of BC cells to DOX were analyzed using a CCK-8 assay.
TGF-β promotes UCA1 expression and UCA1 was increased in BC cells
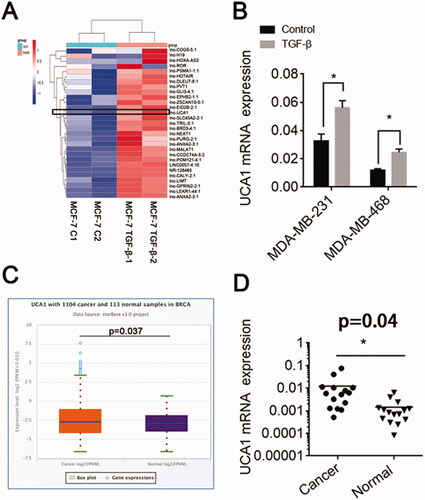
Next, we detected the level of expression of a series of LncRNAs in TGF-β-stimulated MCF-7 cells by qRT-PCR. We found that UCA1 mRNA expression was markedly increased following TGF-β treatment (). The level of lncRNA-UCA1 expression in the other two BC cells was also increased (). The Starbase database revealed that UCA1 was highly expressed in BC (). Moreover, in 15 paired BC samples, a high level of UCA1 mRNA expression was observed in the BC tissues (). Together, these results show that TGF-β up-regulated UCA1 mRNA expression in BC cells.
Figure 2. TGF-β promotes UCA1 expression. (A) MCF-7 cells were incubated with either a vehicle or TGF-β (10 ng/mL) for 24 h. A series of LncRNAs associated with chemoresistance was detected by qRT-PCR. (B) UCA1 expression in the MDA-MB-231 and MDA-MB-468 cell lines following TGF-β1 stimulation. (C) UCA1 expression in BC by Starbase. (D) The level of UCA1 mRNA expression in BC patients.
UCA1 siRNA promotes BC cell sensitivity to DOX
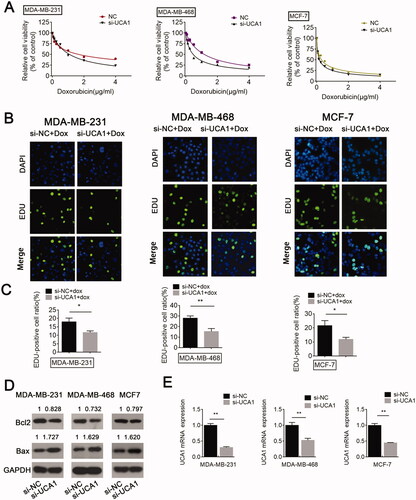
The above data indicate that TGF-β enhances DOX resistance and induces UCA1 up-regulation. To validate the function of UCA1 in chemoresistance, the cells were grouped into a negative control (NC) group and a knockdown group (si-UCA1 group). As expected, an inhibition in lncRNA-UCA1 could markedly enhance the inhibitory effect of DOX on cell viability compared to the NC siRNA group (). Moreover, the EdU assay showed that the knockdown of lncRNA-UCA1 enhanced the inhibition of BC cell proliferation by DOX compared to the NC siRNA group. The EdU-positive cell ratios in the UCA1 siRNA groups were dramatically inhibited compared with the NC groups (). In addition, UCA1 silencing increased cell apoptosis in BC cells compared with the NC group under DOX treatment (). Collectively, these data imply that a lncRNA-UCA1 knockdown might contribute to the sensitization of BC cells to DOX.
Figure 3. UCA1 siRNA promotes BC cell sensitivity to DOX, inhibits cell proliferation, and enhances cell apoptosis. BC cells transfected with or without NC or UCA1 siRNA received DOX treatment. (A) Cell viability was measured by a CCK-8 assay. (B and C) Cell proliferation was detected using an EdU assay. (D) Cell apoptosis was examined by Western blot. (E) Inhibitory efficiency was verified using real-time PCR.
