Abstract
We present calculations for Ti adatoms and interstitials at the (110) surface of rutile TiO2, where these species are known to play a crucial role in surface chemistry. We review structural calculations performed using the DFT+U methodology, which have been benchmarked using controlled self-doping experiments on thin rutile films. The ab initio results have further been used to assess the ability of empirical charge equalisation (QEq) potentials to correctly predict the energetics of these structures. A simple modification to the potential, whereby the oxygen charge is fixed while allowing charge redistribution between Ti ions, has been shown to greatly improve its performance in terms of the energy landscape of the Ti adatoms and interstitials. In this paper, we extend the QEq calculations to consider the diffusion pathways and barriers in the surface using nudged elastic band calculations. We find that key barriers involved in the diffusion of Ti interstitials to adatom sites are much lower with the modified potential, implying that the diffusion is active on experimental time scales at temperatures where the regrowth of reduced rutile crystals exposed to oxygen has been observed.
1. Introduction
Titania is an important technological material with a range of applications in catalysis, sensing and functional coatings among others [Citation1]. The rutile (110) surface has been the subject of intensive investigation as a model reducible system Citation2-12. It has been found that Ti adatoms and interstitials play a crucial role in surface chemistry and properties of the reduced bulk crystal. Scanning tunnelling microscopy (STM) has shown how the (110) surface of a reduced rutile crystal regrows at elevated temperatures above ∼473 K when exposed to an oxygen atmosphere [Citation5,Citation8]. There is compelling evidence that the crystal accommodates its non-stoichiometry through a solid solution of Ti interstitials, which are able to diffuse to the surface at these temperatures to react with the impinging oxygen. Recent STM work has also focused on the surface chemistry of the oxidation process of the reduced crystal [Citation13]. Again, it is shown that the key to a proper understanding is how the material accommodates interstitials, and how they diffuse through the surface region to adatom sites.
A combined experimental and density functional theory (DFT) study has recently yielded detailed information on the structure and energetics of Ti adatoms on rutile (110) [Citation14]. In this work, controlled doping of crystals and epitaxial ultrathin films has been studied using photoemission spectroscopy. Upon vapour deposition of 1/8 ml of Ti, the UPS spectra reveal a defect-induced state in the band gap, about 1 eV below the Fermi level. Furthermore, the valence band states associated with the two-coordinated bridging oxygen at the surface are also altered by the Ti deposition, indicating that deposited Ti adsorb as adatoms that preferentially sit next to two bridging oxygen and one in-plane oxygen. This state disappears upon annealing, and oxidation of the surface, indicating that the deposited species are adatoms at the surface.
Detailed explanation of these results is achieved using the DFT compensated for on-site Coulomb interactions (DFT+U) (see below). However, it is also desirable to have reliable atomistic potential approaches so that the growth dynamics can be studied. The modelling of reducible transition metal oxides presents a great challenge to empirical potential schemes. Since the non-stoichiometry makes the application of fixed charge models problematic, the charge equilibration (QEq) scheme of Rappe and Goddard has an obvious appeal Citation15-18. In this approach, the electronegativity of the ions is used to adjust the charge distribution in an adaptive manner. In this work, we focus on the QEq potential developed by Hallil et al. [Citation19] for rutile TiO2, with parameters fitted to the bulk structure and elastic properties and tested on the bulk O vacancy and low-index surfaces. The potential is used to calculate the diffusion pathways and barriers for Ti interstitials and adatoms with the nudged elastic band (NEB) method [Citation20]. In addition, we investigate the effect of a modification to the potential suggested by the recent analysis of the charge distributions found in the DFT+U calculations, which show that the QEq approach overestimates the role of the oxygen ions in accommodating charge around these point defects [Citation21]. QEq calculations with the oxygen charge fixed to bulk values, but allowing charge transfer between the Ti ions as in the Hallil model, show that the energy landscape of the defects is changed and comes into line with the DFT+U results [Citation21].
In this paper, we review the DFT+U results and the comparisons with the QEq results. We then explore whether this modified potential yields diffusion barriers that accord with the experimental facts for the reoxidation processes discussed above.
