
Abstract
In the immunopathogenesis of systemic lupus erythematosus (SLE), there is a dysregulation of specific immune cells, including T cells. The metabolic reprogramming in T cells causes different effects. Metabolic programs are critical checkpoints in immune responses and are involved in the etiology of autoimmune disease. For instance, resting lymphocytes generate energy through oxidative phosphorylation (OXPHOS) and fatty acid oxidation (FAO), whereas activated lymphocytes rapidly shift to the glycolytic pathway. Specifically, mitochondrial dysfunction, oxidative stress, abnormal metabolism (including glucose, lipid, and amino acid metabolism), and mTOR signaling are hallmarks of T lymphocyte metabolic dysfunction in SLE. Herein it is summarized how metabolic defects contribute to T cell responses in SLE, and some epigenetic alterations involved in the disease. Finally, it is shown how metabolic defects could be modified therapeutically.
Introduction
Systemic lupus erythematosus (SLE) is a chronic autoimmune disease characterized by abnormal responses to self-antigens resulting in multi-organ involvement and diverse clinical manifestations [Citation1]. It is caused by a complex combination of genetic, epigenetic, and environmental factors. Specific autoantibodies secreted from B cells are the main factor that causes tissue damage. However, other immune cells, such as T cells, neutrophils, macrophages, and plasmacytoid dendritic cells (pDC), are involved in SLE [Citation2]. T cells incite and amplify the inflammatory process in SLE through direct contact with other immune cells. Besides, T cells produce pro-inflammatory cytokines or mediate direct effects on target tissues. Aberrations in T cells have been linked to the immunopathogenesis of SLE, including biochemical, molecular, and metabolic abnormalities [Citation3].
Many studies in SLE have shown abnormal cytokine production by pDC, like type I interferon (IFN-I) and aberrant T cell signaling, which leads to abnormalities in T cell differentiation and excessive activation of mature autoreactive B cells [Citation3]. Further, IFN-α induces BlyS/B cell-activating factor (BAFF), which in turn promotes peripheral mature B cells survival [Citation4]. Mature autoreactive B cells produce autoantibodies that bind to nuclear antigens form immune complexes, generating innate immune responses. Those nuclear particles mimic viral fragments and activate Toll-like receptors (TLR) on antigen-presenting cells (APC), mainly dendritic cells (DC) and promote their maturation [Citation5]. Moreover, IFN-α upregulates TLR7 and interferon regulatory factor 7 (IRF7) expression in DC and monocytes, thus enhancing the immune response to nucleic-acid-containing immune complexes. The persistent activation of DC by lupus autoantigens induces T cell activation and proliferation [Citation6].
Abnormal activation of the T Cell Receptor (TCR) and Phosphoinositide-3 Kinases (PI3K)-Akt-mTOR signaling pathways affect T cells' function and differentiation. Besides, aberrant cytokine production and the activation of Janus kinase and signal transducer and activator of transcription (JAK-STAT) pathways are also involved in the differentiation of pathogenic effector T cells (Teff) and impaired regulatory T cells (Treg) [Citation3]. In addition to the aberrant pathways, T cell mitochondrial dysfunction has been associated with SLE disease progression [Citation7], and metabolomic has demonstrated altered intermediates related to main metabolic pathways in patients with SLE [Citation8]. Immunometabolism has become a promising branch of research to understand immune cell differentiation and function, and it could be deployed therapeutically [Citation9].
Previous knowledge allowed the development of therapeutic options or repurposing of recognized therapies for SLE treatment. However, the understanding of metabolic control of T cell function is partial and is currently investigating the metabolism of T cells and its implication in different autoimmune diseases [Citation10]. This narrative review will first be described the normal metabolism of the T cell and later the current understanding of T cell metabolism in SLE. Finally, some new findings of epigenetic control of T cell metabolism and possible therapeutic targets in SLE according to metabolism alterations will be discussed.
Methods
Search strategy
A literature search was conducted, the primary database was PubMed. However, other databases also used were: ScienceDirect and Google Scholar. Search terms included ‘T cells metabolism,’ ‘systemic lupus erythematosus,’ ‘glycolysis,’ ‘glutaminolysis,’ ‘fatty acid oxidation,’ ‘lipid metabolism,’ ‘mitochondrial dysfunction,’ ‘oxidative stress,’ ‘mTOR signaling,’ ‘epigenetic control,’ and these were used in different combinations. English and Spanish-language papers were reviewed.
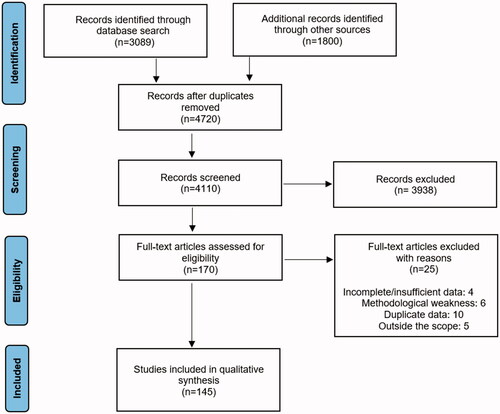
Articles were read and assessed for relevance. Papers with accessible full text were included, and there was no filter regarding the publication dates. shows the PRISMA flow diagram of the search. Finally, 145 manuscripts were chosen to be included in the review. Five major themes were identified, all of them focused on the T cell metabolism in SLE: mitochondrial dysfunction, glutamine and glucose metabolism, lipid metabolism, mTOR signaling, and epigenetic alterations.
Figure 1. PRISMA flow diagram of the review. The flow diagram template was adopted from the PRISMA statement.
T cell metabolism
Immune cells use different metabolic pathways to provide energy for cell survival and synthesize numerous effector molecules. The processing of nutrients like glucose, glutamine, and fatty acids produces ATP to meet energy requirements [Citation11]. Metabolic reprogramming occurs when immune cells are activated by intrinsic or extrinsic signals, changing from oxidative phosphorylation (OXPHOS) that is time-consuming to rapid aerobic glycolysis. Glucose is involved in both glycolysis and OXPHOS [Citation12].
Glycolysis converts glucose to pyruvate and generates ATP (2 molecules). Pyruvate is converted to either lactate or acetyl coenzyme A (acetyl-CoA), at rest, it is more likely to be converted to lactate in the cytoplasm via lactate dehydrogenase, leading to the production of two molecules of ATP per molecule of glucose, rather than to enter the tricarboxylic acid (TCA) cycle (or Krebs cycle) as acetyl-CoA in the mitochondria via pyruvate dehydrogenase (PDH). Acetyl-CoA enters the TCA cycle and produces ATP (36 molecules) via OXPHOS [Citation11]. Glycolysis commonly refers to the lactate end-point branch of glycolysis, while the other is called glucose oxidative or mitochondrial metabolism.
Mitochondrial metabolic pathways are required for T cell proliferation to generate ATP for biosynthesis. Moreover, mitochondria are required for T cell activation to produce reactive oxygen species (ROS) for activation of nuclear factor of activated T cells (NFAT), which functions as a transcription factor for the production of interleukin (IL)-2, an important cytokine that acts as an activating factor for T cells [Citation13].
There is noticeable diversity between T cell subgroups concerning which metabolic pathway predominates in energy production. The differentiation of the naïve T cells is regulated by cytokines and their metabolic reprogramming [Citation14]. Naive CD4+ and CD8+ T cells at rest utilize low rates of OXPHOS. Treg cells and memory CD4+ use OXPHOS and fatty acid oxidation (FAO) to produce energy. Upon activation, naive T cells rapidly shift metabolism from OXPHOS to aerobic glycolysis with large glucose consumption; nevertheless, OXPHOS might also be involved. Differentiated effector CD4+ cells utilize glutaminolysis, rapid glycolysis, and fatty acid synthesis [Citation15].
Immune cells can utilize the TCA cycle to generate energy by metabolizing glutamine through glutaminolysis or fatty acids through β-oxidation. Further, some molecules of the TCA cycle serve as precursors to synthesize fatty acids and lipids, amino acids, nucleotides (for example, via the pentose phosphate pathway or citrate), and other molecules [Citation16]. The pentose phosphate pathway (PPP) branches from the glycolysis pathway and converts glucose-6-phosphate to ribose-5-phosphate to generate reducing equivalents, including NADPH, for the synthesis of nucleic acids and amino acids during cell activation [Citation17].
In naive T cells, upon engagement of the TCR, an anabolic metabolism phenotype is adopted to permit the fast-shifting from quiescence to activation, proliferation, gaining of effector functions, and rise the mitochondrial mass for the energy supply. T lymphocytes activated after antigen presentation proliferate and migrate to the site of damage via the interaction of the TCR complex with the MHC and the co-stimulation between CD28 in T cells and CD80/86 (B7.1/2) in DC. Then three different transduction pathways could be activated: (a) the calcium-calcineurin pathway, which activates NFAT; (b) the RAS-MAPK pathway, which activates the transcription factor AP-1 (activator protein 1); and (c) the transcription factor NF-kB. In turn, NFAT, AP-1, and NF-kB promote the expression of IL-2, which binds to IL-2R (CD25). This receptor can activate three pathways (PI3K/Akt/mTOR, JAK/STAT, and MAPK) that promote cell proliferation [Citation18]. The opposite occurs with the engagement of cytotoxic T lymphocyte-associated protein 4 (CTLA-4) and programmed death 1 (PD-1) [Citation19].
Four major metabolic checkpoint pathways have been proposed that orchestrate T-cell metabolism and differentiation. AMPK (adenosine monophosphate-activated protein kinase), mTOR (mammalian target of rapamycin), HIF-1α (hypoxia-inducible factor 1α), and Myc (myelocytomatosis oncogene). AMPK is a sensor of the AMP/ATP ratio that inhibits mTOR activity. In turn, mTOR is activated through the PI3K/AKT pathway and glucose, and a low amino-acid concentration and AMPK inhibit its activity. mTOR activates the transcription factors HIF-1α (inhibited by oxygen) and Myc. AMPK and mTOR act as post-transcriptional factors. Meanwhile, HIF-1α and Myc induce a transcriptional regulation [Citation18]. For instance, Myc regulates proteins involved in glycolysis and glutaminolysis, and HIF-1α enhances glycolysis [Citation20,Citation21].