TGF-β mediates chemoresistance through UCA1
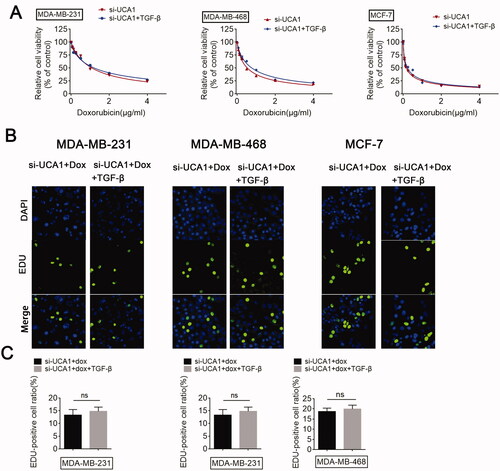
Since we demonstrated that UCA1 siRNA promotes BC cell sensitivity to DOX and TGF-β treatment promoted UCA1 expression, we subsequently investigated the detailed function of UCA1 in TGF-β-treated BC cells. In agreement with the above results, a knockdown in UCA1 could increase the sensitivity of BC cells to DOX. However, the promotional function of UCA1 down-regulation on DOX sensitivity was hardly reversed by TGF-β (). Moreover, TGF-β failed to reverse the inhibitory role of UCA1 inhibition on cell proliferation (). Collectively, these results reveal the regulatory effect of UCA1 on TGF-β-induced chemoresistance in BC cells.
Figure 4. UCA1 is required for TGF-β-induced chemoresistance. Si-UCA1-transfected BC cells were treated with or without TGF-β, respectively, and (A) a CCK-8 assay was performed to monitor the chemosensitivity of si-UCA1 cells to DOX. (B and C) An EdU assay was performed to analyze the proliferation of si-UCA1 cells in response to DOX.
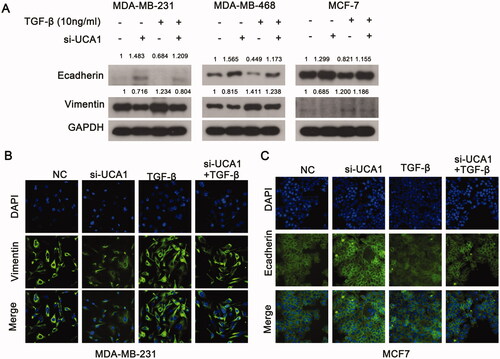
UCA1 affects TGF-β-induced vimentin and E-cadherin expression in BC cells
An important mechanism for TGF-β to induce resistance is the promotion of EMT. Therefore, we investigated the regulation of UCA1 on TGF-β-induced EMT. TGF-β treatment significantly elevated the level of vimentin protein and decreased the level of E-cadherin protein expression, whereas si-UCA1 had an opposite effect compared to the NC group (). However, the promotion of TGF-β on EMT disappeared when combined with si-UCA1. MDA-MB-231 cells exhibited low levels of E-cadherin expression. Conversely, MCF-7 cells expressed minimal levels of vimentin. Therefore, we selected MDA-MB-231 and MCF-7 cells to detect the changes in E-cadherin and vimentin levels with an immunofluorescence assay. These results were consistent with those in the Western blot (). Collectively, these findings demonstrate that UCA1 may be involved in TGF-β-induced EMT in BC.
Figure 5. UCA1 modulates TGF-β-induced EMT. (A) Western blotting was used to analyze the expression of E-cadherin and vimentin in MDA-MB-231 and MCF7 cells following TGF-β stimulation for 24 h. (B) The level of vimentin expression was higher in the MDA-MB-231 siRNA group compared with the negative control cells following TGF-β stimulation for 24 h. (C) The expression of E-cadherin was higher in the MCF-7 siRNA group compared to the negative control cells following TGF-β stimulation for 24 h.
Discussion
Although we observed an improvement in tumor treatment using drug-targeting strategies, drug resistance remains the main cause of cancer treatment failure and death. In the current study, we found that UCA1 was increased in TGF-β-treated BC cells and UCA1 mediated TGF-β-induced resistance through regulating EMT. These findings help us to understand the molecular mechanism of drug resistance and imply that UCA1 functions as novel targeted therapeutic strategies to overcome therapy resistance.
EMT has become a major factor for treating drug resistance [Citation19]. During this process, epithelial cells lose cell-cell contact and apico-basal polarity, as well as obtain additional mesenchymal properties through cytoskeleton rearrangement, adhesion, and changes to the cell structure and morphology [Citation20]. TGF-β has been established as a major inducer and component of the EMT. TGF-β plays different roles at various stages of tumor formation. During the early stage of tumorigenesis, TGF-β, as a tumor suppressor, induces the apoptosis of both normal and precancerous cells. However, during advanced stages when cancer cells acquire carcinogenic mutations and/or lose the function of tumor suppressor genes, TGF-β plays a role as a tumor promoter by stimulating tumor cells to induce the EMT. In this study, we confirmed that TGF-β stimulation altered the morphology of MCF-7 cells from epithelioid to mesenchymal. Moreover, TGF-β treatment led to a decrease in the level of E-cadherin and an increase in vimentin in BC cells, indicating that it is a tumor promoter. Furthermore, we also demonstrated that TGF-β enhanced DOX resistance in BC. The effect of TGF-β signaling on drug resistance is similar to that reported in a previous study [Citation21].
