Abstract
We review state-of-the-art Monte Carlo (MC) techniques for computing fluid coexistence properties (Gibbs simulations) and adsorption simulations in nanoporous materials such as zeolites and metal–organic frameworks. Conventional MC is discussed and compared to advanced techniques such as reactive MC, configurational-bias Monte Carlo and continuous fractional MC. The latter technique overcomes the problem of low insertion probabilities in open systems. Other modern methods are (hyper-)parallel tempering, Wang–Landau sampling and nested sampling. Details on the techniques and acceptance rules as well as to what systems these techniques can be applied are provided. We highlight consistency tests to help validate and debug MC codes.
Introduction
To study thermodynamic properties at the molecular level, one needs to collect information of the positions of the atoms averaged over a long time. In molecular dynamics (MD) simulations,Citation1Citation2Citation3Citation4 successive configurations of the system are generated by integrating Newton's laws of motion, which then yields a trajectory that describes the positions, velocities and accelerations of the particles as they vary with time. MD is conceptually easy to understand. The complexity of MD codes is in speed optimisation such as parallelisation and in implementing derivatives of the energy to get the forces. The derivatives can be tedious to derive and implement, especially for bend–torsion cross potentials and anisotropic sites.
Monte Carlo (MC) takes a similar approach, but focuses on static properties. Therefore, there is no requirement that the systems should evolve in time. In MD, each state of the system depends on the previous state, related in time as a trajectory. But in MC, there is no such connection between ‘snapshots’ (states) of the system. Similar to MD, average properties are computed as averages over all the states of the system.[Citation5] The method is much more amenable to other ensembles than just the canonical ensemble, as long as each state can be generated with the proper weight. In principle, each molecular state can be created from scratch. However, for efficiency, most MC algorithms base a new snapshot on the modification of the current snapshot by performing changes called ‘moves’. Common moves are to translate and/or rotate a molecule. Such an attempt can be ‘accepted’ or ‘rejected’ by an ‘acceptance rule’, which means that the state has changed or that the new state is simply equal to the old state. All these snapshots form a chain, called a Markov chain, and averages are computed as averages of this Markov chain. Only static properties can be computed in MC, because there is no time involved in an MC move. This might seem non-physical or unnatural, but it is in fact where the real power of MC lies. There are no constraints on MC moves other than that they generate the appropriate ensemble. This is guaranteed by the form of the acceptance rules. Of course, the MC moves should ‘sample’ all the relevant states of the system (‘ergodicity’). But, in addition to the required moves, there is an enormous opportunity to devise clever and efficient MC algorithms, sampling techniques and MC moves. For example, MC moves that change the composition or connectivity of the atoms can be devised. The biggest limitation of MC methods is that they are considerably harder to apply to chemically complex molecules than MD. Therefore, MC used to be limited to reasonably small molecules, but the range of systems sizes, molecules, and algorithms is rapidly advancing. For more details, Vitalis et al. published an overview of the state-of-the-art MC methods designed for efficient sampling of biomacromolecules.[Citation6]
MC is all about generating and measuring probability distributions, which provide great opportunities but make MC codes notoriously difficult to debug and test. Errors in algorithms, coding errors and/or sampling errors are difficult to find and analyse. This is largely due to symmetry and cancellation where errors are in many systems hidden in the noise, only to show up in certain systems or conditions. Naively, one might think that two different (and both correct) algorithms should give the same result and in a perfect world they would. But, in practice, each method has its own range of applicability and efficiency. In this review, we therefore provide details on many MC algorithms that have been developed to study fluid properties and adsorption, and show many validation tests aimed at checking the correctness of the MC program implementation.
Molecular simulations
Force fields
The basic ideas behind molecular mechanics (MM) date back to the 1930s and 1940s.Citation7Citation8Citation9 MM assumes that matter consists of atoms and for every set of positions of the atoms the potential energy surface (PES) can be defined.[Citation10] In 1950–1970, the advent of computers caused MM methods to grow in popularity at a rapid rate, and currently, MM is one of the standard methods of structural chemistry. The classical molecular energy can be described as a Taylor expansion in bonds, bends, torsions, etc.[Citation11,Citation12]
Just because a model lacks certain key elements does not mean that all results are wrong; similarly, a more sophisticated force field does not necessarily give better results. A well-calibrated simplistic model can often produce better results than a generic, elaborate model. The more parameters a model has to optimise, the harder it becomes. Parameterisation is an art.[Citation13] There have been a number of approaches to parameterise a force field directly from the quantum chemical calculations (see Ref. [Citation14] and references therein). As the true PES can be approximated using quantum mechanical (QM) methods, a force field can be directly fit to a calculated quantum mechanical PES (QM PES) by numerically matching the gradients or energy. Although it is theoretically possible to include non-bonded interactions in the fitting, it is more common to obtain charges and van der Waals parameters separately and use these as input. A common approach is to use genetic algorithms (GA).[Citation15] Tafipolsky et al. parameterised a force field from first principles reference data by optimising a novel objective function with a GA.[Citation15] The GA are able to efficiently parameterise the bonded terms, which reproduce the density functional theory (DFT) results (structure and vibrational modes) as close as possible.
It can be convenient to think of the optimisation process as bottom-up (left-to-right in Equation (1))
Bonds/angles/torsion: Parameters can be obtained from gas-phase QM and spectroscopy.
Point charges: Parameters can be obtained by minimising the difference of the classical electrostatic potential and a QM electrostatic potential over many grid points (ChelPG methods) Citation16Citation17Citation18; REPEAT method [Citation19,Citation20]; partial equalisation of orbital electronegativity (PEOE) or the Gasteiger method [Citation21] and charge equilibration methods.Citation22Citation23Citation24
Polarisabilities: Parameters can be obtained from gas-phase quantum or from experiment.
van der Waals: Vapour–Liquid Equilibrium (VLE) curves; inflections in isotherms [Citation25,Citation26]; small noble gases such as argon from second virial coefficients; in general, from comparison with experiments (e.g. density, heat capacity and compressibility).
Equation (1) is historically referred to as a force field. The name arose from the lowest order approximation using only springs with force constants. Force fields have matured, and many parameters exist for a wide range of structures. These parameters are crucial and determine the quality of the force field. A few examples of popular generic force fields are AMBER,Citation27Citation28Citation29Citation30Citation31 OPLS,Citation32Citation33Citation34Citation35 AMBER/OPLS,Citation36Citation37Citation38Citation39 CHARMM,Citation40Citation41Citation42 GROMOS,Citation43Citation44Citation45 CVFF,[Citation46] CFF,Citation47Citation48Citation49 MM2,Citation50Citation51Citation52 MM3,[Citation53,Citation54] MM4,Citation55Citation56Citation57Citation58Citation59Citation60 MMFF94,Citation61Citation62Citation63Citation64Citation65Citation66 DREIDING,[Citation67] COMPASS,[Citation68] PCFF [Citation68] and UFF.[Citation69]
A computationally very efficient model is the transferable potentials for phase equilibria (TraPPE) force field by Martin and Siepmann.[Citation70,Citation71] The force field describes, e.g. linear alkanes, mono-branched and di-branched alkanes. Despite the fact that the model lumps CH3, CH2 and CH into single interaction centres, it reproduces the experimental phase diagram and critical points very accurately. This united-atom approach allows for much longer simulation times and larger systems because each of the CHx groups is charge neutral and charge–charge interaction can be omitted. The TraPPE model has been extended over the years to include explicit hydrogen description of benzene and five-membered and six-membered heterocyclic aromatic compounds,[Citation72] thiols, sulphides, disulphides and thiophene,[Citation73] ethers, glycols, ketones and aldehydes,[Citation74] primary, secondary and tertiary amines, nitroalkanes and nitrobenzene, nitriles, amides, pyridine and pyrimidine.[Citation75]
The TraPPE models are calibrated hierarchically using VLE data, e.g. CH4 using methane data, CH3 using ethane data, CH2 using propane data, CH using isobutane and C using neopentane. This then constitutes the ‘calibration set’. With the final parameters in hand, the remaining experimental data, e.g. for heptane or isopentane, can then be used to validate the predictions of the model. If for this ‘validation set’ unsatisfactory results are obtained and the experimental calibration data are not in doubt, then the model needs to be refined. A more sophisticated model could include explicit hydrogens [Citation76] or anisotropic sites.[Citation77,Citation78] However, the united-atom TraPPE model for alkanes is able to accurately describe the properties of alkanes over a wide range of chain lengths, densities, pressures and temperatures.