Glucose metabolism in T cells
Once CD4+ T cells are activated, the engagement of TCR and co-stimulatory receptors (like CD28) leads to the upregulation of the glucose transporter GLUT1 via PI3K-Akt signaling and upregulation of enzymes via HIF-1α and Myc [Citation20]. Glucose transporters allow the importation of glucose into the cell, so they constitute the first step of glycolysis. GLUT1 is the main glucose transporter expressed by T cells. GLUT1 deficiency in T cells of LckCreGlut1fl/fl mice decreased glucose utilization by CD4+ T cells, diminished differentiation to Teff cells, and reduced T cell proliferation [Citation22]. Meanwhile, in Glut1 and myristoylated Akt (mAkt) transgenic mice, the overexpression of GLUT1 in T cells leads to increased glucose uptake and production of IL-2 and IFNγ [Citation23].
HIF-1α regulates glucose metabolism in CD4+ and CD8+ T cells and controls Th1 and Th17 cells differentiation [Citation24]. Glycolysis is increased in Th1, Th17, T follicular helper (TFh) cells, and CD8+ T cells. The importance of glycolysis in Th2 and TFh cell differentiation is unclear [Citation25]. Indeed, a distinct feature of Th17 cells is the overexpression of HIF-1α and reduced PDH activity leading to enhanced pyruvate to lactate production and decreased pyruvate to acetyl-CoA [Citation26]. Otherwise, glycolysis during Th2 cell differentiation requires the Ras homolog gene family member A (RhoA), which is regulated by mTORC2 [Citation27].
Specific enzymes control the activity of PDH in the mitochondria: pyruvate dehydrogenase phosphatase catalytic subunit 2 (PDP2) activates PDH, whereas PDH kinase 1 (PDK1) phosphorylates PDH (active form) to phospho-PDH (inactive form). The gene expression of Pdp2 is suppressed by inducible cAMP early repressor (ICER), an imperative transcriptional factor in Th17 cells [Citation26]. ICER promotes glutaminolysis, suppresses the production of IL-2, and stimulates the production of IL-17 [Citation28]. Th17 cells produce higher lactate levels because they express ICER and increased levels of PDK1. The inhibition of PDK1 prevents the formation of Th17 cells and promotes IFNγ production and Th1 cell differentiation [Citation29]. The cAMP response element modulator (CREM) moderates the transcription of cAMP-responsible genes. CREMα and ICER are increased in Th17 cells [Citation28].
On the other hand, glycolytic activity is required for optimal memory CD8+ T cell function and is promoted by serum acetate accumulation following systemic infections [Citation30]. Glycolysis is also needed for Treg cell activation and proliferation, especially in the presence of abundant pro-inflammatory signals. However, this metabolic pathway interrupts Treg cell function, and during its proliferation, the suppressive capacity of Treg cells is reduced [Citation31]. During Treg cell induction in the periphery, increased expression of FOXP3, that represses the expression of Myc, reprograms the cell to function in low-glucose environments, achieving a lipid metabolism instead of one based on glucose, all of which maintains the suppressive function of peripherally induced T cells (pTreg), and peripheral immune tolerance during tissue injury [Citation32].
Glucose deprivation leads to decreased cellular ATP levels and AMPK activation, enhancing Glut 4 transcription and translocation and promoting glucose intake. AMPK accelerates catabolism and negatively modulates some critical proteins in ATP-consuming reactions, such as mTOR and glycogen synthase, leading to inhibition of gluconeogenesis and glycogen, lipid, and protein synthesis. AMPK inhibits mTOR activity and vice versa [Citation33]. Inhibition of AMPK and the downstream mTORC1 activation promotes TFh cell differentiation and a lupus-prone phenotype [Citation34]. TFh cells are increased in patients with SLE and lupus-prone mice, and their numbers correlate with disease activity [Citation35].
Amino acid metabolism in T cells
Amino acids and their metabolism play a crucial role in immune function. Mainly, glutamine catabolism regulates immune cell function. Amino acids are required in inflammation, protein and nucleic acid synthesis, the regulation of mTORC1 signaling, and the control of stress pathways in T cells. Glutamine is necessary for IL-1 induction by macrophages upon lipopolysaccharide stimulation. Glutamine metabolism also modulates the immune responses of T cells and B cells [Citation36].
Glutamine is a non-essential and the most abundant amino acid in the circulation. Glutamine can enter the cell through the alanine, serine, cysteine-preferring transporter 2 (ASCT2) [Citation37]. T cell activation involves large glutamine consumption and requires glutamine in response to antigen receptor stimulation. Glutaminase (the first enzyme in the glutaminolysis pathway, which generates glutamate from glutamine) leads to increased ROS levels, which are increased upon hypoxia, so glutamine metabolism controls ROS stress [Citation38]. ICER induced glutaminase transcriptionally, promoting glutaminolysis and Th17 cell generation. Th17 cells depend on glutaminolysis more than Th1, Th2, and Treg cells [Citation39].
Glutamine is hydrolyzed to glutamate during glutaminolysis. Glutamate is metabolized to α-ketoglutarate, an intermediate of the TCA cycle, and a substrate for histone demethylases and DNA demethylases [Citation40]. Glutaminolysis is essential for maintaining T cell activation and proliferation. Moreover, glutamine deprivation, deficiency of the transporter ASCT2, and inhibition of α-ketoglutarate production promote Treg cell induction and reduce Th1 and Th17 differentiation [Citation37]. Glutaminase inhibition results in glutamate deficiency, endorses Th1 differentiation, and impairs mTORC1-dependent Th17 differentiation but does not alter Treg cell differentiation [Citation41].
Catabolism of tryptophan produces metabolic intermediates that endorse pTreg cell induction. Besides, pTreg cells express high levels of enzymes involved in amino acid catabolism, and the reduction of some amino acids can significantly affect Treg cell function and maintenance [Citation42]. Otherwise, leucine stimulates mTOR signaling and T cell activation. Leucine deficiency prevents the activity of mTORC1 and limits T cell activation and differentiation into Teff cells [Citation43]. Leucine restriction declines Th17 cell differentiation without affecting Th1 and Th2 cell polarization [Citation44]. Serine is a non-essential amino acid synthesized from 3-phosphoglycerate (a glycolysis intermediate). Serine is a crucial contributor to the one-carbon metabolism cycle, and its metabolites are essential for the purine synthesis pathway. It is also indispensable for Teff cell proliferation [Citation45].
Lipid metabolism in T cells
Lipids play fundamental roles in cell membrane composition, membrane receptor signaling, and energy storage. The essential lipids for cellular function include cholesterol, phospholipids, fatty acids, triglycerides, and glycosphingolipids (GSL). Biosynthesis of fatty acids and cholesterol is essential for T cell proliferation and differentiation in Teff cells, especially Th17 cells. Fatty acids synthesis occurs from acetyl CoA in the cytosol due to the mediation of Acetyl-CoA carboxylase 1 (ACC1). ACC1 promotes metabolic reprograming after TCR stimulation and enhances Th1 and Th17 cell differentiation [Citation46]. FAO is favorably utilized by non-inflammatory and tolerogenic immune cells, while inflammatory responses are characterized by fatty acid synthesis [Citation47].
Meanwhile, cholesterol is synthesized by endoplasmic reticulum-bound 3-hydroxy-3-methylglutaryl-CoA reductase (HMGCR). Statins that inhibit HMGCR reduce the differentiation of Th17 cells [Citation48]. Apolipoprotein E (ApoE) and low-density lipoprotein receptor (LDLR) allow cholesterol recycling and intercellular transport. ApoA1 and high-density lipoproteins (HDL) facilitate the efflux of cholesterol from immune cells via liver X receptor (LXR)-regulated genes [Citation49]. Cholesterol regulates immune responses through at least three mechanisms. First, the cell needs cholesterol for membrane synthesis. Secondly, cholesterol is a key component of lipid rafts [Citation50]. Finally, I IFN reprograms lipid metabolism, including an increased import of cholesterol [Citation51].
Quiescent T cells and Treg cells use mainly FAO, a mitochondrial aerobic process responsible for producing acetyl CoA from fatty acids that enter the TCA cycle [Citation52]. AMPK increases the expression of carnitine palmitoyl transferase I (CPT I) that promotes FAO, whereas AMPK-dependent phosphorylation of ACC1 inhibits fatty acid synthesis [Citation53]. Treg cells have high expression levels of CPT I [Citation54]. ACC1 controls the Th17/Treg cell ratio, might also promote the development of autoimmune pathology, and is hence a potential therapeutic target [Citation17].
Tissue-resident Treg cells use exogenous fatty acids for their induction and maintenance. Consequently, adding fatty acids to cells in culture increases Treg cell, but not Teff cell differentiation [Citation52]. Treg cells in visceral adipose tissue show high levels of CD36 (the scavenger receptor that acts as lipid transport), which facilitates the ingress of exogenous fatty acids [Citation55]. Otherwise, the type of diet-derived lipids can mark T cell differentiation. Western diets are rich in long-chain fatty acids (LCFAs), and fiber-rich diets have abundant short-chain fatty acids (SCFAs). LCFAs endorse the generation of pathogenic Th1 and Th17 cells, whereas SCFAs promote Treg cells in the intestine [Citation56].
Finally, lipid metabolism is primordial in central memory T cells, which utilize endogenous lipids and exogenous glycerol for lipid biosynthesis to produce the substrates needed for FAO [Citation57]. Meanwhile, tissue-resident memory T cells depend on the presence of exogenous fatty acids for their survival and maintenance [Citation58]. Indeed, in long-lived memory T cells, regulation of FAO and mitochondrial biogenesis allows their longevity. However, after the re-stimulation, the memory T cell modifies its metabolism to glycolysis and facilitates the maturation of memory T cells into Teff cells [Citation59].
mTOR pathway in T cells
mTOR is a serine/threonine kinase. Two multimeric complexes mediate its signaling, mTORC1, and mTORC2, which share the catalytic subunit mTOR but are distinguished by the scaffold proteins regulatory-associated protein of mTOR (RAPTOR) and rapamycin-insensitive companion of mammalian target of rapamycin (RICTOR), respectively [Citation49]. These complexes mediate distinct signaling pathways to permit metabolic reprogramming in T cells, generating glycolysis and glutaminolysis instead of FAO and pyruvate oxidation [Citation60].mTOR helps preserve cell homeostasis by regulating metabolic signals and nutrient availability to drive genetic programs involved in cell growth, activation, energy use, proliferation, and survival [Citation61]. mTORC1 activation changes T cell metabolism to deliver the constituents required for Th1, Th17, and TFh proliferation and differentiation. However, this signaling is also essential for the function of Treg [Citation62]. Mitochondrial dysfunction and PPP overactivation can activate mTORC1, which correlates with the metabolic need of activating T cells [Citation63].