2. DFT+U and QEq defect structure calculations
The DFT-PW91+U calculations reported in [Citation14] allow interpretation of the experimental results and show how the charge is distributed around the defect. The calculations have an empirical element through the selection of the value of U for the Ti d orbitals: large values (U∼6 eV) strongly localise the extra electrons on the adatom and surface Ti; zero U delocalises the extra electrons through the surface region and intermediate values yield a picture between these two extremes. It is the last case, specifically with U = 3 eV, that gives the best agreement with the spectroscopy of the gap states induced by the adsorbed adatom [Citation14]. This result is consistent with the value of U assigned in the calculations of oxygen vacancies in the (110) surface [Citation22], which also reduce surface Ti and create a defect state in the band gap. It has been shown that these latter calculations yield a charge distribution consistent with hybrid B3LYP calculations [Citation23]. This implies that the DFT+U can be used reliably to assess the charge distributions around the adatom and interstitials, allowing comparisons with those found in the QEq calculations.
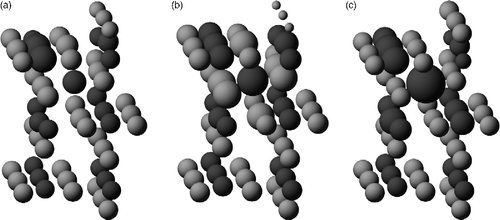
Figure shows a picture of a top-down view of the (110) surface, indicating the adsorption sites for the Ti adatom and interstitial and the labelling we use in this paper. We first look at the distribution of charges observed around the Fi interstitial site in our calculations. The DFT+U charge is portioned to the ions using the Bader analysis [Citation24]. Figure (a) shows the results, where the size of the ions represents how many excess electrons they have over their charges in the DFT+U bulk rutile crystal [Citation21]. Figure (b) shows the equivalent result from the QEq model of Hallil et al. [Citation19]. Note that the bridging oxygens in this latter image are rather smaller than those in Figure (a); in QEq, the under-coordinated oxygen tends to become less negative and lose charge to neighbours, a feature not apparent in the DFT+U results. Furthermore, the Ti interstitial's nearest neighbour oxygen all acquires excess charge from the interstitial, again in contrast to the DFT+U behaviour. To quantify the effect, in the QEq calculations, it is found that the oxygen gain/lose up to ± 0.4e depending on their neighbourhood, whereas the DFT+U changes are much smaller, up to ± 0.1e.
Figure 1 A plan view of the surface indicating the adatom and interstitial sites used in this paper. Ti atoms are dark grey and O atoms light grey, with larger atoms closer to the surface. The bridging oxygen rows are indicated with the dashed lines. Site A (and equivalents A′ and D) is the adatom in its most favourable site, next to two bridging oxygens and one in-plane oxygen. Site B (and its equivalent E) is the adatom in its second favourable binding site next to one bridging and two in-plane oxygens. Site Fi is an interstitial site one O–Ti–O layer down; there is an alternative interstitial site Bi, underneath B. Interstitials another O–Ti–O layer down are labelled Fii and Bii, respectively.
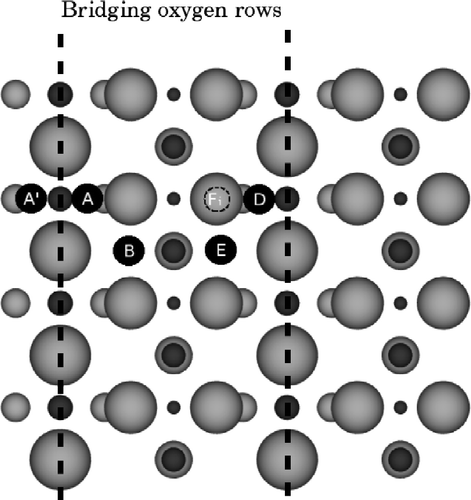
Figure 2 Charge distributions induced by the layer 1 interstitial site Fi: (a) DFT+U calculations using Bader analysis; (b) the original Hallil QEq potential and (c) the fixed oxygen-charge variant of the QEq potential. O is light grey and Ti dark grey. The radius of the ions reflects the excess charge ( − e) the ions have compared with the bulk environment (referred to the ions at the bottom of the picture). To set the scale, the interstitial in (b) has an excess of 0.42e− , and its six nearest neighbour oxygens each have an excess of ∼0.3e− , over bulk values. Only a section of the full cell used in the calculations is shown for clarity, with the upper surface being the free one with the exposed bridging oxygen.
This charging of the oxygen results in a dramatically different energy landscape for the points defects, as shown in Figure . The DFT+U results show that the layer 1 interstitials, Fi and Bi, are favoured over the most stable adatom-binding site A by about 0.62 and 0.53 eV, respectively. The QEq energy differences are much higher at 2.48 and 2.41 eV, respectively. The cause of this dramatic favouring of the interstitials over adatoms in the QEq model can be traced to the elastic energy gains due to the lattice distorting in response to the oxygen nearest neighbour charging. While the finding that Ti interstitials are more energetically favourable than adatoms agrees with the experimental results for adatoms diffusing down into the bulk upon annealing [Citation14], trapping them in the bulk by such a large amount as 2.5 eV does not accord with the regrowth of reduced rutile at elevated temperature, even if oxygen adspecies promote the growth [Citation5,Citation8,Citation13]. It appears that the empirical QEq potential overestimates the adsorption energy of the interstitials with respect to the adatom energy.