Adjusting these force field parameters to VLE data is currently a cumbersome and computationally expensive task. For alkanes, a hierarchical optimisation (e.g. first CH3 from ethane, then CH2 from propane and so on) is possible due to the simplistic nature of alkanes, but this is an exception rather than the rule. A force field optimisation via the VLE of CO2 involves optimising the van der Waals interactions of the carbon and oxygen atom type simultaneously, therefore requiring many time-consuming iterations. An objective function can be defined as the squared deviation between the computed and the experimental data. Current fitting strategies involve lowering this function below a defined threshold using Simplex- or gradient-based methods. Each iteration point requires a full molecular simulation run. Van Westen et al. developed a method to overcome this limitation.[Citation79] The actual optimisation is performed in a faster, different framework than molecular simulation, i.e. using the perturbed-chain statistical associating fluid theory (PC-SAFT) equation of state.[Citation80] The PC-SAFT is able to sufficiently capture the size and shape of molecules and the dispersion interactions of fluids to reduce the amount of iterations to only 2 or 3.
Molecular simulation codes
A list of molecular codes that employ force fields for classical molecular simulations is (by no means complete) given in the following:
DLPOLY: DLPOLY is an MD software for macromolecules, polymers, ionic systems, solutions and other molecular systems. It can also perform MC simulations (DL MONTE) but only metropolis sampling.[Citation81]
TINKER: The TINKER molecular modelling software is a complete and general package for MM and dynamics, with some special features for biopolymers. The software by Jay Ponder performs energy minimisations, molecular, stochastic and rigid body dynamics with different periodic boundaries, normal mode vibrational analysis, simulated annealing, free energy calculations and global optimisation among other things. It can use a large array of common force fields (e.g. Amber, CHARMM, Allinger MM and OPLS).[Citation82]
DYNAMO: Library of modules developed for the simulation of molecular systems using hybrid QM and MM potentials. The library supports a range of different types of molecular calculation including geometry optimisations, reaction-path determinations, MD and MC simulations.[Citation83,Citation84]
GULP (the General Utility Lattice Program): It is a powerful classical simulation code written by Julian Gale for performing a wide range of calculations on 3D periodic solids, 2D surfaces, gas-phase clusters and isolated defects in a bulk material.[Citation85] In particular, the code has a large number of material-specific force fields, such as the shell model for simulating ionic materials. It can also be used to fit and develop new force fields with the wide range of potentials. GULP is integrated in the Materials Studio package from Accelrys.
Materials Studio (forcite): It is an advanced classical MM tool that allows fast energy calculations and reliable geometry optimisation of molecules and periodic systems. For crystal structures, geometry optimisation with Forcite retains the crystal symmetry. Forcite provides the user with great flexibility, offering a range of force fields and charging methods.[Citation86,Citation87]
LAMMPS (Large-scale Atomic/Molecular Massively Parallel Simulator): It is a classical MD code with very good performance and scalability.[Citation88] LAMMPS has potentials for soft materials (biomolecules and polymers) and solid-state materials (metals and semi-conductors) and coarse-grained or mesoscopic systems.
GROMACS (GROningen MAchine for Chemical Simulations): It is a MD package primarily designed for simulations of proteins, lipids and nucleic acids.[Citation89,Citation90]
BOSS (Biochemical and Organic Simulation System): Bill Jorgensen's general purpose molecular modelling system that performs MM calculations, MC statistical mechanics simulations and semi-empirical AM1, PM3 and PDDG/PM3 QM calculations.[Citation91] The MM calculations cover energy minimisations, normal mode analysis and conformational searching with the OPLS force field. The MC simulations can be carried out for pure liquids, solutions, clusters or gas-phase systems.
MCPRO: Performs MC statistical mechanics simulations of peptides, proteins and nucleic acids in solution; it was derived from BOSS, but makes extensive use of the concept of residues. The MC simulations can be performed in a periodic solvent box, in a solvent cluster or in a dielectric continuum including the gas phase.[Citation92]
Etomica: Java-based MC package by David Kofke and Andrew Schultz originally designed for use as a teaching tool, but potentially powerful enough for research grade application. The code is object oriented and can be used independent of development environment.[Citation93]
Towhee: An MC molecular simulation code originally developed in the group of J. Ilja Siepmann and designed for the prediction of fluid-phase equilibria using atom-based force fields and the Gibbs ensemble with particular attention paid to algorithms addressing molecule conformation sampling.[Citation94] The code is now mainly developed and maintained by Marcus G. Martin and has subsequently been extended to several ensembles, many different force fields and solid (or at least porous) phases.
MedeA® Gibbs: Gibbs ensemble MC package from the group of Alain Fuchs to compute the thermophysical properties of single and multi-phase fluids as well as adsorption isotherms of fluids on solids.[Citation95]
BIGMAC: General purpose configurational-bias Monte Carlo (CBMC) code developed in the group of Berend Smit for grand canonical and Gibbs ensemble simulations of linear alkanes.[Citation96,Citation97]
OPENMD: An open source MD engine, created mostly by graduate students in the Gezelter group at the University of Notre Dame, which is capable of efficiently simulating liquids, proteins, nanoparticles, interfaces and other complex systems. Proteins, zeolites, lipids and transition metals (bulk, flat interfaces and nanoparticles) have all been simulated using force fields included with the code.
Cassandra: an open source MC package being developed in the Edward Maginn group at the University of Notre Dame.[Citation98] Cassandra is capable of simulating any number of molecules composed of rings, chains or both, such as small organic molecules, oligomers and ionic liquids. Cassandra can simulate the following ensembles: canonical (NVT), isothermal–isobaric (NpT), grand (μVT), osmotic (μpT), Gibbs (NVT and NpT versions) and reactive (RxMC). Cassandra is parallelised with openMP.
MUSIC (Multipurpose Simulation Code): Object-oriented code developed in the group of Randall Snurr the major application of which is in the computation of diffusion and adsorption in zeolites. It can also perform liquid and gas simulations, as well as GCMC, NVT-MC, NpT-MC, Hybrid MC, MD and NEMD simulations.[Citation99,Citation100]
RASPA: Simulation package (MC, MD and optimisation) developed in the group of Randall Snurr at Northwestern University in collaboration with Universidad Pablo de Olavide with emphasis on adsorption and diffusion simulations in flexible nanoporous materials. It includes CBMC, CFMC, Gibbs, for single and multicomponents that can be flexible and/or rigid and can handle many ensembles.[Citation101] All simulations in this work are performed with this code.
Three main types of parallel molecular dynamics simulations have been developed: the replicated data decomposition, the spatial decomposition, and the force decomposition.[Citation102] Similar techniques can also be used to parallelise MC simulations.[Citation103] MC is more amenable to parallelisation than MD, because most MC algorithms have at least some degree of inherent parallelism. Parallel tempering runs multiple synchronised Markov chains simultaneously. Only when random pairs of systems exchange information is communication necessary. Conventional MC moves in the canonical ensemble do not lend themselves to efficient parallelisation, but more advanced moves are straightforward to parallelise (e.g. in biasing moves ‘trial positions’ can be computed independently). Such parallel algorithms are described in Refs Citation104Citation105Citation106. The multiple first-bead method of Esselink et al. places many first beads and chooses the most favourable.[Citation104,Citation105] The method increases the probability of successful insertions especially at higher densities.
The effort of parallel implementation is significant, and there are two good reasons to do it: (1) memory problems (i.e. the memory would not fit onto a single processor) and (2) to get the final results faster. Both cases can occur for very large systems. If these situations do not apply, then MC is more amenable to a ‘task-farming approach’. Each node and, therefore, each simulation runs totally independently of all other nodes. Instead of running a single serial code 10 times longer (e.g. to get more accurate results), one can just run 10 independent serial runs on different nodes and combine the results for these 10 runs. It is for this reason that parallel MC codes are relatively scarce.
Ensembles
To describe the microscopic state of a system, classically, at some instant of time, we need 6 N variables; for each of the N atoms we have three positions and three velocities. This state is a point in a 6 N space, called phase space. The state evolves through phase space according to the laws of mechanics. The measurements of macroscopic variables such as temperature T, volume V and pressure p involve taking time averages over the phase-space curve of the system. This is in fact the basis of the MD technique. Around 1900, Gibbs introduced the concept of ‘ensembles’, in which the time averaging is replaced by averaging over a group of microstates with the same macroscopic state (e.g. N, V and T). The ergodic principle states that this ensemble averaging and time averaging are equivalent (when simulated infinitely long). The phase-space points Γ are distributed according to a probability density . The functional form depends on the chosen ensemble.
For single components, there is one fundamental thermodynamic function
Transformation of the internal energy U
where the functionis known as the Hill energy.
Transformation of the enthalpy H
where the functionTransformation of the Helmholtz function A
where the functionTransformation of the Gibbs function G
where the function
All seven functions ,
,
,
,
,
and
are derivable from a Legendre transformation of the fundamental law of the energy conservation expressed in the internal energy function
(Equation (2)). Figure , taken from Ref. [Citation108], shows the ensemble and their connection to the reservoirs. The reservoirs impose constant temperature, tension and pressure and chemical potential. The ensembles in the left-hand column in Figure are constant energy ensembles, whereas the ensembles in the right-hand column have constant temperature.