Signaling through mTORC1 shifts the balance of CD4+ T cell polarization away from Treg development and toward Th1 and Th17 phenotype by increasing glycolysis (in these subgroups) triggers retinoic acid-related orphan receptor gamma t (RORγt) and STAT3 phosphorylation [Citation64]. Protein phosphatase 2A (PP2A) controls mTORC1 signaling in Treg cells. Moreover, glycolysis, OXPHOS, and expression of the large neutral amino acids transporter small subunit 1 (LAT1) increase in the absence of PP2A in Treg cells. All of which leads to the deviation in the development of Treg cells toward Teff cells. In Treg cells, PP2A is specially activated by ceramide, whose concentration is increased thanks to FOXP3 [Citation65].
Treg cells have high levels of activated AMPK, which inhibits mTORC1, declines GLUT1 expression, and helps FAO and Treg cell induction [Citation66]. Upstream of AMPK, the liver kinase B1 (LKB1) acts independently of AMPK and stabilizes FOXP3 expression, and promotes immunosuppressive transforming growth factor β (TGF-β) signaling, so LKB1 is essential for the maintenance of the Treg cell lineage [Citation67]. However, some signaling via the PI3K–mTORC1 axis is obligatory in T cells for CTLA-4 expression, control cholesterol, lipid metabolism, and the inhibition of the mTORC2 pathway [Citation68]. Mechanistically, PI3K-mTORC2 activation in Treg cells induces the expression of glucokinase, which binds actin and promotes the cytoskeletal remodeling necessary for cell migration [Citation49].
Metabolic abnormalities in T cells in lupus
T cell hyperactivity, mitochondrial dysfunction, and oxidative stress
T cells from mice and patients with lupus are chronically active with up-regulation of calcium signaling, TCA cycle activity, and dependency on OXPHOS for their energetic needs [Citation69]. Mitochondria is an essential organelle in cellular metabolism and immune responses. It is the primary energy-producing organelle through the TCA cycle production of ATP and OXPHOS, but it is also a signal-transducing organelle [Citation70]. Mitochondria in mice and patients with lupus are hyperpolarized toward augmented glucose-derived OXPHOS, which results in T cells with depleted antioxidant capacity associated with lower glutathione and NADPH levels [Citation71]. CD4+ T cells isolated from people with SLE have hyperpolarized megamitochondria due to increased mitochondrial biogenesis and defective mitophagy, with paradoxically decreased ATP production and marked escape of ROS outside of the organelles [Citation72].
The oxidative stress resulting from all the described alterations contributes to pathogenesis in SLE by increasing apoptosis, decreasing the clearance of apoptotic bodies, and through oxidative modification of many biomolecules, such as DNA and enzymes [Citation73]. Furthermore, the surface glycoprotein CD3ζ chain is degraded and replaced by the FcεRIγ chain in the TCR due to its oxidative stress, a process that is promoted by PP2A, which is increased in SLE T cells. FcεRIγ can promote tyrosine-protein kinase SYK recruitment, resulting in T cell hyperactivation due to both increased sensitivity of the receptor and increased downstream signaling [Citation74]. TCR signaling hyperactivity results in mitochondria hyperpolarization, culminating in increased production of ROS and more oxidative stress [Citation17].
ROS is a group free radical generated during mitochondrial respiration as the result of incomplete reduction of oxygen. Under controlled physiological circumstances, ROS plays a positive role in CD4+ T cell proliferation, differentiation, and cytokine production [Citation13]. Indeed, low ROS production leads to reduced production of IL-2 and IL-4 [Citation75]. Additionally, ROS activates mTOR which is involved in maintaining and promoting increased mitochondrial biomass by decreasing mitophagy [Citation76]. However, a study in patients with SLE demonstrated that ribonucleoprotein immune complexes induced mitochondrial membrane hyperpolarization and ROS generation, resulting in both neutrophil extracellular trap (NET) formation and oxidation of mitochondrial DNA (mtDNA) [Citation77].
A study with SLE patients demonstrated that, in vitro, extracellular oxidized mtDNA is a potent proinflammatory mediator and induces the IFN-I signaling pathway, which in turn depends on the DNA sensor STING. On the other hand, an in vivo lupus mouse model (MRL/lpr mice) confirmed that mitochondrial ROS inhibition reduces disease severity and attenuates IFN-I responses [Citation77,Citation78]. Oxidized mtDNA in a normal state is disassociated from a protein involved in the maintenance and compaction of the mitochondrial genome into nucleoids, called transcription factor A mitochondria (TFAM). The dissociation from TFAM permits that the oxidized mtDNA goes to lysosomal degradation. In SLE, disassociation of TFAM is inhibited due to downregulated phosphorylation, leading to the accumulation of oxidized mtDNA and IFN-I production [Citation78]. So elevated ROS cause multiple organ injury, which produces cell apoptosis again, leading to a vicious circle in SLE patients [Citation79].
Hypoxia regulates immunometabolism in numerous ways depending on the transcription factor HIF-1α [Citation80]. HIF-1α is accumulated under hypoxic conditions, and it increases levels of multiple genes implicated in cell adaptations to hypoxia. HIF-1α has been demonstrated to be essential for Th1 and Th17 cell differentiation [Citation21]. Nevertheless, HIF-1α has positive and negative effects on Treg cell differentiation [Citation81]. Otherwise, ROS modulate the HIF pathway, although the exact mechanism remains unclear [Citation82].
In addition to chronic stimulation and dependence on OXPHOS, genetics could have a vital role in altered mitochondrial homeostasis. Studies from lupus-prone mice expressing the susceptibility locus Sle1c2 (B6.Sle1c mice), involved in T cell mitochondrial metabolism, have demonstrated that those mice develop spontaneous lupus-like autoimmunity. B6.Sle1c mice express decreased levels of estrogen-related receptor γ (ERRγ), a nuclear receptor that negatively regulates T cell activation, and the T cells from that mice have a decreased mitochondrial mass and chronic mitochondrial hyperpolarization [Citation83]. Moreover, in humans, an SNP variant of the ATP6 or F0F1-ATPase gene has been related to SLE development. Inhibition of this ATPase generates mitochondrial hyperpolarization and ATP depletion, just as it happens in SLE [Citation84,Citation85].
Glutamine and glucose metabolism
Glucose metabolism
Chronically activated CD4+ T cells from SLE patients or lupus-like mice models have higher oxygen consumption [Citation69]. Chronic stimulation of T cells in lupus depends on OXPHOS, whereas the acute activation of T cells is maintained by aerobic glycolysis. The higher rates of glycolysis also result in increased mTORC1 signaling and promote autoimmunity [Citation86]. OXPHOS fails to produce sufficient ATP in SLE T cells even when T cells have enlarged mitochondrial biomass. Then, in SLE, secondary glycolysis is observed [Citation29].
The overexpression of GLUT1 augments the activation of murine T cells. It generates an accumulation of effector and follicular T cells, which have signs of autoimmunity, including the production of autoantibodies and deposition of immune complexes in the glomeruli [Citation23]. Meanwhile, in people with active and inactive SLE, Koga et al. found GLUT1 overexpression in effector memory CD4+ T cells. The inhibition with KN‐93 of the calcium/calmodulin-dependent protein kinase IV (CaMK4) can reverse the increased Glut1 expression. CaMK4 is a multifunctional kinase that regulates the expression of several genes and is overexpressed in SLE T cells [Citation87]. CaMK4 can also endorse glycolysis, promoting the activity of pyruvate kinase M2, the final enzyme in glycolysis, which is requisite for Th17 differentiation, an important T cells subset in the SLE pathogenesis [Citation88].
As previously mentioned, PDH promotes the OXPHOS over the lactate glycolytic pathway, and PDP2 activates PDH. Th17 cells have increased ICER activity (compared with both Th17 cells from healthy individuals and other T cell subsets), decreasing PDP2 [Citation26]. Then, memory Th17 cells from patients with SLE have a decreased PDP2 expression. Forced expression of PDP2 suppressed Th17 differentiation in lupus-prone MRL/lpr mice and patients with SLE. These results are consistent with the glycolytic requirements of Th17 cells [Citation25], and the expansion of Th17 cells in SLE patients [Citation89]. In this same sense, manipulate PDP2 could be a potential therapeutic strategy in SLE.
Glutamine metabolism
Glutaminolysis is essential for T cell activation and proliferation. Whether the rates of glutaminolysis are increased in SLE is undetermined. Nevertheless, enzymes involved in glutaminolysis are significantly elevated in CD4+ T cells from the B6.Sle1.Sle2.Sle3 (TC, for triple congenic) model of lupus [Citation90] and decreasing the rate of glutaminolysis in lupus-prone MRL/lpr mice, through glutaminase 1 deletion or inhibition, decrease Th17 cell differentiation and reduces disease activity due to HIF-1α and glycolysis downregulation [Citation91]. Besides, the glutamine analog 6-diazo-5-oxo-L-norleucine (DON) impedes glutaminolysis, which inhibits T cells' activation-induced proliferation in vitro [Citation20].
Under ischemic conditions, pTreg cells cannot function with low-glucose conditions as ischemia-mediated HIF-1α production endorses Th17 differentiation and can lead to SLE development [Citation16]. The ICER/CREMα-suppressive splice variants of CREM are highly expressed in SLE CD4+ T cells [Citation28] and enhance the transcription of glutaminase, promoting glutaminolysis and Th17 cell differentiation [Citation39]. Moreover, glutaminolysis regulates TFh, the inhibition of glutaminolysis reduces TFh cells, and the production of the dsDNA antibody [Citation35].