Figure 3 The energy landscape of the adatom and interstitials in the B/Bn (o) and A/Fn sites (*),where layer 0 corresponds to the adatoms and layer n to the interstitials below n O–Ti–O surface layers. The energies are given relative to the most stable adatom site for the model, which is site A in all cases.
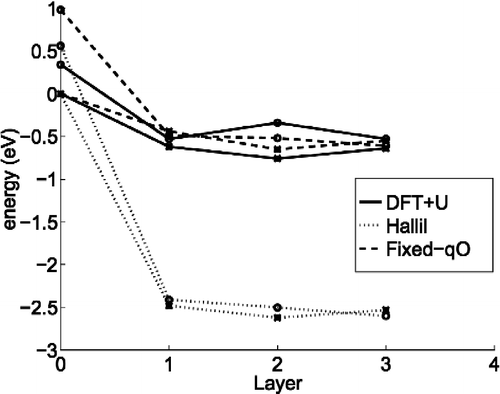
The conclusion about the adverse role played by the charging of the oxygen in the QEq model has been tested in a simple way by modifying the QEq scheme so that charge transfer only occurs between Ti species, with the oxygen charge fixed to the bulk rutile crystal values [Citation21]. The other parameters in the model all stay the same, so that the bulk properties such as optimised lattice parameters are not changed. However, surface calculations will of course be affected. Figure (c) shows the charge distribution around the relaxed Fi interstitial in this modified QEq scheme, and the impact on the energy landscape is shown in Figure . As can be seen, the landscape now compares much more favourably with the DFT+U results, implying that the oxygen charging does indeed play an adverse role in the behaviour of the original Hallil QEq model.
3. Ti adatom and interstitial diffusion pathways
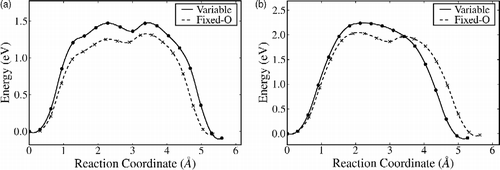
The diffusion pathways for adatoms and interstitials are calculated using the NEB method. Two types of pathways are possible: direct moves passing through the more open channels in the crystal structure, and exchange (or interstitialcy) moves that involve the displacement of neighbouring Ti ions during the move, with the original adatom or interstitial finishing up in the vacated lattice site. Figure (a) shows the results for the transition A–Fi, where the adatom exchanges with its nearest neighbour Ti, which lies beneath the two bridging oxygen next to site A. The displaced Ti ends up in the interstitial site Fi on the opposite side of the bridging row one O–Ti–O layer down. A movie of this transition is provided in the supporting information. As can be seen, the barrier for the forward move is slightly higher in the fixed oxygen-charge QEq variant (Table ), but much lower at 1.41 eV (cf. 3.31 eV) for the reverse move. This model implies that the diffusion of Ti interstitials to the surface will be active on experimental time scales at elevated temperature above ∼500 K.
Figure 4 NEB pathways for (a) A–Fi via exchange with a surface sixfold coordinated Ti; (b) B–Bi via exchange with a fivefold coordinated surface Ti and (c) B–Bi direct move without exchange. Here, the reaction coordinate is the Euclidian distance along the path from the initial state.
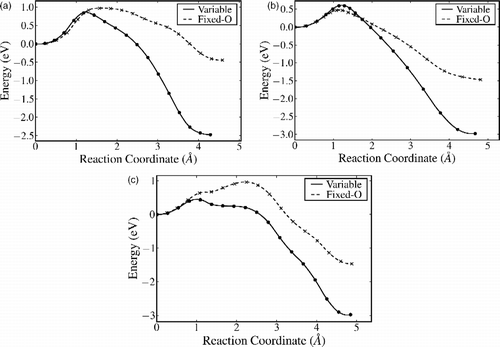
Table 1. Energy barriers (eV) for the forward and backward moves found using the NEB method using the fixed oxygen-charge variant of the QEq model.