![Figure 1 (Colour online) Ensembles: Shown are the eight ensembles for a single component system. The systems interact through a combined temperature, pressure and chemical potential reservoir. The ensembles on the left are adiabatically insulated from the reservoir, whereas those on the right are in thermal contact with the reservoir. Pistons and porous walls allow for volume and particle exchange. Adiabatic walls are shown cross-hatched, whereas dithermal walls are shown as solid lines. Ensembles on the same height are related by Laplace and inverse Laplace transformations. The pressure stands for the pressure and the tension. Picture taken from Ref. [Citation108].](/cms/asset/d1e102e0-862b-4389-93e6-b6dbc4038086/gmos_a_819102_f0001_oc.gif)
All eight ensembles (for a single component system) may be simulated using either MD or MC simulations. The probability distributions are exponentials for the isothermal ensembles and power laws for the adiabatic ensembles.[Citation109] For example, for the NVT ensemble the probability density has the Boltzmann form with U(r) the potential energy and C a constant. For the (Ht and pN) ensemble, the trial MC moves involve particle moves and simulation cell shape/size moves.[Citation110] For the (Rt and pμ) ensemble, MC moves involve particle moves, shape/size moves and insertion and deletion of particles.[Citation111] For MC simulations of these ensembles, one uses the probability density directly in the simulation, whereas for MD simulations, ordinary differential equations of motion are solved for equations arising from Hamilton's equations.
System set-up
The simulation cell and boundary conditions
Computer simulations are limited to a number of molecules that can be efficiently stored in memory. Although simulations of hundreds of thousands of atoms have been reported and will likely increase by orders of magnitude in the future, this number is still far removed from the thermodynamic limit. To be able to extrapolate the results for a finite system to macroscopic bulk values one usually employs periodic boundary conditions.[Citation1] Periodic boundary conditions are commonly applied to overcome the problems of surface effects. The original simulation box, including all the atoms within it, is replicated throughout the space.[Citation112] When a molecule in the original box moves, its periodic images in each of the surrounding boxes moves in exactly the same way. If a molecule leaves the central box, one of its images will enter the box through the opposite face. It is not necessary to store the coordinates and momenta of all the images, only those in the central box are needed, because the images can be obtained from translation operators. Usually, one imposes the minimum image convention: the distance between two particles is taken to be the shortest distance between their periodic images. The boundary of the periodic box does not have any physical significance, only the shape and orientation.[Citation3] The use of periodic boundary conditions inhibits the occurrence of long-wavelength fluctuations. It is, for example, not possible to simulate a liquid close to the glass-liquid critical point where the range of critical fluctuations is macroscopic.[Citation3]
In general, the unit cell is defined by the cell lengths a, b, c and angles α, β, γ and by the fractional coordinates s of the atoms within the unit cell. These coordinates form an orthonormal dimensionless space. The transformation from fractional space to Cartesian space can be carried out by the matrix h:
In general, it is prudent to use a sufficiently large simulation cell (and a large amount of particles) to avoid ‘finite size’ effects. The error of thermodynamic properties in periodic systems with only a few hundred particles can be quite large,[Citation113] especially in the vicinity of critical points. It is advisable to study the system properties as a function of system size to estimate the finite size effects. Also, sometimes theoretical corrections can be applied to the simulation results.Citation114Citation115Citation116
Initialisation, equilibration and production runs
To prepare the system at the desired temperature in an equilibrium configuration for MD, we initialise the system by the following procedure:
N molecules are inserted into the simulation box at random positions as long as no overlaps occur with a framework and/or other particles, and as long as the positions are accessible from the main cages and channels (when a framework solid is present). Inaccessible pockets need to be blocked.[Citation117] One can use a list of volume shape/sizes that are automatically considered an overlap, either computed in advance or on-the-fly.[Citation118]
During the initialisation period, we perform an NVT MC simulation to rapidly achieve an equilibrium molecular arrangement.
After the initialisation step, we assign all the velocities of atoms from the Maxwell–Boltzmann distribution at the desired average temperature. The total momentum of the system is set to zero (to avoid centre-of-mass drift of the system). Next, we equilibrate the system further by performing an NVT MD simulation using a thermostat.
The equilibration is completed and during the production run we collect statistics using either the NVE or NVT ensemble. Following this equilibration procedure, the average temperature using NVE over the entire production period is usually within a few Kelvin of the desired average temperature, while NVT would give the exact desired average temperature if simulated sufficiently long.
After these steps, the procedure for MC and MD differs. For MD, the next step is to equilibrate the velocities. Atoms are assigned velocities from a Maxwell–Boltzmann distribution, and the integration of the equations of motion is started. Due to previous equilibration of positions, the actual time to properly equilibrate the velocities (and also again the positions) is significantly reduced. The final step for MD is to start the actual ‘production run’ where properties of interest are measured. For MC, the production run can immediately be started after the equilibration of the positions.
MD is counted in MD steps, i.e. single integration steps, from which the total simulations time in ps or ns can be deduced. For MC, the duration of the simulation is measured in ‘MC steps’ or ‘MC cycles’. An MC step is one performed MC move, either accepted or rejected. The MC moves are chosen in random order with a preset probability. An MC cycle takes the number of particles into account, and in each cycle on average one MC move has been attempted per particle. The reason behind this is that one needs to sample longer if there are more molecules in the system. Therefore, the number of cycles is less dependent on the system size. To avoid poor sampling at low densities, the number of steps per cycle can have a set of lower limit of 20. A cycle is then, for example, defined as [Citation96]
Long-range interactions
van der Waals
The most common repulsion/dispersion functional form for the van der Waals interaction is the Lennard-Jones potential (see Figure )
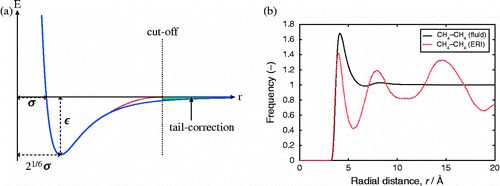
To make the simulations tractable, the van der Waals potentials are truncated at a certain distance where the interactions are considered sufficiently small. In MC, the truncated potential can be used, and the energy correction due to this truncation, called the ‘tail correction’, can be approximated (see Figure ). The truncation distance and whether to use tail correction or not should be considered as part of the force field. For MD, the truncation in the energy leads to a divergence in the forces. Common approaches include to shift the van der Waals potential to zero at the cut-off and the use of a switching function where the energy is forced to smoothly go to zero Citation120Citation121Citation122
The cut-off is usually chosen as the distance where the radial distribution function (RDF) approaches unity. The requirement that the smallest perpendicular distance of the simulation cell has to be larger than twice the cut-off distance rc determines the minimum amount of crystallographic unit cells to be used in adsorption simulations. If we assume the RDF for
we can write
Coulombic interactions
The Coulombic potential decays as r− 1 and the tail correction diverges. The total electrostatic potential energy of interaction between point charges qi at the positions ri is given by
The basic idea of the Ewald approach is as follows. The error function erf(x) and its complement are defined as:
Ewald noted that
The vectors , which need not be orthogonal, form the edges of the unit cell. The conjugate reciprocal vectors
are defined by the relations
The Ewald expression for the Coulombic energy in a truly periodic system reads
For a net-charged system, the fully periodic Coulombic energy diverges. This does not happen with the Ewald method, as the divergence catastrophe is avoided by a ‘neutralising plasma’ – a uniform distribution of charge equal and opposite to the net charge of the system – which is usually said to be added to the system. The neutralising plasma is effectively represented in Ewald summations by the omission of the k = 0 term in the reciprocal space sum. Systems with a net charge possess a finite non-zero energy and pressure that may lead to artefacts in such systems. Bogusz et al. have derived corrections to the Ewald expressions to remove pressure and free energy artefacts in charged periodic systems.[Citation129] The net-charge correction term can be logically broken into five terms: a reciprocal space term, a real space term, a self term, a surface dipole term and a system-dependent correction, such as a Born or Poisson–Boltzmann term (which corrects for missing long-range polarisation):
The Ewald method can be implemented very efficiently for MC methods as only the change for moving atoms needs to be computed.[Citation130] The real part and the imaginary part
of the structure factor can be expressed as a summation over all particles in the system. It is convenient to store
and
in the memory of the computer (one complex number for each k). For particle displacements, rotations, regrows, (test) insertions, deletions and identity changes, one can easily calculate the new values for
and
by subtracting the contributions of the old configuration and by adding the contributions of the new configuration. This has to be done only for atoms that have a different position in the old and new configuration.