CD8+CD38high T cell population has decreased cytotoxic capacity, degranulation, and expression of cytolytic enzymes. Those cells are expanded in patients with SLE who have more infections. CD38 is an ectonucleotidase that degrades NAD and suppresses cytotoxicity-related molecules through the histone methyltransferase EZH2, so it is a marker of T cell exhaustion [Citation92]. Indeed, CD38 is overexpressed in CD4+, CD8+, or CD25+ SLE T cells [Citation93] and, in vitro generated CD38- T cells have enhanced OXPHOS and higher glutaminolysis rates [Citation94].
Lipid metabolism
Lipid metabolism also has a role in the pathogenesis of autoimmunity. Indeed, a high-fat diet in mice provokes cholesterol accumulation in the spleen and lymph nodes as well as autoantibody production [Citation95]. Lipid rafts of the cell plasma membrane are composed, in a great measure, of cholesterol and GSL. Inside them, signaling-related molecules store to allow cell activation. SLE CD4+ T cells have an increased lipid rafts formation compared with healthy individuals, which contributes to increasing TCR signaling [Citation74,Citation96]. Besides, SLE CD4+ T cells generate alterations into the lipid rafts that evoke earlier and higher calcium responses, which result in heightened and accelerated T cell signaling [Citation96]. Enhanced lipid raft aggregation makes a more fluid membrane, and molecules move around faster [Citation70].
In part, alterations in TCR signaling in SLE could be attributed to lipid alterations in the plasma membrane. Specifically, the GSL in resting T cells from patients with SLE have an altered pattern, resembling activated cells. An in vitro study demonstrated that the inhibition of GSL synthesis reduced T cell activation and decreased anti-dsDNA antibody titers in SLE patients [Citation97]. Meanwhile, in the renal specimen of MRL/lpr mice and patients with lupus nephritis has been demonstrated an over-expression of β-1,4-galactosyltransferase 5 (β4GalT-5) and neuraminidase 1 (NEU1) related with increased synthesis of GSL [Citation98].
The LXR is a member of the nuclear receptor family of transcription factors. There are two isoforms: LXRα and LXRβ. LXRs regulate lipid metabolism and promotes GSL synthesis. LXR has proinflammatory and anti-inflammatory functions, and high levels of LXRβ have been designated as responsible for GSL accumulation in SLE CD4+ T cells [Citation97,Citation99]. The use of an LXR antagonist can normalize the production of GSL by CD4+ T cells and their function in vitro [Citation97].
Friend leukemia integration 1 (FLI1) is another transcription factor expressed in mature T and B cells and some kidney cells, it regulates GSL synthesis, and elevated FLI1 levels predispose to SLE. Hence, FLI1-haplodeficiency reduced levels of GSL, decreased T cell activation, ameliorated disease, and promoted survival in MRL/lpr mice [Citation100]. In contrast, the overexpression of FLI1 in mice generates progressive renal disease and renal failure due to tubulointerstitial nephritis and immune-complex glomerulonephritis [Citation101]. FLI1 upregulates the expression of CXCR3 on T cells and the expression of chemokines in the kidneys, which enhances T cell migration to the kidneys [Citation102].
Besides, activated T cells from SLE patients show a reduced induction of B and T lymphocyte attenuator (BTLA), an inhibitory receptor-like CTLA-4 and PD-1. In SLE T cells, the capacity of BTLA to detain activation is impaired, which could be associated with deficient BTLA recruitment to the immunological synapse. A study with SLE patients supports this hypothesis, and the authors used N-butyldeoxynojirimycin (NB-DNJ), a glucosylceramide synthase inhibitor that normalizes lipid metabolism in CD4+ T cells from SLE and restored BTLA inhibition. The dissociation of TCR molecules in the presence of this inhibitor allowed BTLA recruitment to TCR clusters that link GSL homeostasis to intracellular trafficking of components of the TCR [Citation103].
Regarding the impact of diet on the pathogenesis of SLE, a Western diet associated with atherogenic dyslipidemia enhanced the production of autoantibodies and the severity of lupus by expanding the number of TFh cells in ApoE or LDLR-deficient lupus-prone BXD2 mice. In the same study, authors reported that dyslipidemia induces IL-27 production by DCs, which in turn expands TFh cells while inhibiting the differentiation of follicular Treg cells. Blocking IL-27 in atherogenic mice reduced autoantibodies and TFh cells to levels similar to those of control mice [Citation104]. On the other hand, a link between autoimmunity and cellular cholesterol accumulation has been suggested. It was demonstrated in a study in which an almost complete absence of HDL due to ApoA1 deletion induced autoimmune phenotypes, characterized by cholesterol-engorged, enlarged lymph nodes, anti-dsDNA autoantibodies, and increased T cell activation [Citation105].
mTOR signaling and interconnection of the metabolic pathways in SLE
As was mentioned, mTOR is an axis in the cellular metabolic signal network, regulating cellular growth and energy utilization. CD4+ T cells from SLE patients and lupus-prone mice showed increased mTOR activation [Citation90]. The oxidative stress with the subsequent ROS production, the overactivity of PPP, and the upregulation of CaMK4 led to enhanced mTOR activity in SLE [Citation63,Citation106]. High mTOR expression is a relevant signaling mechanism leading to abnormal T cell metabolism and Th17/Treg imbalance in SLE patients [Citation107]. More specifically, increased activation of mTORC1 is observed in CD4+ T cells obtained from SLE patients and lupus-prone mice, leading to elevated IL-17 and IL-4, producing double-negative (CD3+CD4−CD8−) T cells expansion and Treg depletion [Citation62]. Double-negative T cells stimulate B cells to produce autoantibodies in SLE [Citation108].
Unrestricted mTORC1 signaling provokes severe SLE-related manifestations, demonstrated in patients with mutations in tuberous sclerosis complex genes that are known suppressors of mTORC1 signaling [Citation109]. The inhibition of mTORC1 with rapamycin leads to Treg cell expansion, contraction of Th17 cells, and suppression of STAT3 signaling [Citation108]. Further, the inhibition of mTORC1 activity through metformin (that promotes AMPK activation) alleviates disease manifestations in lupus-prone mice [Citation110].
PP2A is involved in SLE development by regulating the production of IL-2 and IL-17 in CD4+ T cells [Citation111]. The deletion of PP2A in Tregs resulted in increased mTORC1 activation and a severe autoimmune disorder [Citation65]. Then, mTOR activity is necessary for Treg development and function, but its activation level must be kept in check by PP2A. Besides, the function and differentiation of follicular Treg cells that suppress GC B cells and TFh cells are also mTORC1-dependent [Citation112]. The previous demonstrates the importance of mTOR in the differentiation of T cell subgroups and the delicate spectrum that separates normality from autoimmunity when given by aberrant expressions of mTOR.
The glucose, fatty acid, and amino acid metabolisms are interlinked and can be co-regulated. mTOR regulates glycolysis and fatty acid synthesis in activated T cells [Citation113]. Additionally, mTOR promotes mRNA translation and lipid synthesis and senses amino acids and growth factors [Citation114]. Kynurenine (a tryptophan metabolite) can also stimulate mTORC1 activity as its accumulation promotes oxidative stress. Kynurenine is elevated in SLE patients due to impaired degradation, which is associated with low levels of NADPH-dependent kynurenine hydrolase [Citation115]. In turn, highly activated mTOR promotes Th17, Th2, and Th1 cell lineages differentiation [Citation116].
Otherwise, a study in TC mice showed that TFh cells had a high level of mTORC1 activation, which was reduced by the inhibition of glucose metabolism, and in this way also with the decrease in TFh cells, GC B cells, and autoantibody production [Citation35]. This links glycolysis, mTORC1 activation, and TFh expansion in lupus. Otherwise, evidence implicates mTORC2 with TFh cell differentiation, and germinal center (GC) formation depends on TFh, which are expanded in people with SLE [Citation24]. In summary, metabolic changes are fundamental to cell immune function, and mTOR is a master regulator that senses and integrates diverse nutritional and environmental signals, including growth factors, amino acids, energy levels, and cellular stress [Citation117]. So, it is a potential therapeutic target that could act in different points of the T cell metabolism.
Epigenetic control of metabolism in SLE
Epigenetics can modify gene expression through processes like methylation/demethylation of DNA, acetylation/deacetylation of histone and nonhistone proteins, and microRNA (miRNA) regulations, thus controlling immune response in lupus. However, these epigenetic regulations are reversible and are influenced by the presence of metabolic intermediates [Citation118]. For example, the rapid maturation of memory T cells into Teff cells upon stimulation, associated with a rapid switch to glycolysis, is a process mediated by mTORC2–AKT signaling and epigenetic changes, such as deacetylation mediated by NAD+ sensitive class III histone deacetylases [Citation119]. Additionally, 2-hydroxyglutarate (a glutamine metabolite) could hypermethylate the Foxp3 gene locus and inhibit Foxp3 transcription, thus enhancing Th17 cell differentiation, regulating Th17/Treg balance by an epigenetic mechanism [Citation120].
Both oxidative stress (consequently ROS) and PP2A inhibit the expression of DNA methyltransferase 1 (DNMT1) in T cells [Citation121,Citation122], thus leading to DNA hypomethylation and upregulation of genes associated with the pathogenesis of SLE [Citation123]. Consequently, targeting hypomethylation in specific cell subgroups could be a potential therapeutic strategy in SLE. Indeed, inhibition of DNA methylation in CD4+ T cells enables the expansion of Treg cells. In contrast, inhibition of DNA methylation in CD8+ T cells stimulates cytotoxicity and prevents the expansion of pathogenic double-negative T cells in lupus-prone mice [Citation124].
The JMJC domain-containing histone demethylases regulate gene expression too. ROS can decrease JMJC domain-containing histone demethylases activity [Citation125], resulting in activated T and B cells in SLE patients [Citation126]. Cheng et al. demonstrated that the histone demethylase activity is influenced by mTOR through modulation of HIF-1α expression, promoting JMJC demethylase activity [Citation117]. mTOR is sensitive to hyperglycemia and amino acids and therefore enhances HIF-1α transcription [Citation16]. In conclusion, demethylase enzyme activity is regulated by metabolic intermediates, and its activity is also influenced by mitochondrial dysfunction through ROS production [Citation7].