Figure (b) shows the pathway for the diffusion between adatom site B and interstitial Bi via exchange with the nearest neighbour Ti, which is an in-plane fivefold coordinated surface site. The forward movement has a slightly lower activation energy, but again the reverse movement is much lower in the fixed oxygen-charge model. The direct path movement for B–Bi is shown in Figure (c) and the relevant energies given in Table . The Hallil model favours the direct B–Bi move over the exchange, in contrast to the fixed oxygen-charge model. Movie files for these two transitions are also provided in the supporting information.
To complete the discussion of the adatom/interstitial diffusion pathways, we must consider the diffusion parallel to the surface. Table shows the results for various barriers. The transition A–B is of particular interest to us here. Taken together, we can now consider the barrier for the diffusion of the adatom from its most stable site A to any interstitial site. For both the fixed oxygen-charge and original Hallil models, A–Fi with exchange is the lowest energy pathway at 0.97 and 0.85 eV, respectively. The alternative pathway, A–B followed by B–Bi as a direct move or exchange, has higher barriers of 1.45 (exchange) and 1.00 eV (direct), respectively. The reverse process of an interstitial diffusing back to the surface also follows the Fi–A via exchange route.
Figure (a) shows the pathway for the diffusion of the interstitial from site Bi–Bii via exchange with an intervening nearest neighbour Ti, and in Figure (b) by the direct route. The fixed oxygen-charge variant has a slightly lower barrier than the original Hallil model, and is lower in both models than the transition Fi–A (Table ). Thus, the latter is the rate-limiting step for the diffusion of deep-lying interstitials to the free surface. Note how the barrier for the direct Bi–Bii diffusion is much higher than that for the direct B–Bi, the latter move being enabled at the surface by the greater freedom for the surrounding ions to move away during the process to accommodate the movement.
Figure 5 NEB pathways for (a) Bi–Bii via exchange with a bulk Ti and (b) Bi–Bii direct move without exchange. Here, the reaction coordinate is the Euclidian distance along the path from the initial state.
4. Summary and conclusions
In summary, we have modified the Hallil QEq model [Citation19] in the light of the DFT+U calculations for the adatom and interstitial energy landscape [Citation14] and the Bader analysis of the charge distributions around the point defects [Citation21]. This modification keeps the oxygen charge fixed to bulk values while allowing the usual charge transfer between the Ti ions only. While this scheme might not be the last word on the construction of QEq potentials for titania and other reducible transition metal oxides, we believe that it convincingly demonstrates how the original model allows too much charge transfer to and from the oxygen.
We have investigated the impact of the modified potential on the diffusion pathways of the adatoms and interstitials in the surface region. Our NEB results have shown that the diffusion of interstitials to the free surface will be accomplished via exchange with a sixfold coordinated surface Ti in the QEq models. However, the activation barrier in the original Hallil model is very high at 3.31 eV, whereas in the fixed oxygen-charge model it is much lower at 1.41 eV. Given the Arrhenius nature of the diffusion, the latter seems more reasonable result, given the experimental evidence for the regrowth of the (110) surface of a reduced rutile crystal with oxygen exposure above ∼473 K [Citation5,Citation8]. However, further work is required, notably the impact surface oxygen has on these results, and is the subject of ongoing work.
gmos_a_360905_sm2743.zip
Download Zip (3.7 MB)Acknowledgements
This work was supported by the UK Engineering and Physical Sciences Research Council through grant no. EP/C524349 within the Materials Modelling Consortium on Functional Coatings. The authors are grateful to Louis Vernon and Ed Sanville of Loughborough University for helpful discussions.
References
- Diebold , U. 2003 . The surface science of titanium dioxide . Surf. Sci. Rep. , 48 : 53 – 229 .
- Guo , Q. , Cocks , I. and Williams , E.M. 1996 . Surface structure of (1 × 2) reconstructed TiO2(110) studied using electron stimulated desorption ion angular distribution . Phys. Rev. Lett. , 77 : 3851 – 3854 .
- Bennett , R.A. , Poulston , S. , Stone , P. and Bowker , M. 1999 . STM and LEED observations of the surface structure of TiO2(110) following crystallographic shear plane formation . Phys. Rev. B , 59 : 10341 – 10346 .
- Bennett , R.A. , Stone , P. , Price , N.J. and Bowker , M. 1999 . Two (1 × 2) reconstructions of TiO2(110): surface rearrangement and reactivity studied using elevated temperature scanning tunneling microscopy . Phys. Rev. Lett. , 82 : 3831 – 3834 .
- Stone , P. , Bennett , R.A. and Bowker , M. 1999 . Reactive re-oxidation of reduced TiO2(110) surfaces demonstrated by high temperature STM movies . New J. Phys. , 1 : 1.1 – 1.12 .