The Ewald summation is superior to the recently proposed Wolf method [Citation131] and other direct summation methods, both in accuracy and in speed.[Citation130] It is well known that a direct r− 1 summation only converges for very large values of the cut-off radius,[Citation3] for example more than 40 Å for neutral molecules. Damping the Coulombic potential reduces this range, although still a cut-off of at least 25 Å seems to be the minimum requirement,[Citation130] but at the cost of uncertainty in the damping constant α. Different values for α are recommended in the literature implying that this damping is system specific and possibly dependent on the molecule type, framework topology, temperature, etc. The Ewald summation is able to compute the electrostatic energy exactly (to within any specified numerical precision) for a system of charges in an infinitely periodic structure at a cost less than quadratic with the number of particles (N3/2 for Ewald). The energy and forces computed with the Ewald summation are well defined and unique. They do not depend on a judicious choice of damping parameters and cut-off values, and Ewald type methods are at the same level of accuracy significantly cheaper.
The MM2 and MM3 force fields calculate the electrostatic interaction in non-charged polar molecules by using bond dipoles. The electrostatic interaction potential is then given by
Units
An excellent discussion of the handling of units is given in the manual of DLPOLY.[Citation81] A small set of internal units needs to be chosen. A convenient set that is chosen in DLPOLY, RASPA and many other codes is given in the following:
The unit of length l0 is chosen as Å, i.e. l0 = 10− 10 m.
The unit of time t0 is chosen as picoseconds, i.e. t0 = 10− 12 s.
The unit of mass m0 is chosen as atomic mass units, i.e.
The unit of charge q0 is chosen as units of proton charge, i.e.
The unit of energy
The unit of pressure
The unit of diffusion
Methodology
Monte Carlo
Statistical mechanics formulate the following postulates [Citation4,Citation132]:
Postulate of ensemble averaging.
The average behaviour of a macroscopic system in equilibrium is given by the average taken over a suitable ensemble consisting of an infinite number of randomised mental copies of the system of interest.
Postulate of equal a priori probabilities.
In a state of macroscopic equilibrium, all stationary quantum states of equal energy have equal a priori probability (in the micro-canonical ensemble).
Postulate of equilibrium state.
Equilibrium state is the one that occupies the maximum volume in the phase space.
The method of calculation is statistical in nature.
The predictions are regarded as true on average rather than precisely expected for any particular system.
The probability of finding a system in a given state being proportional to the phase-space volume associated with it, the most probable state would be one that occupies the maximum volume in the phase space. It follows that the equilibrium state is the state of maximum probability.
In its simplest form, the MC method is nothing more than a computer-based exploitation of the law of large numbers to estimate a certain probability or expectation. At the heart of the algorithm lies the ‘random numbers generator’, and therefore, it is advisable to use a high-quality generator such as the ‘Mersenne Twister’.[Citation133,Citation134] The Markov Chain Monte Carlo (MCMC) method is an important tool to estimate the average properties of systems with a very large number of accessible states.[Citation135,Citation136] The system evolves from state to state (possibly the same state), and averages of a property are computed as the average over the elements of the Markov chain. For an infinite Markov chain the expression is exact. The scheme makes use of the fact that only the relative probability of visiting points in configuration space is needed, not the absolute probability. To visit points with the correct frequency, the MCMC algorithm generates random trial moves from the current (‘old’) state (o) to a new state (n). To show that an arbitrary initial distribution eventually relaxes to the equilibrium distribution, it is often convenient to apply the condition of detailed balance (as is used in the original Metropolis scheme). If and
denote the probability of finding the system in states (o) and (n), respectively, and
and
denote the conditional probability to perform a trial move from
and
, respectively, then the condition called ‘detailed balance’ can be written as
Let us recall for example the canonical partition function [Citation137]
In MC, only the positions are used. The momenta can be integrated analytically by making use of
The de Broglie wavelength Λ is the wavelength of a gas particle with momentum determined by the average thermal kinetic energy per degree of freedom . If we use the de Broglie relation
, then from
we have
. The condition for the applicability of classical or Boltzmann statistics is equivalent to the condition
, where Λ represents the critical length scale at which interactions are neglected. Closely related to h divided by the momentum, the de Broglie wavelength is defined as
For ensembles where the simulation cell is allowed to change, it is more convenient to redefine the positions in fractional coordinates using .[Citation138] In fractional coordinates, it is easier to describe a volume change while leaving the particle positions the same. The partition function in fractional coordinates is related to the Cartesian version by a factor
Some of the most commonly used ensembles are the canonical ensemble, the isothermal–isobaric ensemble, the grand canonical ensemble, the Gibbs ensemble and the ensemble.
Canonical MC
In the canonical ensemble, the number of particles N, the temperature T and the volume V are constant. The partition function Q is Citation137Citation139Citation140Citation141
The particle moves such as translation and rigid rotation have the following acceptance rule:
NpT ensemble
The MC extension to the NpT ensemble is by Woods [Citation142,Citation143] and samples the phase space of a constant N, constant p, constant T ensemble with the appropriate phase-space probability. In addition to the particle moves performed at constant T, changes in the volume are attempted and accepted/rejected according to an evaluation of the enthalpy change. The NpT ensemble has volume trial moves which implies implicit particle scaling using a factor
. The ΔV is a random number uniformly distributed over the interval
. The isothermal–isobaric ensemble partition function, introduced by Guggenheimer,[Citation144] is given in fractional positions by [Citation138,Citation145]
However, often an alternative move is used, where the volume is changed not in V but in . The partition function is
The change in enthalpy for a volume change
The particle moves have the same acceptance rules in the NpT ensemble as in the NVT ensemble
Gibbs ensemble
The Gibbs ensemble MC simulation technique allows direct simulation of VLE curves of fluids.[Citation150,Citation151] In some sense, it combines the NVT, the and the NpT ensembles and therefore provides a generic theoretical framework. Gibbs ensemble simulations are performed in two separate microscopic regions, each within periodic boundary conditions (Figure (a)). An n-component system at constant temperature T, total volume V and total number of particles N is divided into two regions, with volumes
and
and number of particles
and
. The partition function is given by
displacement of a randomly selected particle.
Figure 3 (Colour online) Gibbs, adsorption using Gibbs, the osmotic and the grand canonical ensemble, (a) in the Gibbs ensemble the total volume and the total number of molecules are fixed. The volume move makes one box bigger and the other box smaller which leads to pressure equilibration. The exchange of particles between the boxes leads to equal chemical potential and in both boxes the same temperature is imposed. (b) Adsorption isotherms can be computed in the Gibbs ensemble where the fluid phase is explicitly simulated. (c) The osmotic ensemble replaces the explicit fluid phase by an imaginary reservoir. (d) The grand canonical ensemble also uses the imaginary reservoir but in addition keeps the volume fixed. 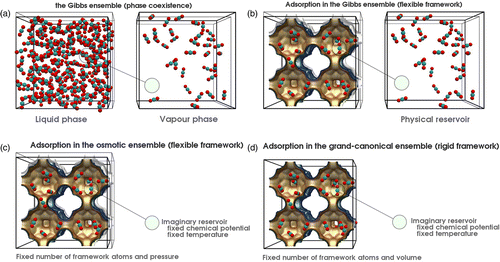
The acceptance rule is identical to that used in a conventional NVT ensemble simulation
change in box volumes while keeping the total volume constant.
The acceptance rule for a random walk in
transfer of a randomly selected particle from one box to the other.
For pure component systems, the phase rule requires that only one intensive variable (the temperature) can be independently specified when two phases coexist. The vapour pressure is obtained from the simulation. By contrast, for multicomponent systems pressure can be specified in advance, with the total system being considered at constant NpT. The only change necessary is that the volume changes in the two regions are now independent. The acceptance rule for a random walk in of region I, while the
volume remains unchanged which is given by
Adsorption simulation
In adsorption studies, one would like to know the amount of materials adsorbed as a function of pressure and temperature of the reservoir with which the adsorbent is in contact. Adsorption simulations can be performed in the Gibbs ensemble (adsorption equilibrium between a bulk fluid phase and the interior of an adsorbing material).[Citation152] The reservoir is explicitly simulated from which the pressure can be obtained (see Figure (b)). Conversion using an equation of state is not necessary between pressure and fugacity, albeit that the fugacity coefficient is one of the models (but hopefully close to the experimental fugacity coefficient if the fluid model is sufficiently accurate). Two other ensembles for computing adsorption are the (Figure (c)) and grand canonical ensemble (Figure (d)). Gibbs ensemble simulations of adsorption are almost identical to grand canonical simulations. The only difference is that the total composition of the system is imposed, rather than the chemical potentials in the exterior of the pore.