Possible therapeutic targets in SLE according to metabolism alterations
Targeting metabolic pathways to manipulate autoreactive T cells in SLE is a promising strategy taking more and more interest. Different molecules are in the course of experiments. Some others have already shown some benefit in rearranging the altered metabolic pathways of lupus. Some of the therapeutic molecules will be listed in this part of the text. They have been tested in murine models or humans, some already available in clinical practice and others still in studies, with promising results. Several molecules act at different points in the metabolic gear in T cells simultaneously. shows a general scheme of the therapeutic targets described in the text.
Figure 2. Therapeutic targets that module different metabolic pathways in autoreactive SLE T cells. 2-DG: 2-deoxy-D glucose; AKT: protein kinase B (PKB), also known as AKT; AMPK: adenosine monophosphate-activated protein kinase; BPTES: bis-2-(5-phenylacetamido-1,3,4-thiadiazol-2-yl) ethyl sulfide; CaMK4: calcium/calmodulin-dependent protein kinase 4; FLI1: friend leukemia integration 1; HIF-1α: hypoxia-inducible factor 1α; IL: interleukin; MMF: mycophenolate mofetil; mTORC1: mammalian target of rapamycin complex 1; Myc: myelocytomatosis oncogene; NB-DNJ: N-butyldeoxynojirimycin; PI3K: Phosphoinositide-3 Kinases; ROS: reactive oxygen species; TCR: T cell receptor. Created with BioRender.com (accessed on June 2021).
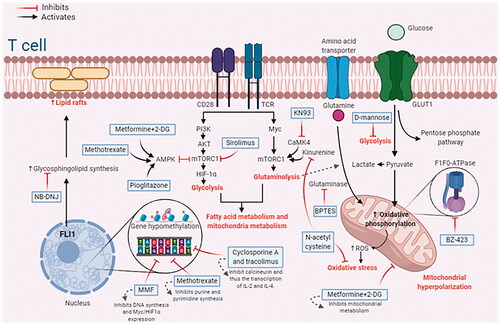
Metformin and 2-deoxy-D glucose (2-DG)
Metformin acts primarily on T lymphocytes' mitochondrial dysfunction and oxidative stress in SLE. Treatment of TC lupus-prone mice with metformin inhibits mitochondrial metabolism due to the inhibition of complex I of the mitochondrial electron transport chain, fixed aberrant T cell metabolism, decreased IFNγ production, and reestablished the IL-2 production [Citation90]. Metformin also normalized in vitro IFNγ production in CD4+ T cells isolated from patients with SLE [Citation127]. The normalization of T cell metabolism through the dual inhibition of glycolysis and mitochondrial metabolism has been proven using a combination of metformin plus glycolytic inhibitor 2-DG. The study demonstrated a synergic effect in vivo and decreased serological markers of SLE disease activity and improved nephritis [Citation90]. Meanwhile, treatments of TC mice with either 2DG or metformin prevent autoimmune activation, while their combination reverses the autoimmune pathology [Citation29].
Moreover, in vivo treatment of lupus-prone mice with 2-DG normalized TFh cell numbers and reversed serological markers of lupus. This glycolytic requirement is limited to autoreactive TFh cells, as TFh cells induced by immunization or infection with influenza virus were not altered by 2DG [Citation35]. This implies that the metabolic requirements of autoreactive CD4+ T cells are distinctive, which could be an important consideration in terms of therapeutic targets [Citation49]. On a bridge between the experimental and the clinical, a proof-of-concept trial showed that the addition of metformin to standard-of-care treatment decreased the frequency of clinical flares, enabled prednisone dose reduction, and reduced body weight in patients with mild-to-moderate SLE [Citation128]. More recently, a study reporting post-hoc pooled analyses from the trial mentioned above [Citation128], and placebo-controlled ‘Met Lupus’ trial suggested that metformin reduced subsequent disease flares in patients with SLE with low disease activity. Subgroup analysis showed that patients with negative anti-dsDNA antibody and normal complement at baseline and a disease duration <5 years with concomitant use of hydroxychloroquine had a better response to metformin [Citation129].
Bz-423 (benzodiazepine) and rapamycin
This molecule impedes mitochondrial metabolism by inhibiting F1F0-ATPase, an ATP synthase whose expression and activity are augmented in autoreactive lymphocytes from mice with lupus. The administration of Bz-423 suppressed disease manifestations like glomerulonephritis and arthritis in MRL/lpr mice. Regarding the way this molecule achieves its action, it is considered that the administration of Bz-423 increased the levels of ROS, which ultimately generates apoptosis of autoreactive cells [Citation85,Citation130]. In the meantime, in vitro treatment with rapamycin decreases glycolysis and mitochondrial potential and corrects the replacement of the CD3ζ chain on CD4+ T cells [Citation90].
mTORC1 and calcineurin inhibitors
The inhibition of mTORC1 blocks T-cell activation and proliferation, prevents Teff-cell metabolism and differentiation, and promotes the generation of Treg cells to maintain its suppressive effects [Citation18]. Some clinical trials showed that sirolimus, a mTORC1 inhibitor, could reduce disease activity in refractory lupus patients, expand their Foxp3+ Treg cells, and inhibit the secretion of cytokines, such as IL-17 [Citation131,Citation132]. This is associated with the inhibition of Th17 and double-negative T cells and promoting Treg cell differentiation and function in patients with SLE [Citation133].
Pioglitazone is a PPAR-γ (a transcription factor for adipogenesis) agonist, activates AMPK, inhibits mTORC1, and promotes the expansion of SLE Treg cells, which express high concentrations of PPAR-γ in vitro [Citation134]. Furthermore, pioglitazone promotes lipid uptake by Tregs by upregulating the expression of CD36 and activating fatty acid oxidation [Citation135].
Calcineurin is a serine-threonine phosphatase that dephosphorylates NFAT, leading to its translocation to the nucleus. NFAT binds to the Myc promoter, increasing cell proliferation via Myc upregulation. Tacrolimus and CsA bind the immunophilins FKBP12 and cyclophilin, respectively, which inhibit calcineurin activity. Consequently, NFAT is not translocated into the nucleus and thus cannot induce the transcription of critical genes like IL-2 and IL-4, leading to immunosuppressive effects [Citation18]. CsA treatment reduces Myc expression in murine CD4+ T cells [Citation136]. Moreover, CsA blocks the expression of LAT1 mRNA following TCR activation, reducing the level of leucine uptake by T cells, leading to a state of anergy and inhibiting CD4+ and CD8+ differentiation [Citation137].
More recently, voclosporin, an analog of cyclosporine with a modification at the amino acid-1 position, has been studied in patients with lupus nephritis. This pharmacologic agent has been designed to improve potency against calcineurin inhibition and better metabolic stability than cyclosporine. Based on positive results from the pivotal phases II and III trials, oral voclosporin was approved in some countries for use in combination with a background immunosuppressive therapy regimen for adults with active lupus nephritis [Citation138,Citation139].
Mycophenolate mofetil (MMF) and methotrexate
MMF is a pro-drug that inhibits DNA synthesis and restricts T cell expansion. This molecule also inhibits Myc and HIF-1α expression and signaling via the PI3K–AKT–mTOR pathway in CD4+ T cells to reduce glycolysis and oxidative stress in CD4+ T cells [Citation140]. On the other hand, methotrexate impedes folate metabolism, essential for cell growth [Citation45]. Methotrexate can also increase adenosine and AMP production, which activates AMPK, and in this way, mTOR signaling is inhibited [Citation18].
KN93-calbiochem and D-mannose
Regarding the therapeutic targets in glycolysis, the pharmacological inhibition of CaMK4 decreases glycolysis and ameliorates disease activity in MRL/lpr mice. Moreover, delivering a CaMK4 inhibitor (KN93-Calbiochem) to podocytes preserved their ultrastructure, averted immune complex deposition and crescent formation, and suppressed proteinuria in lupus-prone mice [Citation141,Citation142]. Otherwise, a study has shown that D-mannose, a C-2 epimer of glucose, natural in plants and fruits and found in human blood at concentrations much lower than glucose, induced Treg differentiation in mice. In vitro, D-mannose stimulated Treg cell differentiation in human and mouse cells by promoting TGF-β activation as an indirect consequence of the inhibitory effect of D-mannose on glycolysis. In contrast, D-mannose increased FAO in T cells [Citation143].
A selective glutaminase 1 inhibitor and N-acetyl cysteine (NAC)
The selective glutaminase 1 inhibitor bis-2-(5-phenylacetamido-1,3,4-thiadiazol-2-yl) ethyl sulfide (BPTES) improved autoimmune pathology in a Th17-dependent manner in MRL/lpr mice and decreased Th17 differentiation in patients with SLE. Moreover, BPTES reduced glycolysis and the expression of HIF-1α protein [Citation91]. Another potential therapy in amino acid metabolism is the cysteine analog, NAC. Cysteine is required for glutathione synthesis, and cysteine is depleted in SLE T cells. Glutathione has antioxidative properties that counter the oxidative stress observed in SLE [Citation115]. Treatment with NAC proved beneficial in SLE patients. It reversed the expansion of double-negative T cells by blocking mTORC1 activity (due to its capacity to disrupt mitochondrial hyperpolarization), stimulated Foxp3 expression, and decreased dsDNA levels [Citation144].
NB-DNJ and nitro-fatty acids
As was mentioned previously, NB-DNJ (inhibits the synthesis of GSL) restores TCR signaling and normalizes lipid metabolism in CD4+ T cells from patients with SLE in vitro [Citation97]. Furthermore, NB-DNJ treatment restores the functionality of BTLA, which improves lipid metabolism in CD4+ T cells in MRL/lpr mice [Citation103]. Nitro-fatty acids also represent potential therapy in SLE. Nitro-fatty acids are endogenously formed fatty acids that inhibit signaling via the adaptor molecule stimulator of IFN genes (STING) in T cells (and in other cells), which is required for the excessive production of type I interferons in SLE [Citation145].
Lipid metabolism plays an essential role in autoimmunity, and the therapeutic targeting of lipid metabolism is promising. It will be necessary to differentiate the distinct roles of lipid metabolism across immune cell subgroups. This includes the possibility of modulating diet to modify lipid metabolism as a potential treatment for SLE [Citation9].