This study sought to investigate the mechanism of TGF-β-mediated resistance. We found that a series of LncRNAs were overexpressed following TGF-β treatment, among which UCA1 was more prominent. Moreover, UCA1 was increased in the tumor samples and growing evidence indicates that abnormal LncRNA contributes to drug resistance in tumors. For example, LncRNA MACC1-AS1 and FOXD2-AS1 mediate NSD2-induced cisplatin resistance in esophageal squamous cell carcinoma [Citation22]. In addition, exosomal HOTTIP contributed to cisplatin resistance in gastric cancer cells by regulating the miR-218/HMGA1 axis [Citation23]. Similarly, we found that the inhibition of UCA1 promoted BC cell sensitivity to DOX. Importantly, the effect of TGF-β on resistance disappeared following combination treatment with si-UCA1, which indicated that UCA1 impacts TGF-β-mediated resistance.
It was subsequently found that the changes in the level of vimentin and E-cadherin induced by TGF disappeared following the deletion of UCA1, implying that UCA1 is involved in TGF-β-mediated resistance by regulating EMT. Consistent with our results, the study by Zhong et al. demonstrated that the downregulation of UCA1 inhibited TGFβ1-induced EMT in lung cancer [Citation24]. Although others have reported the function of UCA1 on drug resistance in various malignant tumors [Citation25–27], the effect of UCA1 in TGF-β-mediated resistance in BC has not been previously reported. Our data suggest that UCA1 might represent a potential therapeutic target against drug resistance as a downstream component of TGF-β signaling.
There are still some shortcomings in our research. While we have demonstrated that UCA1 regulates DOX resistance mediated by TGF-β through the EMT pathway, there is a lack of data on the associated regulatory molecules or mechanisms used to verify these findings. Recent studies suggest that UCA1 could act as ‘ceRNA’ to promote 5-fluorouracil resistance in colorectal cancer [Citation14]. Therefore, we used a public database, a dual-luciferase reporter assay, and qRT-PCR to predict ‘ceRNA’ (Figure S1). The results showed that UCA1 negatively regulates miR-455-5p, which negatively regulates Notch-1. Future experiments should be performed to confirm these findings.
In conclusion, UCA1 is upregulated in TGF-β-treated BC cells and contributes to TGF-β-mediated DOX resistance via the EMT signaling pathway, indicating that UCA1 represents a promising target for improving chemotherapy against BC.
Author contributions
HZ designed the research study. KW wrote the manuscript. BZ and XY performed the research. ZG, MZ, QW, and ZL analyzed the data.
Ethics approval
All experiments regarding patient samples conducted in this study were approved by the Ethics Committee of Zhejiang Chinese Medicine University (Approval number: 2021-KL-081-01).
Supplemental Material
Download PDF (333.3 KB)Disclosure statement
The authors declare that there are no conflicts of interest.
Data availability statement
The datasets used and/or analyzed during the current study are available from the corresponding author upon reasonable request.
Additional information
Funding
Related Research Data
References
- Bray F, Ferlay J, Soerjomataram I, et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424.
- Shafei A, El-Bakly W, Sobhy A, et al. A review on the efficacy and toxicity of different doxorubicin nanoparticles for targeted therapy in metastatic breast cancer. Biomed Pharmacother. 2017;95:1209–1218.
- Nicolini A, Giardino R, Carpi A, et al. Metastatic breast cancer: an updating. Biomed Pharmacother. 2006;60(9):548–556.
- Gonzalez-Angulo AM, Morales-Vasquez F, Hortobagyi GN. Overview of resistance to systemic therapy in patients with breast cancer. Adv Exp Med Biol. 2007;608:1–22.
- Wijdeven RH, Pang B, van der Zanden SY, et al. Genome-Wide identification and characterization of novel factors conferring resistance to topoisomerase II poisons in cancer. Cancer Res. 2015;75(19):4176–4187.
- Du B, Shim JS. Targeting Epithelial-Mesenchymal transition (EMT) to overcome drug resistance in cancer. Molecules. 2016;21(7):965.
- Marcucci F, Stassi G, De Maria R. Epithelial-mesenchymal transition: a new target in anticancer drug discovery. Nat Rev Drug Discov. 2016;15(5):311–325.