Grand canonical ensemble
The most common ensemble for adsorption is the grand canonical ensemble or ensemble.[Citation153] In this ensemble, the chemical potential μ, the volume V and the temperature T are fixed. MC simulations can be performed in the grand canonical ensemble by a direct generalisation of the NVT ensemble.[Citation154,Citation155] The partition function is given by
The acceptance rule can be derived
The pressure p in the reservoir is related to the chemical potential
The acceptance rules for insertion and deletion in the grand canonical ensemble are as follows:
μ1N2pT ensemble
The N2pT ensemble [Citation158] is the natural ensemble to compute adsorption for flexible frameworks. The system is considered as two components, where the chemical potential of component 1 is kept constant (and has variable particle number), while the component 2 has a constant particle number. In a constant pressure ensemble, the volume will automatically adjust to hold the pressure constant as the composition or temperature change. For a single component system, it is not possible to vary three intensive variables independently because of the Gibbs–Duhem relation which relates them. However, for two (or more) species systems, it is possible to derive, rigorously, a statistical ensemble in which T, P,
and
are held fixed. For this ensemble,
is the chemical potential of the adsorbate and
is the fixed number of atoms of the framework (host). This is a hybrid statistical ensemble which has some properties similar to the single species (NpT) and (
) ensembles.
The form of the probability distribution for the ensemble can be derived by performing three Laplace–Legendre transforms from the microcanonical (EVNadsNhost) ensemble. The probability that the system with temperature T, pressure P, chemical potential of the adsorbate and
host atoms has volume V and Nads atoms,
, where s stands for the positions of all the atoms in the system, is given by [Citation158]
In (N2pT) MC simulation, one carries out (at least) four distinct types of trial moves.[Citation158]
Configurational-bias Monte Carlo
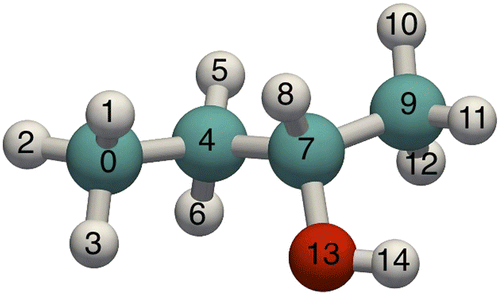
Conventional MC is time consuming for long-chain molecules. Moreover, the configurations of long molecules in the framework become increasingly different from the gas phase. The fraction of successful insertions into the sieve becomes too low. To increase the number of successfully inserted molecules, the CBMC technique was developed. Figure shows the definition of S2-butanol which will be used here to illustrate the method, and in a later section to validate the generated bond/bend/torsion distributions. Instead of generating ideal gas configurations and trying to insert the molecule as a whole, the CBMC method inserts chains part by part biasing the growth process towards energetically favourable configurations, and therefore significantly reduces overlap with the framework and other particles. The CBMC framework is based on the work by Rosenbluth and Rosenbluth [Citation162] and developed by a variety of researchers.Citation163Citation164Citation165Citation166Citation167Citation168Citation169 Later, CBMC was extended to include growpaths for branched molecules,[Citation3,Citation71,Citation76,Citation123,Citation170,Citation171] cyclic molecules Citation172Citation173Citation174Citation175Citation176 and reactive CBMC.[Citation177]
Growing a chain molecule
Let us first tackle the problem of how to generate a molecule with an appropriate intra-molecular energy (bond, bend and torsion). We can choose any of the atoms as a start point. Let us assume to start our growth process from atom 4 (the labelling is shown in Figure ). The starting atom is connected to atoms 0, 5, 6 and 7. One of these can be chosen randomly, for example atom 7. The position of atom 7 lies on a sphere with a radius depending on the bond-length distribution (Figure (a)), and is determined by computing a random vector on a unit sphere adjusted in length for the bond potential. The chosen bond length can be generated either using an ‘acceptance–rejection’ scheme or using a small MC routine. Having grown atoms 4 and 7 one can continue to grow on either side but let us assume to continue along the atom 7 path. Atom 7 is connected to atom 4 (already grown) and atoms 8, 9 and 13. Vlugt et al. noted that atoms 8, 9 and 13 must be grown simultaneously.[Citation97,Citation170] That is, it would be wrong to first place atom 13, next atom 8 and then atom 9. Because of the bond-bending potentials, the branches connected to the same central atom cannot be added independently. Vlugt et al. developed an MC procedure that can be used to generate the positions of the branch atoms using the following moves:
Changing the bond length
Figure 5 (Colour online) Growing (branched) molecules, (a) bonds are grown by choosing random positions on a sphere; bond lengths (b) and bend angles (c and d) between atoms of a branch are changed by an MC algorithm. The MC moves displace atoms along the bond vectors, or change the bend angle, or rotate the branch atoms around the axis of the bond that was already grown. 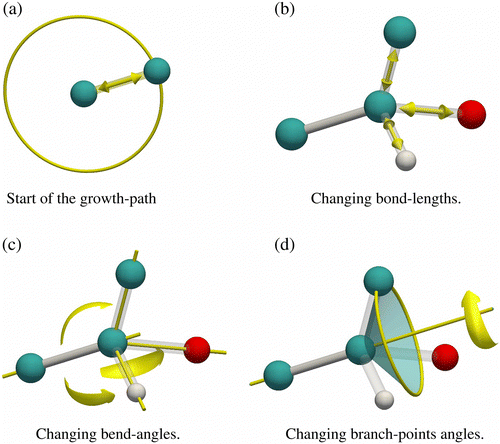
One of the branch atoms is randomly chosen and an attempt is made to change the bond length (see Figure (b)). The probability of generating a branch position b with bond length l is given by
and the acceptance rule for a change in bond length fromChanging the bend angle
One of the branch atoms is randomly chosen and an attempt is made to change the bend angle (see Figure (c)). The probability of generating a branch position b with bend angle θ is given by
and the acceptance rule for a change in bend angle fromRotation on a cone
One of the branch atoms is randomly chosen and rotated randomly on a cone (see Figure (d)). This move changes the bend angle between the branch atoms. The acceptance rule for a rotation on a cone is
The scheme of Vlugt et al. handled torsion potentials by including the torsion energy in Equation (92). Unfortunately, this method still fails to generate the proper distribution when there are multiple torsional angles that share the same two central atoms because the bond bending and torsional angle distributions are no longer independent. Vlugt et al. modified the force field for branched alkanes such that only one torsion potential was defined over a central bond. For multiple torsions over a central bond all of the atoms connected to those central atoms must be generated simultaneously in order to get the correct distribution. This implies that the conformation of the entire 2,3-dimethylbutane molecule must be generated in a single step in order to obtain the correct distribution. One of the methods developed to solve this problem is the coupled–decoupled CBMC algorithm of Martin and Siepmann.[Citation71,Citation171]
The bond angles are selected based solely on the bond angle energies and the phase-space terms, and then those angles are used in all subsequent selections (torsion and non-bonded). Thus, the bond angle selection is decoupled from the other selections. In contrast, for each non-bonded trial a full selection is done to generate torsional angles, so these two selections are coupled. Before explaining this further, let us see how to actually increase acceptance of insertion using more than one set of ‘trail positions’.
Trial positions
So far we have shown how to generate a molecule. In order to be able to steer (to ‘bias’) the growth, at each step, k sets of branch atoms called ‘trial positions’ are generated of which one is chosen with the appropriate probability. The growth control is largely based on the ‘external’ environment of the molecule, for example the framework and/or other molecules that are present in the system. In the CBMC scheme, it is therefore convenient to split the total potential energy U of a trial site into two parts.
Coupled–decoupled CBMC
The Vlugt CBMC scheme and other schemes for branched alkanes generate conformations according to the probability
Decoupled selections
In the decoupled selection, the probability to generate a given configuration and the corresponding Rosenbluth weights are given by
whereBend angles are chosen in a biased fashion and used as input for the subsequent biased selection of the torsional angles. These are then used as input for the Lennard-Jones selection, but the main drawback of this method is that once the torsional angles have been chosen in a biased fashion, there is only one possible trial site available for the Lennard-Jones selection (Coupled selections
In a coupled biased selection, the selections are coupled such that each biased selection sends multiple possible conformations to the next selection step. In this case
Handling multiple torsions and intra-molecular VDW interactions
A combination of the coupled- and decoupled-biased selections allows a great deal of flexibility when designing CBMC schemes. To handle multiple torsions over a single bond and to handle Lennard-Jones interactions, we can use decoupled CBMC and generate trial vectors for the torsional selection, of which
are selected for the Lennard-Jones step. The intra-molecular Lennard-Jones interactions are treated here as ‘external’ Lennard-Jones interactions. The CBMC scheme can also be used when the atoms of the molecule have charges, for molecules that have anisotropic sites and for molecules with cross terms such as bond–bond, bond–bend and bend–torsion potential. The molecule is grown without these terms but afterwards the weight is corrected (see section ‘Advanced bias corrections’).