As described during this section, we already have pharmacological agents in clinical practice that could modulate the immunometabolic alterations of T cells in SLE, most notably metformin, pioglitazone, tacrolimus, CsA, voclosporin, MMF, methotrexate, or NAC. Some of these drugs even with human research and positive findings, as mentioned. Other molecules are still in experimental phases in murine or human T cells but with quite promising mechanisms of action, modulating dysfunctional SLE T cells by different metabolic pathways.
Conclusions
T cell metabolism plays a fundamental role in the immunopathogenesis of SLE, with alterations in the mitochondrial function, abnormal metabolism (including glucose, lipid, and amino acid metabolism), and signaling pathways that direct mTOR. These are related and modulated by four metabolic checkpoints, Myc, HIF1, mTOR, and AMPK, which act anabolically or catabolically to reprogram cell metabolism. Although, the knowledge of different signaling pathways and the metabolites involved in the entire process becomes relevant for the pathophysiological understanding of SLE. The subject's relevance increases when it is proposed that therapeutic targets can be generated from this knowledge. These therapeutic molecules would not only act through the metabolic control in the active T cell but also through the cellular differentiation between the cell subsets, which would also allow increasing differentiation toward a T cell linage at the expense of others, for example, elevation in Treg cells instead of Th17 cells.
It is hoped that soon, metabolomics, together with other omics, will be able to clarify many gaps in the understanding of the immunopathogenesis of different pathologies, such as SLE and that a personalized therapeutic option can be offered to patients. Although there is much studied and known, which has been possible especially in the last two decades thanks to advances in different aspects of research in basic sciences, there are still several gaps in knowledge that must be addressed. Research in immunometabolism is expected to offer novel opportunities for monitoring and treating immune-mediated diseases. In this sense, the study in murine models has been very supportive as a basis for the subsequent interpretation of human immunopathological alterations. Nevertheless, interpretations should always be cautious when overlapping the findings in murine and humans. Animal models have allowed us to take a big step forward in understanding human diseases.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Tsokos GC, Lo MS, Costa Reis P, et al. New insights into the immunopathogenesis of systemic lupus erythematosus. Nat Rev Rheumatol. 2016;12(12):716–730.
- Herrada AA, Escobedo N, Iruretagoyena M, et al. Innate immune cells' contribution to systemic lupus erythematosus. Front Immunol. 2019;10:772.
- Katsuyama T, Tsokos GC, Moulton VR. Aberrant T cell signaling and subsets in systemic lupus erythematosus. Front Immunol. 2018;9:1088.
- Jego G, Palucka AK, Blanck JP, et al. Dendritic cells induce plasma cell differentiation through type I interferon and interleukin 6. Immunity. 2003;19(2):225–234.
- Mistry P, Kaplan MJ. Cell death in the pathogenesis of systemic lupus erythematosus and lupus nephritis. Clin Immunol. 2017;185:59–73.
- Ganguly D, Chamilos G, Lande R, et al. Self-RNA-antimicrobial peptide complexes activate human dendritic cells through TLR7 and TLR8. J Exp Med. 2009;206(9):1983–1994.
- Lee HT, Wu TH, Lin CS, et al. The pathogenesis of systemic lupus erythematosus – from the viewpoint of oxidative stress and mitochondrial dysfunction. Mitochondrion. 2016;30:1–7.
- Guleria A, Pratap A, Dubey D, et al. NMR based serum metabolomics reveals a distinctive signature in patients with lupus nephritis. Sci Rep. 2016;6:35309.
- Chris Wincup GR, Luong S-N, McDonnell T. Immunometabolism in rheumatic disease: the role in pathogenesis and implications for treatment. EMJ Rheumatol. 2019;6(1):81–89.
- Wu T, Xie C, Han J, et al. Metabolic disturbances associated with systemic lupus erythematosus. PLOS One. 2012;7(6):e37210.
- O'Neill LAJ, Kishton RJ, Rathmell J. A guide to immunometabolism for immunologists. Nat Rev Immunol. 2016;16(9):553–565.
- Pearce EL, Poffenberger MC, Chang CH, et al. Fueling immunity: insights into metabolism and lymphocyte function. Science. 2013;342(6155):1242454.
- Sena LA, Li S, Jairaman A, et al. Mitochondria are required for antigen-specific T cell activation through reactive oxygen species signaling. Immunity. 2013;38(2):225–236.
- Loftus RM, Finlay DK. Immunometabolism: cellular metabolism turns immune regulator. J Biol Chem. 2016;291(1):1–10.
- Fox CJ, Hammerman PS, Thompson CB. Fuel feeds function: energy metabolism and the T-cell response. Nat Rev Immunol. 2005;5(11):844–852.
- MacIver NJ, Michalek RD, Rathmell JC. Metabolic regulation of T lymphocytes. Annu Rev Immunol. 2013;31:259–283.
- Sharabi A, Tsokos GC. T cell metabolism: new insights in systemic lupus erythematosus pathogenesis and therapy. Nat Rev Rheumatol. 2020;16(2):100–112.
- Fernández-Ramos AA, Poindessous V, Marchetti-Laurent C, et al. The effect of immunosuppressive molecules on T-cell metabolic reprogramming. Biochimie. 2016;127:23–36.
- Chi H. Regulation and function of mTOR signalling in T cell fate decisions. Nat Rev Immunol. 2012;12(5):325–338.
- Wang R, Dillon CP, Shi LZ, et al. The transcription factor myc controls metabolic reprogramming upon T lymphocyte activation. Immunity. 2011;35(6):871–882.
- Choudhry H, Harris AL. Advances in Hypoxia-Inducible factor biology. Cell Metab. 2018;27(2):281–298.
- Macintyre AN, Gerriets VA, Nichols AG, Michalek RD, et al. The glucose transporter Glut1 is selectively essential for CD4 T cell activation and effector function. Cell Metab. 2014;20(1):61–72.
- Jacobs SR, Herman CE, Maciver NJ, et al. Glucose uptake is limiting in T cell activation and requires CD28-mediated Akt-dependent and independent pathways. J Immunol. 2008;180(7):4476–4486.
- Zeng H, Cohen S, Guy C, et al. mTORC1 and mTORC2 kinase signaling and glucose metabolism drive follicular helper T cell differentiation. Immunity. 2016;45(3):540–554.
- Shi LZ, Wang R, Huang G, et al. HIF1alpha-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and treg cells. J Exp Med. 2011;208(7):1367–1376.
- Kono M, Yoshida N, Maeda K, et al. Pyruvate dehydrogenase phosphatase catalytic subunit 2 limits Th17 differentiation. Proc Natl Acad Sci USA. 2018;115(37):9288–9293.
- Yang J-Q, Kalim KW, Li Y, et al. RhoA orchestrates glycolysis for TH2 cell differentiation and allergic airway inflammation. J Allergy Clin Immunol. 2016;137(1):231–245.e4.
- Yoshida N, Comte D, Mizui M, et al. ICER is requisite for Th17 differentiation. Nat Commun. 2016;7:12993.
- Yin Y, Choi SC, Xu Z, et al. Glucose oxidation is critical for CD4+ T cell activation in a mouse model of systemic lupus erythematosus. J Immunol. 2016;196(1):80–90.
- Balmer ML, Ma EH, Bantug GR, et al. Memory CD8(+) T cells require increased concentrations of acetate induced by stress for optimal function. Immunity. 2016;44(6):1312–1324.
- Gerriets VA, Kishton RJ, Johnson MO, et al. Foxp3 and toll-like receptor signaling balance Treg cell anabolic metabolism for suppression. Nat Immunol. 2016;17(12):1459–1466.
- Pacella I, Procaccini C, Focaccetti C, et al. Fatty acid metabolism complements glycolysis in the selective regulatory T cell expansion during tumor growth. Proc Natl Acad Sci USA. 2018;115(28):E6546–E6555.
- Cantó C, Auwerx J. AMP-activated protein kinase and its downstream transcriptional pathways. Cell Mol Life Sci. 2010;67(20):3407–3423.
- Ramiscal RR, Parish IA, Lee-Young RS, et al. Attenuation of AMPK signaling by ROQUIN promotes T follicular helper cell formation. Elife. 2015;4:e08698.
- Choi S-C, Titov AA, Abboud G, Seay HR, et al. Inhibition of glucose metabolism selectively targets autoreactive follicular helper T cells. Nat Commun. 2018;9(1):4369.
- Cruzat V, Macedo Rogero M, Noel Keane K, et al. Glutamine: metabolism and immune function, supplementation and clinical translation. Nutrients. 2018;10(11):1564.
- Nakaya M, Xiao Y, Zhou X, et al. Inflammatory T cell responses rely on amino acid transporter ASCT2 facilitation of glutamine uptake and mTORC1 kinase activation. Immunity. 2014;40(5):692–705.
- Le A, Lane AN, Hamaker M, et al. Glucose-independent glutamine metabolism via TCA cycling for proliferation and survival in B cells. Cell Metab. 2012;15(1):110–121.
- Kono M, Yoshida N, Maeda K, et al. Transcriptional factor ICER promotes glutaminolysis and the generation of Th17 cells. Proc Natl Acad Sci USA. 2018;115(10):2478–2483.
- Nakajima H, Kunimoto H. TET2 as an epigenetic master regulator for normal and malignant hematopoiesis. Cancer Sci. 2014;105(9):1093–1099.
- MO, Johnson MMW, MZ, Madden ML, Davila, et al. Distinct regulation of Th17 and Th1 cell differentiation by glutaminase-dependent metabolism. Cell Metab. 2018;175(7):1780–1795.
- Buck MD, O'Sullivan D, Pearce EL. T cell metabolism drives immunity. J Exp Med. 2015;212(9):1345–1360.
- Wolfson RL, Chantranupong L, Saxton RA, et al. Sestrin2 is a leucine sensor for the mTORC1 pathway. Science. 2016;351(6268):43–48.
- Sundrud MS, Koralov SB, Feuerer M, et al. Halofuginone inhibits TH17 cell differentiation by activating the amino acid starvation response. Science. 2009;324(5932):1334–1338.
- Ma EH, Bantug G, Griss T, et al. Serine is an essential metabolite for effector T cell expansion. Cell Metab. 2017;25(2):345–357.