- Wang P, Chen W, Ma T, et al. lncRNA lnc-TSI inhibits metastasis of clear cell renal cell carcinoma by suppressing TGF-β-induced epithelial-mesenchymal transition. Mol Ther Nucleic Acids. 2020;22:1–16.
- Syed V. TGF-β signaling in cancer. J Cell Biochem. 2016;117(6):1279–1287.
- Xu X, Zhang L, He X, et al. TGF-β plays a vital role in triple-negative breast cancer (TNBC) drug-resistance through regulating stemness, EMT and apoptosis. Biochem Biophys Res Commun. 2018;502(1):160–165.
- Bhan A, Soleimani M, Mandal SS. Long noncoding RNA and cancer: a new paradigm. Cancer Res. 2017;77(15):3965–3981.
- Li J, Meng H, Bai Y, et al. Regulation of lncRNA and its role in cancer metastasis. Oncol Res. 2016;23(5):205–217.
- Fang Z, Zhao J, Xie W, et al. LncRNA UCA1 promotes proliferation and cisplatin resistance of oral squamous cell carcinoma by sunppressing miR-184 expression. Cancer Med. 2017;6(12):2897–2908.
- Xian Z, Hu B, Wang T, et al. lncRNA UCA1 contributes to 5-Fluorouracil resistance of colorectal cancer cells through miR-23b-3p/ZNF281 axis. Onco Targets Ther. 2020;13:7571–7583.
- Yang A, Liu X, Liu P, et al. LncRNA UCA1 promotes development of gastric cancer via the miR-145/MYO6 axis. Cell Mol Biol Lett. 2021;26(1):33.
- Li Y, Zeng Q, Qiu J, et al. Long non-coding RNA UCA1 promotes breast cancer by upregulating PTP1B expression via inhibiting miR-206. Cancer Cell Int. 2019;19:275.
- Tuo YL, Li XM, Luo J. Long noncoding RNA UCA1 modulates breast cancer cell growth and apoptosis through decreasing tumor suppressive miR-143. Eur Rev Med Pharmacol Sci. 2015;19(18):3403–3411.
- Liu C, Jiang F, Zhang X, et al. Long Non-Coding RNA UCA1 modulates paclitaxel resistance in breast cancer via miR-613/CDK12 axis. Cancer Manag Res. 2020;12:2777–2788.
- De Las Rivas J, Brozovic A, Izraely S, et al. Cancer drug resistance induced by EMT: novel therapeutic strategies. Arch Toxicol. 2021;95(7):2279–2297.
- Brabletz T, Kalluri R, Nieto MA, et al. EMT in cancer. Nat Rev Cancer. 2018;18(2):128–134.
- Zhuang J, Shen L, Yang L, et al. TGFβ1 promotes gemcitabine resistance through regulating the LncRNA-LET/NF90/miR-145 signaling axis in bladder cancer. Theranostics. 2017;7(12):3053–3067.
- Xue W, Shen Z, Li L, et al. Long non-coding RNAs MACC1-AS1 and FOXD2-AS1 mediate NSD2-induced cisplatin resistance in esophageal squamous cell carcinoma. Mol Ther Nucleic Acids. 2021;23:592–602.
- Wang J, Lv B, Su Y, et al. Exosome-Mediated transfer of lncRNA HOTTIP promotes cisplatin resistance in gastric cancer cells by regulating HMGA1/miR-218 axis. Onco Targets Ther. 2019;12:11325–11338.
- Zuo ZK, Gong Y, Chen XH, et al. TGFβ1-Induced LncRNA UCA1 upregulation promotes gastric cancer invasion and migration. DNA Cell Biol. 2017;36(2):159–167.
- Huang G, Li L, Liang C, et al. Upregulated UCA1 contributes to oxaliplatin resistance of hepatocellular carcinoma through inhibition of miR-138-5p and activation of AKT/mTOR signaling pathway. Pharmacol Res Perspect. 2021;9(1):e00720.
- Yao Q, Zhang L, Wang Y, et al. LncRNA UCA1 elevates the resistance of human leukemia cells to daunorubicin by the PI3K/AKT pathway via sponging miR-613. Biosci Rep. 2021;41(6):BSR20201389.
- Pan J, Li X, Wu W, et al. Long non-coding RNA UCA1 promotes cisplatin/gemcitabine resistance through CREB modulating miR-196a-5p in bladder cancer cells. Cancer Lett. 2016;382(1):64–76.