Chemical potential reference state
The excess chemical potential is defined as the difference in chemical potential of the interacting chain and a chain in the ideal gas state. The Rosenbluth weight
of the reference state of the ideal gas is needed when comparing with real experimental data. When CBMC is used, it is straightforward to show that
has to be replaced by
for inserting a particle and by
for the deletion of a particle. Detailed balance is followed when
is replaced by
, i.e. the average Rosenbluth weight of a chain in the reservoir. This implies that
has to be computed only once for a given molecule and temperature.[Citation97]
The reference state is important and enters in the acceptance rules for CBMC insertion and deletion moves
Advanced bias corrections
Dual-cut-off CBMC uses an additional bias in the selection of trial segments.[Citation178] The motivation is that hard-core (repulsive) interactions are more important than long-range interactions in the selection of trial segments. Therefore, a second smaller cut-off radius can be used for the selection of trial positions (for LJ fluids one can use cut-off of around σ Å). The potential is split in two parts
Similarly, for anisotropic alkane models the chain can be grown as an ordinary united-atom model, but by correcting with the difference between the anisotropic and the approximate potential in the acceptance rule the correct distribution is recovered.[Citation179]
For computational efficiency, for molecules with partial charges it is advantageous to grow beads only with the LJ part plus the real part of the Ewald energy, and correct the final configuration for the Ewald Fourier energy using Equation (111). The more expensive Fourier part of the Ewald summation then only has to be computed once (and not for all the trial positions of all the beads).[Citation97] This also avoids the problem that during the growth the already grown part is not charge neutral.
Continuous fraction Monte Carlo
All open ensemble methods suffer from a major drawback: the insertion probability becomes vanishingly low at high densities. One of the schemes to remedy this problem is the CFMC method of Shi and Maginn.[Citation180,Citation181,Citation182] The system is expanded with an additional particle scaled in interactions using a parameter λ. Various choices for the scaling are possible, but for example the Lennard-Jones and charge–charge interactions are scaled as
CFMC uses conventional MC for thermalisation (such as translation, rotation moves and/or MC–MD hybrid moves), but in addition attempts to change λ of the fractional molecule using . The
is chosen uniformly between
and
and scaled to achieve around 50% acceptance. However, many systems show behaviour where λ changes are hard. An additional bias η on λ can be used, where each λ has an associated biasing factor η. This bias will be removed by the acceptance rules. A careful calibration of η can make the λ histograms flat and hence can avoid the system getting stuck in a certain λ-range. There are three possible outcomes of a change from
to
λ remains between 0 and 1.
The change in energy of the particle with the new
There is no change in the number of particle, nor in the positions, nor in the intra-molecular energies. Only λ and the inter-molecular energy have changed.λ becomes larger than 1.
When λ exceeds unity,
λ becomes smaller than 0.
When λ falls below 0,
The fractional molecule has interactions with the systems but its influence vanishes in the thermodynamic limit. A scheme that completely avoids the influence of the fractional particle is to make discrete changes in λ and only take thermodynamic averages when or
. However, this significantly reduces the amount of sampling points. A continuous distribution of couplings is therefore more convenient, and as we will show later, the influence of the fractional particle is in general small due to symmetry/cancellation.
The method is readily extended to other ensembles like the Gibbs ensemble. Here, an additional constraint is applied: the total lambda is unity, but the lambdas of the individual system are allowed to change, i.e. for box I and box II we require . The acceptance rule for a λ change which remains between 0 and 1 is
Insertions become particularly difficult in strongly interacting fluids such as ionic liquids and water. At the low temperatures, the efficiency of Gibbs exchange moves was found to be two orders of magnitude higher than that obtained by state-of-the-art rotational and configurational bias moves. It is therefore a very powerful method to handle open systems at high densities. A downside of CFMC compared to CBMC is that it takes longer time to equilibrate at lower densities. Only at or
, an attempt is made to delete or insert a molecule, respectively. Even when the λ moves are made equally likely using biasing, the diffusive nature of λ in the range
makes the insertion/deletion rate lower. For this reason, Shi and Maginn used a relatively high percentage of λ moves in comparison to the other MC moves. To overcome this (small) issue in a practical way, one can first use CBMC or restart from a CBMC simulation.
The nature of the algorithm can lead to blocking issues. For example, in CFMC the potentials are scaled such that particle can overlap. Shi and Maginn noted that for water models the lack of Van der Waals interaction for the hydrogens leads to overlap of the negatively charged oxygen with the positively charged hydrogen. The repulsive force that normally prevents this has been reduced by the potential scaling. A simple solution is to reject all MC moves where atoms get closer, for example 1 Å.[Citation180]
Reactive Monte Carlo
Many methods have been proposed to compute the equilibrium properties of chemically reacting or associating fluids.[Citation183,Citation184] The earlier methods required specification of chemical potentials or chemical potential differences. One of the most popular recent methods is the reactive Monte Carlo (RCMC) algorithm which samples reactions directly [Citation185,Citation186] (without going through the transition states). Therefore, only equilibrium properties can be computed. The method is conceptually simple, convenient and versatile. No chemical potentials or chemical potential differences need to be specified for the reaction steps, just the stoichiometry of the reactions. Essentially, the method enhances the GCMC with a ‘forward’ and ‘backward’ reaction step, which ensures that the chemical reaction equilibria between the reactants and the products are maintained. For a chemical reaction at equilibrium in a single- or a two-component system, the stoichiometric coefficient of each component i times the chemical potential of each component must be zero
RCMC simulations can be performed in the canonical,[Citation185,Citation186] grand canonical,[Citation187,Citation188] isothermal–isobaric,Citation189Citation190Citation191 Gibbs [Citation192] and other ensembles. When RCMC is used in conjunction with an ensemble that conserves the number of particles, the number of atoms rather than the number of molecules is held fixed.[Citation183] The RCMC method has also been combined with the replica exchange scheme,[Citation193] MD,[Citation194] the CBMC scheme [Citation177] and the CFMC scheme.[Citation182]
MC moves
The two most common MC moves are rigid translation and rotation. The molecule is displaced or rotated by a small modification. The modification is usually scaled to achieve around 50% acceptance. In a strongly interacting fluid (e.g. water), the acceptance ratio of the rigid rotation becomes low and it might be better to do a full random rotation. The ‘reinsertion’ move removes a randomly selected molecule and reinserts it at a random position. For rigid molecules it uses orientational biasing, and for chains the molecule is fully regrown (the internal configuration is modified). The reinsertion move bypasses (free) energy barriers and is particularly useful to redistribute molecules over cages in nanoporous materials. At high densities, the acceptance ratio of the reinsertion move becomes vanishingly low. To properly sample the internal structure (i.e. bond/bend/torsions) the ‘partial reinsertion’ move is useful. Several atoms of the molecules are kept fixed, while others are regrown. Because there is already space for the atoms the acceptance ratios are high. For mixtures, especially at higher density, the ‘identity switch’ move becomes crucial. The identity-change trial move Citation152Citation195Citation196Citation197 is called semi-grand ensemble,[Citation198] but it can also be seen as a special case of the Gibbs ensemble. One of the components is selected at random, and an attempt is made to change its identity. The acceptance rule is given by [Citation195]
To sample concerted motions of atoms the hybrid MC/MD can be used. To obtain a new configuration, a short NVE MD of M time steps is run and accepted or rejected.[Citation203,Citation204] The starting velocities are chosen from a Maxwell–Boltzmann distribution at the desired temperature. The acceptance rule is
The chiral inversion move is especially aimed to improve the sampling of chiral molecules.[Citation208,Citation209] In this operation, all atoms are transposed by a mirror operation that leaves the fixed framework invariant. This operation applies, in principle, to all atoms in the system. However, it only affects the orientation and position of guest molecules as zeolite atoms are just moved to a position that was previously occupied by an atom of exactly the same type. If chiral molecules are involved, the MC move changes every enantiomer to its opposite form in one single step, which can dramatically improve the ergodicity of the sampling. For MFI, for example, equals
, with
as the dimensions of the periodic simulation box. Let ξ denote the fractional content of the species in the bulk fluid phase such that
. The acceptance rule is given by
To overcome the low insertion rates at low temperature and/or high densities several advanced MC moves were developed. Uhlherr and Theodorou [Citation210] developed a generic framework dubbed ‘MinMap Monte Carlo’. Insertions were performed over a series of m stages, with a separate minimisation after each stage. Initially, the atom was inserted as a repulsive soft sphere. Later stages slowly inflate the atom to a full Lennard-Jones interaction of the same size, complete with attractive tail. The main purpose of the quench is to relax the configuration by removing unrealistic interactions such as atom overlaps. Atom deletion moves followed the reverse procedure to deflate the selected atom prior to removal.