- Ricciardi S, Manfrini N, Alfieri R, et al. The translational machinery of human CD4+ T cells is poised for activation and controls the switch from quiescence to metabolic remodeling. Cell Metab. 2018;28(6):895–906.e5.
- Qian X, Yang Z, Mao E, et al. Regulation of fatty acid synthesis in immune cells. Scand J Immunol. 2018;88(5):e12713.
- Zhang X, Tao Y, Troiani L, et al. Simvastatin inhibits IFN regulatory factor 4 expression and Th17 cell differentiation in CD4+ T cells derived from patients with multiple sclerosis. J Immunol. 2011;187(6):3431–3437.
- Teng X, Li W, Cornaby C, et al. Immune cell metabolism in autoimmunity. Clin Exp Immunol. 2019;197(2):181–192.
- Ito A, Hong C, Oka K, et al. Cholesterol accumulation in CD11c+ immune cells is a causal and targetable factor in autoimmune disease. Immunity. 2016;45(6):1311–1326.
- York AG, Williams KJ, Argus JP, et al. Limiting cholesterol biosynthetic flux spontaneously engages type I IFN signaling. Cell. 2015;163(7):1716–1729.
- Michalek RD, Gerriets VA, Nichols AG, et al. Estrogen-related receptor-α is a metabolic regulator of effector T-cell activation and differentiation. Proc Natl Acad Sci USA. 2011;108(45):18348–18353.
- Kono M, Yoshida N, Tsokos GC. Metabolic control of T cells in autoimmunity. Curr Opin Rheumatol. 2020;32(2):192–199.
- Procaccini C, Carbone F, Di Silvestre D, et al. The proteomic landscape of human ex vivo regulatory and conventional T cells reveals specific metabolic requirements. Immunity. 2016;44(3):712.
- Burzyn D, Benoist C, Mathis D. Regulatory T cells in nonlymphoid tissues. Nat Immunol. 2013;14(10):1007–1013.
- Haghikia A, Jörg S, Duscha A, et al. Dietary fatty acids directly impact central nervous system autoimmunity via the small intestine. Immunity. 2015;43(4):817–829.
- O'Sullivan D, van der Windt GJ, Huang SC, et al. Memory CD8(+) T cells use cell-intrinsic lipolysis to support the metabolic programming necessary for development. Immunity. 2014;41(1):75–88.
- Pan Y, Tian T, Park CO, et al. Survival of tissue-resident memory T cells requires exogenous lipid uptake and metabolism. Nature. 2017;543(7644):252–256.
- Phan AT, Doedens AL, Palazon A, Tyrakis PA, et al. Constitutive glycolytic metabolism supports CD8+ T cell effector memory differentiation during viral infection. Immunity. 2016;45(5):1024–1037.
- Kishore M, Cheung KCP, Fu H, et al. Regulatory T cell migration is dependent on glucokinase-mediated glycolysis. Immunity. 2017;47(5):875–889.e10.
- Morel L. Immunometabolism in systemic lupus erythematosus. Nat Rev Rheumatol. 2017;13(5):280–290.
- Delgoffe GM, Pollizzi KN, Waickman AT, et al. The kinase mTOR regulates the differentiation of helper T cells through the selective activation of signaling by mTORC1 and mTORC2. Nat Immunol. 2011;12(4):295–303.
- Fernandez DR, Telarico T, Bonilla E, et al. Activation of mammalian target of rapamycin controls the loss of TCRzeta in lupus T cells through HRES-1/Rab4-regulated lysosomal degradation. J Immunol. 2009;182(4):2063–2073.
- Nagai S, Kurebayashi Y, Koyasu S. Role of PI3K/Akt and mTOR complexes in Th17 cell differentiation. Ann N Y Acad Sci. 2013;1280:30–34.
- Apostolidis SA, Rodríguez-Rodríguez N, Suárez-Fueyo A, et al. Phosphatase PP2A is requisite for the function of regulatory T cells. Nat Immunol. 2016;17(5):556–564.
- Michalek RD, Gerriets VA, Jacobs SR, et al. Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J Immunol. 2011;186(6):3299–3303.
- Yang K, Blanco DB, Neale G, et al. Homeostatic control of metabolic and functional fitness of Treg cells by LKB1 signalling. Nature. 2017;548(7669):602–606.
- Zeng H, Chi H. Metabolic control of regulatory T cell development and function. Trends Immunol. 2015;36(1):3–12.
- Wahl DR, Petersen B, Warner R, et al. Characterization of the metabolic phenotype of chronically activated lymphocytes. Lupus. 2010;19(13):1492–1501.
- Vukelic M, Kono M, Tsokos GC. T cell metabolism in lupus. Immunometabolism. 2020;2(2):e200009.
- Perl A. Oxidative stress in the pathology and treatment of systemic lupus erythematosus. Nat Rev Rheumatol. 2013;9(11):674–686.
- Perl A, Gergely P Jr., Banki K. Mitochondrial dysfunction in T cells of patients with systemic lupus erythematosus. Int Rev Immunol. 2004;23(3–4):293–313.
- Lightfoot YL, Blanco LP, Kaplan MJ. Metabolic abnormalities and oxidative stress in lupus. Curr Opin Rheumatol. 2017;29(5):442–449.
- Tsokos GC. Systemic lupus erythematosus. N Engl J Med. 2011;365(22):2110–2121.
- Kaminski MM, Sauer SW, Klemke CD, et al. Mitochondrial reactive oxygen species control T cell activation by regulating IL-2 and IL-4 expression: mechanism of ciprofloxacin-mediated immunosuppression. J Immunol. 2010;184(9):4827–4841.
- Bartolomé A, García-Aguilar A, Asahara SI, et al. MTORC1 regulates both general autophagy and mitophagy induction after oxidative phosphorylation uncoupling. Mol Cell Biol. 2017;37(23):e00441–17.
- Lood C, Blanco LP, Purmalek MM, et al. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat Med. 2016;22(2):146–153.
- Caielli S, Athale S, Domic B, et al. Oxidized mitochondrial nucleoids released by neutrophils drive type I interferon production in human lupus. J Exp Med. 2016;213(5):697–713.
- Takeshima Y, Iwasaki Y, Fujio K, et al. Metabolism as a key regulator in the pathogenesis of systemic lupus erythematosus. Semin Arthritis Rheum. 2019;48(6):1142–1145.
- Eckardt KU. Immunometabolism: oxygen sensing and cell metabolism in inflammation. Nat Rev Nephrol. 2017;13(12):727–728.
- Feldhoff LM, Rueda CM, Moreno-Fernandez ME, et al. IL-1β induced HIF-1α inhibits the differentiation of human FOXP3+ T cells. Sci Rep. 2017;7(1):465.
- Movafagh S, Crook S, Vo K. Regulation of hypoxia-inducible factor-1a by reactive oxygen species: new developments in an old debate. J Cell Biochem. 2015;116(5):696–703.
- Perry DJ, Yin Y, Telarico T, et al. Murine lupus susceptibility locus Sle1c2 mediates CD4+ T cell activation and maps to estrogen-related receptor γ. J Immunol. 2012;189(2):793–803.
- Vyshkina T, Sylvester A, Sadiq S, et al. Association of common mitochondrial DNA variants with multiple sclerosis and systemic lupus erythematosus. Clin Immunol. 2008;129(1):31–35.
- Bednarski JJ, Warner RE, Rao T, et al. Attenuation of autoimmune disease in fas-deficient mice by treatment with a cytotoxic benzodiazepine. Arthritis Rheum. 2003;48(3):757–766.
- Guma M, Tiziani S, Firestein GS. Metabolomics in rheumatic diseases: desperately seeking biomarkers. Nat Rev Rheumatol. 2016;12(5):269–281.
- Koga T, Sato T, Furukawa K, et al. Promotion of calcium/calmodulin-dependent protein kinase 4 by GLUT1-dependent glycolysis in systemic lupus erythematosus. Arthritis Rheumatol. 2019;71(5):766–772.
- Kono M, Maeda K, Stocton-Gavanescu I, et al. Pyruvate kinase M2 is requisite for Th1 and Th17 differentiation. JCI Insight. 2019;4(12):e127395.
- Suárez-Fueyo A, Bradley SJ, Tsokos GC. T cells in systemic lupus erythematosus. Curr Opin Immunol. 2016;43:32–38.
- Yin Y, Choi SC, Xu Z, et al. Normalization of CD4+ T cell metabolism reverses lupus. Sci Transl Med. 2015;7(274):274ra18.
- Kono M, Yoshida N, Maeda K, et al. Glutaminase 1 inhibition reduces glycolysis and ameliorates lupus-like disease in MRL/lpr mice and experimental autoimmune encephalomyelitis. Arthritis Rheumatol. 2019;71(11):1869–1878.
- Katsuyama E, Suarez-Fueyo A, Bradley SJ, et al. The CD38/NAD/SIRTUIN1/EZH2 axis mitigates cytotoxic CD8 T cell function and identifies patients with SLE prone to infections. Cell Rep. 2020;30(1):112–123.e4.
- Pavón EJ, Zumaquero E, Rosal-Vela A, et al. Increased CD38 expression in T cells and circulating anti-CD38 IgG autoantibodies differentially correlate with distinct cytokine profiles and disease activity in systemic lupus erythematosus patients. Cytokine. 2013;62(2):232–243.
- Chatterjee S, Daenthanasanmak A, Chakraborty P, et al. CD38-NAD + axis regulates immunotherapeutic anti-tumor T cell response. Cell Metab. 2018;27(1):85–100.e8.
- Chiurchiù V, Leuti A, Maccarrone M. Bioactive lipids and chronic inflammation: managing the fire within. Front Immunol. 2018;9:38.
- Krishnan S, Nambiar MP, Warke VG, et al. Alterations in lipid raft composition and dynamics contribute to abnormal T cell responses in systemic lupus erythematosus. J Immunol. 2004;172(12):7821–7831.
- McDonald G, Deepak S, Miguel L, et al. Normalizing glycosphingolipids restores function in CD4+ T cells from lupus patients. J Clin Invest. 2014;124(2):712–724.
- Nowling TK, Mather AR, Thiyagarajan T, et al. Renal glycosphingolipid metabolism is dysfunctional in lupus nephritis. J Am Soc Nephrol. 2015;26(6):1402–1413.