- Bennett , R.A. , Stone , P. and Bowker , M. 1999 . Scanning tunneling microscopy studies of the reactivity of the TiO2(110) surface: re-oxidation and the thermal treatment of metal nanoparticles . Farad. Discuss. , 114 : 267 – 277 .
- Bennett , R.A. 2000 . The re-oxidation of the substoichiometric TiO2(110) surface in the presence of crystallographic shear planes . Phys. Chem. Comm. , 3 : 1 – 14 .
- Smith , R.D. , Bennett , R.A. and Bowker , M. 2002 . Measurement of the surface-growth kinetics of reduced TiO2(110) during reoxidation using time-resolved scanning tunnelling microscopy . Phys. Rev. B , 66 035409
- McCarty , K.F. and Bartlet , N.C. 2003 . Role of bulk thermal defects in the reconstruction dynamics of the TiO2(110) surfaces . Phys. Rev. Lett. , 90 046104
- McCarty , K.F. 2003 . Growth regimes of the oxygen-deficient TiO2(110) surface exposed to oxygen . Surf. Sci. , 543 : 185 – 206 .
- Blanco-Rey , M. , Abad , J. , Rogero , C. , Mendez , J. , Lopez , M.F. , Martin-Gago , J.A. and de Andres , P.L. 2006 . Structure of rutile TiO2(110)-(1 × 2): formation of Ti2O3 quasi-1D metallic chains . Phys. Rev. Lett. , 96 055502
- Park , K.T. , Park , M.H. , Meunier , V. and Plummer , E.W. 2006 . Surface reconstructions of TiO2(110) driven by suboxides . Phys. Rev. Lett. , 96 226105
- Wendt , S. , Sprunger , P.T. , Lira , E. , Madsen , G.K.H. , Li , Z. , Hansen , J.O. , Matthiesen , J. , Blekinge-Rasmussen , A. , Laegsgaard , E. , Hammer , B. and Besenbacher , F. 2008 . The role of interstitial sites in the Ti3d defect state in the band gap of titania . Science , 320 : 1755 – 1759 .
- Nolan , M. , Elliott , S.D. , Mulley , J.S. , Bennett , J.A. , Basham , M. and Mulheran , P.A. 2008 . Electronic structure of point defects in controlled self-doping of the TiO2(110) surface: combined photoemission spectroscopy and density functional theory study . Phys. Rev. B , 77 235424
- Rappe , A.K. and Goddard , W.A. 1991 . Charge equilibration for molecular dynamics simulation . J. Phys. Chem. , 95 : 3358 – 3363 .
- Zhou , X.W. , Wadley , H.N.G. , Filhol , J.-S. and Neurock , M.N. 2004 . Modified charge transfer-embedded atom method potential for metal/metal oxide systems . Phys. Rev. B , 69 035402
- Swamy , V. and Gale , J.D. 2000 . Transferable variable-charge interatomic potential for atomistic simulation of titanium oxides . Phys. Rev. B , 62 : 5406 – 5412 .
- Swamy , V. , Muscat , J. , Gale , J.D. and Harrison , N.M. 2002 . Simulation of low index rutile surfaces with a transferable variable-charge Ti-O interatomic potential and comparison with ab initio results . Surf. Sci. , 504 : 115 – 124 .
- Hallil , A. , Tétot , R. , Berthier , F. , Braems , I. and Creuze , J. 2006 . Use of a variable charge interatomic potential for atomistic simulations of bulk, oxygen vacancies, and surfaces of rutile TiO2 . Phys. Rev. B , 73 165406
- Henkelman , G. and Johnsson , H. 2000 . Improved tangent estimate in the nudged elastic band method for finding minimum energy paths and saddle points . J. Chem. Phys. , 113 : 9978 – 9985 .
- Mulheran , P.A. , Basham , M. , Bennett , R.A. , Sanville , E. , Nolan , M. and Elliott , S.D. Testing QEq potentials: self-doped titania surface defects . Phys. Rev. B , (submitted)
- Morgan , B.J. and Watson , G.W. 2007 . A DFT+U description of oxygen vacancies at the TiO2(110) surface . Surf. Sci. , 601 : 5034 – 5041 .
- Di Valentin , C. , Pacchioni , G. and Selloni , A. 2006 . Electronic structure of defect states in hydroxylated and reduced rutile TiO2(110) surfaces . Phys. Rev. Lett. , 97 166803
- Sanville , E. , Kenny , S.D. , Smith , R. and Henkelman , G. 2007 . Improved grid-based algorithm for Bader charge allocation . J. Comput. Chem. , 28 : 899 – 908 .