Many other moves and methods are published, e.g. energy–cavity biasing,[Citation211] Reptation move,[Citation212] aggregation–volume—bias,[Citation213] as well as MC methods like recoil growth,[Citation214,Citation215] reverse MC,[Citation216] phase switch MC,Citation217Citation218Citation219 kinetic MC,Citation220Citation221Citation222 rotational isomeric state MC,[Citation223] basin-hopping MC,[Citation224] tethered MC,[Citation225] smart walking,[Citation226] self-referential method,Citation227Citation228Citation229 expanded ensembles,[Citation230,Citation231] Fragment regrowth MC [Citation232] and Waste recycling MC.[Citation233,Citation234]
Parallel and hyper-parallel tempering
Parallel tempering, also known as replica exchange, is one of the methods that have been proposed to overcome the barriers facing traditional molecular simulation methods and to improve sampling of configuration space for complex systems.Citation235Citation236Citation237Citation238Citation239 Open ensembles provide an effective means for overcoming some of the problems associated with slow relaxation phenomena. Yan and de Pablo proposed a combination of expanded ensembles (also known as simple tempering) and multidimensional parallel tempering, referred to as ‘hyper-parallel tempering’ Monte Carlo (HPTMC).[Citation240] The hyper-parallel algorithm uses the three following moves:
Conventional canonical MC moves to thermalise the system
Trial shrink/growth moves are used to change the length of the tagged chain in each replica, thereby implementing the underlying expanded grand canonical formalism.
Configuration swaps are proposed between pairs of replicas i and j. To enforce a detailed balance condition, the pair of replicas to be swapped are selected at random, and the trial swap move is accepted with probability
The general idea of parallel tempering is not limited to exchanges or swaps between systems at different temperatures. For example, Van Erp et al. performed parallel tempering in the mole fraction for adsorption of chiral mixtures.[Citation209] The general distribution of an adsorbent that is in contact with a multicomponent gas is given by
Density-of-state (DOS) methods
The canonical partition function can be expressed as [Citation241]
Histogram reweighting method
The original histogram reweighting technique for MC simulations was developed by Ferrenberg and Swendsen for the canonical ensemble.[Citation247,Citation248] It has been extended to the grand canonical ensemble Citation261Citation262Citation263 and isothermal–isobaric ensemble.[Citation264]
With only a few simulations, the entire vapour–liquid coexistence region of a pure fluid can be investigated.Citation264Citation265Citation266Citation267 The histogram methods can produce highly accurate data, especially in the vicinity of critical points.[Citation266]
Histogram reweighting collects data for the probability of occurrence of N particles in the simulation cell with total configurational energy in the vicinity of E. The probability distribution has the following form
In general, it is not possible to cover all thermodynamic states of interest from a single simulation. Instead, multiple simulations are required at various chemical potentials and temperatures. The histograms that result from the simulations can be combined by minimising the differences between predicted and observed histograms.[Citation247,Citation248] Let us assume that there are multiple overlapping runs, . The composite probability
of observing N particles and energy E, assuming equal statistical efficiency, is
To combine multiple histograms, a reasonable amount of overlap must be present between neighbouring histograms. This means that when determining phase coexistence properties, there must be a connection between the liquid and vapour at the conditions of interest. In most cases, it is convenient to bridge the liquid and vapour regions with a run near the critical point. Additional histograms are added on the liquid and vapour sides as one progresses along the coexistence curve.
The methods have been generalised to mixtures.[Citation266,Citation268,Citation269] In practice, it is impractical to work with 3D histograms as the range of phase space covered in a simulation can be quite large. Instead, it is more efficient to record periodically the set of ,
, E values. The required probability distribution is extracted from this list at the end of the simulation.[Citation268]
Wang–Landau sampling
The Wang–Landau sampling method Citation4Citation249Citation250Citation270Citation271Citation272Citation273 has the objective to make all energies equally probable. During a random walk, the weights are iteratively adjusted using importance sampling. The weights that achieve a flat histogram are reciprocal to the DOS. The precision by which the weights are adjusted is iteratively increased until desired precision is reached. The algorithm proceeds as follows:
Choose a set of energy ranges and set the density of states
Start random walk in energy space. The acceptance acceptance probability is
The move is initially accepted with unity probability. Each time an energy level is visited, update the corresponding density of states by multiplying the existing value by a modification factor f>1Do the random walk until the accumulated histogram of energy is flat
Reset the histogram and reduce the modification factor to continue and converge the
Shi and Maginn found that Wang–Landau sampling is very efficient in obtaining the biasing factors for CFMC.Citation180Citation181Citation182 The λ range is for example divided into 10 bins. Initially, all biasing factors are zero. During equilibration, the bin corresponding to the current λ is modified according to after an MC move attempt, where ν is a scaling parameter initially set to 0.01. Histograms are measured, and in every 10,000 attempts the histograms are checked for flatness. The histogram is considered sufficiently flat when all bins are at least 30% as often visited as the most visited bin. If so, then histograms are set to zero and the scaling factor is modified to
. Equilibration of η can be stopped once the value of ν is lower than
.
Nested sampling
A recent method to explore the configurational phase space of systems is NS.[Citation256,Citation258,Citation259,Citation274] It is based on the original nested sampling algorithm proposed in the Bayesian statistics community by Skilling [Citation275,Citation276] and is able to explore the entire PES efficiently in an unbiased way. A unique strength of the method is that the sampling efficiency is not impeded by phase transitions, unlike the parallel tempering and Wang–Landau methods. To illustrate the differences, a hypothetical first-order phase transition is shown in Figure (left), taken from Ref. [Citation256]. Up-down denotes energy E and left–right the logarithm of available phase space . The thick lines in the parallel tempering and Wang–Landau sampling denote the region where a phase transition occurs. In this region, the energy rapidly changes over a small temperature range. This entropy jump makes equilibration and sampling difficult using these methods. NS avoids this by constructing a series of energy levels that are equispaced in
. Hence, the energy level spacings near the phase transition will be narrow. The NS method constructs such a sequence of energy levels in a single top-down pass.
![Figure 6 (Colour online) NS, (left) first-order transitions are dealt with differently by parallel tempering, Wang–Landau sampling, and NS (right). NS is a top-down approach where a set of random walkers converge downwards in energy at a rate equal to the logarithm of the phase space. Left figure taken from Ref. [Citation256].](/cms/asset/83fcc237-08e5-4cea-afee-b455dda71e0e/gmos_a_819102_f0006_oc.gif)
NS is a top-down approach (in energy) where the whole phase space is sampled using N sample points (‘random walkers’) that control the resolution. The error in scales as
. The sampling procedure is as follows.
Generate N random configurations, uniformly distributed over the space of all configurations.
Find the highest energy
Replace the removed configuration with a new one subjected to the following two criteria:
the energy should be lower than the energy
the energy should be uniformly distributed in the regions of the PES that has lower energy than
The new configuration
Check for convergence, if not, go to 2.
After n iterations of the nested sampling procedure beginning with n = 0, the removed energy is assigned a phase-space weight of
where
is the phase-space volume ratio between neighbouring
values, and the nested sampling approximation of the configurational partition function and of an observable is [Citation256]
Since the sampled phase-space volume decreases exponentially with the iteration number n, the NS algorithm is able to sample exponentially small regions of configurational space. Because of the fixed α value, even energy regions where a phase transition occurs are properly sampled. Instead of using a fixed energy spacing, like in Wang–Landau, the NS algorithm automatically has more energy sampling in the region of a phase transition.
More generally, a smaller value of α could be used, but this would entail discarding more than one configuration per NS iteration and the expression to estimate the partition function would need to be modified. The NS is also able to compute free energy differences and rates. For free energy differences, the discarded configurations {x} need to be separated into ‘states’ using an appropriate metric.[Citation256]
There is no temperature β in the sampling algorithm. The sampled weights are independent of β because is a monotonic function of E. Like all DOS methods, any observable can be evaluated at an arbitrary temperature just by re-summing over the same sample set. Of course for lower temperatures one needs more sampling of the energy basins, and for higher temperature the low-energy states contribute less to the partition function. In contrast to other DOS methods, the NS algorithm always converges with a resolution at the basins of the PES determined by the sample size N. High energies are sampled uniformly, with the low-energy samples directly obtained from the high-energy samples.
Pártay et al. demonstrate the new framework in the context of Lennard-Jones (LJ) clusters. For cluster size 2-5, N = 300, while for size 30–40, to achieve an essentially exact heat capacity versus temperature curve.[Citation256] The latter corresponds to about
energy evaluations. For LJ17, the efficiency gain of NS is a factor of 10 over parallel tempering, while for the larger LJ25 cluster, as the entropy jump is larger, the efficiency gain is a factor of 100.
Testing, debugging and verification
MC codes are notoriously difficult to debug. One deals with probabilities and energy distributions instead of generating deterministic trajectories like in MD. In MC, one can have sampling issues without knowing it. Error bars are in that respect not helping, because small error bars for short and intermediate runs usually are suspicious and indicatory of sampling problems. In this section, we describe tests that combine various methods (MC, MD and energy minimisation) and explore limiting cases (e.g. infinite dilution). It is therefore a good approach to not just have a code aimed at MC or only at MD, but have a code that combines all these methods. The advantage is that all energy evaluations and method details are consistent and can be controlled exactly. This combination of methods allows for a wide and various array of stress tests. In this section, we discuss a few examples.