- Waddington KE, Jury EC, Pineda-Torra I. Liver X receptors in immune cell function in humans. Biochem Soc Trans. 2015;43(4):752–757.
- Richard EM, Thiyagarajan T, Bunni MA, et al. Reducing FLI1 levels in the MRL/lpr lupus mouse model impacts T cell function by modulating glycosphingolipid metabolism. PLOS One. 2013;8(9):e75175.
- Zhang L, Eddy A, Teng YT, et al. An immunological renal disease in transgenic mice that overexpress Fli-1, a member of the ets family of transcription factor genes. Mol Cell Biol. 1995;15(12):6961–6970.
- Sundararaj KP, Thiyagarajan T, Molano I, et al. FLI1 levels impact CXCR3 expression and renal infiltration of T cells and renal glycosphingolipid metabolism in the MRL/lpr lupus mouse strain. J Immunol. 2015;195(12):5551–5560.
- Sawaf M, Fauny J-D, Felten R, et al. Defective BTLA functionality is rescued by restoring lipid metabolism in lupus CD4+ T cells. JCI Insight. 2018;3(13):e99711.
- Ryu H, Lim H, Choi G, et al. Atherogenic dyslipidemia promotes autoimmune follicular helper T cell responses via IL-27. Nat Immunol. 2018;19(6):583–593.
- Wilhelm AJ, Zabalawi M, Grayson JM, et al. Apolipoprotein A-I and its role in lymphocyte cholesterol homeostasis and autoimmunity. Arterioscler Thromb Vasc Biol. 2009;29(6):843–849.
- Koga T, Hedrich CM, Mizui M, et al. CaMK4-dependent activation of AKT/mTOR and CREM-α underlies autoimmunity-associated Th17 imbalance. J Clin Invest. 2014;124(5):2234–2245.
- Shan J, Jin H, Xu Y. T cell metabolism: a new perspective on Th17/treg cell imbalance in systemic lupus erythematosus. Front Immunol. 2020;11(1027):1027.
- Kato H, Perl A. Mechanistic target of rapamycin complex 1 expands Th17 and IL-4+ CD4−CD8− double-negative T cells and contracts regulatory T cells in systemic lupus erythematosus. JI. 2014;192(9):4134–4144.
- Henske EP, Jóźwiak S, Kingswood JC, et al. Tuberous sclerosis complex. Nat Rev Dis Primers. 2016;2:16035.
- Lee SY, Moon SJ, Kim EK, et al. Metformin suppresses systemic autoimmunity in Roquinsan/san mice through inhibiting B cell differentiation into plasma cells via regulation of AMPK/mTOR/STAT3. J Immunol. 2017;198(7):2661–2670.
- Katsiari CG, Kyttaris VC, Juang Y-T, et al. Protein phosphatase 2A is a negative regulator of IL-2 production in patients with systemic lupus erythematosus. J Clin Invest. 2005;115(11):3193–3204.
- Xu L, Huang Q, Wang H, et al. The kinase mTORC1 promotes the generation and suppressive function of follicular regulatory T cells. Immunity. 2017;47(3):538–551.e5.
- Murray PJ, Rathmell J, Pearce E. SnapShot: immunometabolism. Cell Metab. 2015;22(1):190–191.e1.
- Zhang CX, Wang HY, Yin L, Mao YY, et al. Immunometabolism in the pathogenesis of systemic lupus erythematosus. J Transl Autoimmun. 2020;3:100046.
- Perl A, Hanczko R, Lai ZW, et al. Comprehensive metabolome analyses reveal N-acetylcysteine-responsive accumulation of kynurenine in systemic lupus erythematosus: implications for activation of the mechanistic target of rapamycin. Metabolomics. 2015;11(5):1157–1174.
- Zeng H, Chi H. mTOR signaling in the differentiation and function of regulatory and effector T cells. Curr Opin Immunol. 2017;46:103–111.
- Cheng SC, Quintin J, Cramer RA, et al. mTOR- and HIF-1α-mediated aerobic glycolysis as metabolic basis for trained immunity. Science. 2014;345(6204):1250684.
- Reid MA, Dai Z, Locasale JW. The impact of cellular metabolism on chromatin dynamics and epigenetics. Nat Cell Biol. 2017;19(11):1298–1306.
- Gubser PM, Bantug GR, Razik L, et al. Rapid effector function of memory CD8+ T cells requires an immediate-early glycolytic switch. Nat Immunol. 2013;14(10):1064–1072.
- Xu T, Stewart KM, Wang X, et al. Metabolic control of TH17 and induced treg cell balance by an epigenetic mechanism. Nature. 2017;548(7666):228–233.
- Sunahori K, Nagpal K, Hedrich CM, et al. The catalytic subunit of protein phosphatase 2A (PP2Ac) promotes DNA hypomethylation by suppressing the phosphorylated mitogen-activated protein kinase/extracellular signal-regulated kinase (ERK) kinase (MEK)/phosphorylated ERK/DNMT1 protein pathway in T-cells from controls and systemic lupus erythematosus patients. J Biol Chem. 2013;288(30):21936–21944.
- Li Y, Gorelik G, Strickland FM, et al. Oxidative stress, T cell DNA methylation, and lupus. Arthritis Rheumatol. 2014;66(6):1574–1582.
- Ray D, Strickland FM, Richardson BC. Oxidative stress and dietary micronutrient deficiencies contribute to overexpression of epigenetically regulated genes by lupus T cells. Clin Immunol. 2018;196:97–102.
- Li H, Tsokos MG, Bickerton S, et al. Precision DNA demethylation ameliorates disease in lupus-prone mice. JCI Insight. 2018;3(16):e120880.
- Niu Y, DesMarais TL, Tong Z, et al. Oxidative stress alters global histone modification and DNA methylation. Free Radic Biol Med. 2015;82:22–28.
- Zhang Q, Long H, Liao J, et al. Inhibited expression of hematopoietic progenitor kinase 1 associated with loss of Jumonji domain containing 3 promoter binding contributes to autoimmunity in systemic lupus erythematosus. J Autoimmun. 2011;37(3):180–189.
- Titov AA, Baker HV, Brusko TM, et al. Metformin inhibits the type 1 IFN response in human CD4+ T cells. J Immunol. 2019;203(2):338–348.
- Wang H, Li T, Chen S, et al. Neutrophil extracellular trap mitochondrial DNA and its autoantibody in systemic lupus erythematosus and a proof-of-concept trial of metformin. Arthritis Rheumatol. 2015;67(12):3190–3200.
- Sun F, Geng S, Wang H, et al. Effects of metformin on disease flares in patients with systemic lupus erythematosus: post hoc analyses from two randomised trials. Lupus Sci Med. 2020;7(1):e000429.
- Johnson KM, Chen X, Boitano A, et al. Identification and validation of the mitochondrial F1F0-ATPase as the molecular target of the immunomodulatory benzodiazepine Bz-423. Chem Biol. 2005;12(4):485–496.
- Yap DYH, Tang C, Chan GCW, et al. Longterm data on sirolimus treatment in patients with lupus nephritis. J Rheumatol. 2018;45(12):1663–1670.
- Lai Z-W, Kelly R, Winans T, et al. Sirolimus in patients with clinically active systemic lupus erythematosus resistant to, or intolerant of, conventional medications: a single-arm, open-label, phase 1/2 trial. The Lancet. 2018;391(10126):1186–1196.
- Lai ZW, Borsuk R, Shadakshari A, et al. Mechanistic target of rapamycin activation triggers IL-4 production and necrotic death of double-negative T cells in patients with systemic lupus erythematosus. JI. 2013;191(5):2236–2246.
- Zhao W, Berthier CC, Lewis EE, et al. The peroxisome-proliferator activated receptor-γ agonist pioglitazone modulates aberrant T cell responses in systemic lupus erythematosus. Clin Immunol. 2013;149(1):119–132.
- Cipolletta D, Feuerer M, Li A, et al. PPAR-γ is a major driver of the accumulation and phenotype of adipose tissue treg cells. Nature. 2012;486(7404):549–553.
- Mognol GP, de Araujo-Souza PS, Robbs BK, et al. Transcriptional regulation of the c-Myc promoter by NFAT1 involves negative and positive NFAT-responsive elements. Cell Cycle. 2012;11(5):1014–1028.
- Sinclair LV, Rolf J, Emslie E, et al. Control of amino-acid transport by antigen receptors coordinates the metabolic reprogramming essential for T cell differentiation. Nat Immunol. 2013;14(5):500–508.
- Li Y, Palmisano M, Sun D, et al. Pharmacokinetic disposition difference between cyclosporine and voclosporin drives their distinct efficacy and safety profiles in clinical studies. Clin Pharmacol. 2020;12:83–96.
- Heo YA. Voclosporin: first approval. Drugs. 2021;81(5):605–610.
- Domhan S, Muschal S, Schwager C, et al. Molecular mechanisms of the antiangiogenic and antitumor effects of mycophenolic acid. Mol Cancer Ther. 2008;7(6):1656–1668.
- Maeda K, Otomo K, Yoshida N, et al. CaMK4 compromises podocyte function in autoimmune and nonautoimmune kidney disease. J Clin Invest. 2018;128(8):3445–3459.
- Ichinose K, Juang YT, Crispín JC, et al. Suppression of autoimmunity and organ pathology in lupus-prone mice upon inhibition of calcium/calmodulin-dependent protein kinase type IV. Arthritis Rheum. 2011;63(2):523–529.
- Zhang D, Chia C, Jiao X, et al. D-mannose induces regulatory T cells and suppresses immunopathology. Nat Med. 2017;23(9):1036–1045.
- Lai ZW, Hanczko R, Bonilla E, et al. N-acetylcysteine reduces disease activity by blocking mammalian target of rapamycin in T cells from systemic lupus erythematosus patients: a randomized, double-blind, placebo-controlled trial. Arthritis Rheum. 2012;64(9):2937–2946.
- Hansen AL, Buchan GJ, Rühl M, et al. Nitro-fatty acids are formed in response to virus infection and are potent inhibitors of STING palmitoylation and signaling. Proc Natl Acad Sci USA. 2018;115(33):E7768–E7775.