Checking energies, gradients and higher derivatives
It is essential to implement the potential energy evaluation correctly. However, there is no real ‘test’ other than (1) checking the source routines carefully and (2) comparing to other codes. The first should be done regardless, but for the second option one has to make sure that all details are handled identically: e.g. cut-off radius, shifted at the cut-off or truncated or smoothed near the cut-off, tail corrections or not, charge method and Ewald precision. The different charge methods include shifted or smoothed Coulomb, Wolf's method, Ewald or Smoothed-Particle Mesh Ewald (SPME). VDW LJ size parameters are usually given in terms of σ, but sometimes the size value should be given as the potential minimum (e.g. default in TINKER). There are also force field conventions that must be handled identically which are as follows: Are 1-2 and 1-3 interactions removed from VDW and electrostatics? Is 1–4 removed or scaled or fully included or only handled when a torsion is also defined? Is the bendangle a conventional angle or handled as an in-plane/out-of-plane pair? and so on. Even codes that handle generic force fields (e.g. CHARMM, AMBER, OPLS, UFF and DREIDING) do not implement these details in exactly the same way or have different default settings. For testing (and other) purposes, it is convenient to have input/output converters for formats to and from other codes such as DLPOLY and TINKER. Table shows a typical energy analysis of a snapshot done with these codes. The energies should be very close, but slight differences can appear due to different handlings of VDW cut-off and charge–charge interactions. For this comparison, these details were handled identically, except that RASPA uses conventional Ewald and TINKER/DLPOLY uses SPME for charge–charge interactions. One can generate snapshots of molecules and compare the energies between codes. One important source of error is the precision of writing/reading floating point numbers. Especially the use of file formats with limited precision such as pdb format is discouraged for this purpose.
Table 1 Energies of a snapshot of 20 CO2 and 10 pentane molecules (TraPPE model) in a Å box computed with RASPA, TINKER and DLPOLY 4.
Once energies are checked, the analytical forces can be compared with numerical estimates using finite difference methods. For example, using a central difference method with four energy evaluations one can obtain the gradient with fourth-order accuracy
In a similar way, it is possible to test the configurational part of the stress tensor and elasticity tensor. The stress tensor in the canonical ensemble is defined as
The instantaneous stress tensor is not necessarily symmetric. For example, for rigid molecules the tensor is asymmetric. The presence of intra-molecular constraints results in a force term to be added to the strain derivative of an atomic system.
Checking boundary conditions
The implementation of boundary conditions can be tested, e.g. by constructing different unit cells of a nanoporous material. The unit cell of the Chabazite zeolite (CHA) is usually defined as a triclinic structure with Å, and
. However, it is possible to transform the unit cell into different (but also periodic) cells. For example, to transform an
,
monoclinic cell to an
,
orthorhombic cell the following transformation matrix can be used
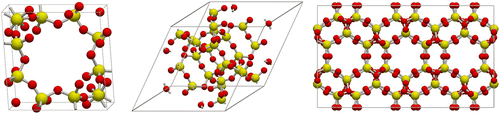
Orthorhombic cells allow for a faster implementation of the periodic boundary condition (Equation (16)), because the off-diagonal elements of the box matrix h are zero. When periodic unit cells are very large, for example MIL-101 with Å and
, the computation can be made tractable by converting the cell to a primitive cell
Å,
.[Citation277] The full cell has 14416 atoms, the primitive cell 3604 (4 times less).
Checking MD
Besides the force routines, MD needs an integration algorithm. In principle, MD is only well defined in the micro-canonical ensemble, but over the years integration algorithms have been developed for other ensembles such as the canonical, constant pressure and constant tension ensembles. These algorithms inevitable modify the trajectories, but only slightly and usually have negligible effects on dynamics properties. A good test is to first use the NVE ensemble as a reference simulation and measure the average temperature. Then an NVT simulation done exactly at that measured temperature can reveal any artefacts due to the thermostat. During an NVE trajectory, the sum of the potential and kinetic energy should be conserved, and this can be checked. But also the extended MD ensembles have a conserved quantity (when one adds the energy of the thermo- and barostats). Newton's equations of motion are time reversible and so should the integration algorithm. More generally, it is highly desirable to have a symplectic or measure-preserving integration algorithm.Citation278Citation279Citation280Citation281Citation282Citation283Citation284 Figure shows the excellent long-term energy conservation that is obtained when such integrators are used. The drift of the energy needs to be sufficiently small (usually smaller than ) for the trajectories to be considered accurate enough. To test the energy drift
of the numerical integration algorithm for a given time step
after N integration steps, one usually computes [Citation278]
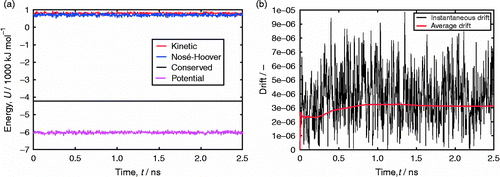
Comparing average energies
For a single molecule, MC is able to compute the average interaction energy of a molecule inside a nanoporous material with high accuracy. MD, however, is not well suited to compute single-particle properties. For example, most thermostats require many particles and the intrinsic error decays inversely with the number of used particles. The low-density limit of MD remains difficult. The best (correct) solution is to view the limit as N very large and
. However, this makes the simulations expensive, and the second best solution is to use, for example, 256 particles but remove all inter-particle interactions in the simulations (the particle only interacts with the framework; all particles feel the same thermostat). Figure shows the comparison of MC and MD computing the average energy of a particle inside a nanoporous material as a function of temperature. The MC simulations use translation/rotation, and the CBMC simulation also includes the reinsertion move. Also included in the graph are the lowest binding energies obtained from energy minimisation. A combined view of all these methods shows that CBMC converges at low temperatures to the minimum energy at 0 K (as it should). Error bars on these simulations are small and the curve over temperature is smooth (as is physically expected). Conventional MC and MD shared the same flow at low temperature, namely these methods tend to get ‘stuck’ in local energy minima. The reinsertion move bypasses these free energy bottlenecks by simply deleting the molecule and randomly placing it elsewhere. To increase the acceptance of the reinsertion move, one can use multiple-first bead and orientational biasing (as is done here using k = 10 positions for the first bead, and k = 10 random orientations, out of which one is selected with the appropriate probability, usually the most energetically favourable one).
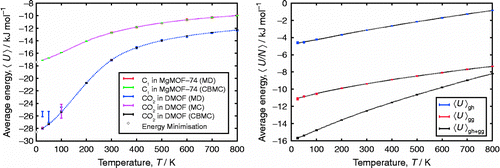
The data with error bars are listed in the Supporting Information (Tables S2 and S3) available via the article webpage. The results at higher temperatures where all used methods should be applicable for these systems show that MC with rotation and translation and MC with translation, rotation and reinsertion move and CBMC with just reinsertion give statistically identical results. There is also no influence (when simulated long enough) of the number k (number of first bead trials and trial orientations). This provides strong evidence that the reinsertion move is correctly implemented.
CBMC is very well suited to obtain the average energy of a single molecule, and this property is directly related to the enthalpy of adsorption at infinite dilution
Figure (right) shows a comparison of the average energy (per molecule) at finite loading. An excellent agreement between CBMC and MD is found (also see Table S4 of the Supporting Information available via the article webpage). Note that the average energy is not directly related to the enthalpy of adsorption, but rather has to be computed for example using the Clausius–Clapeyron equation, which requires the partial derivative of the pressure with respect to temperature at constant loading, or using a fluctuation formula
Validating NpT simulations
It is straightforward to verify NpT MC with NpT MD simulations. For consistency, in both simulation types a shifted potential is used (a cut-off of 12 Å). A starting box size of with 256 propane molecules was used. MC used 30000 initialisation cycles and 50000 cycles for production. The MD started with 10000 MC cycles, then 50 ps of velocity equilibration and 500 ps of time for the production run. The equations of motions were integrated using a measure-preserving integrator for MD in the isothermal–isobaric ensemble.[Citation283,Citation284] Figure shows that both methods give statistically identical results (see Table S5 of the Supporting Information available via the article webpage).
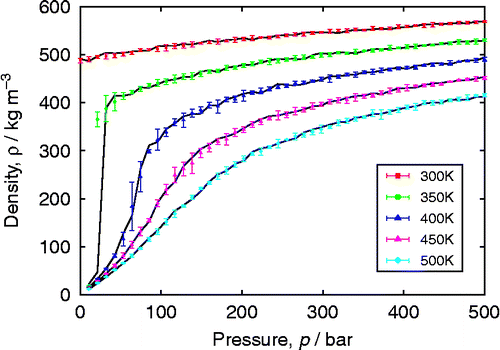
Validating CBMC growth
To validate the growth of CBMC, we examine the bond, bend and torsion distributions of S2butanol. The definition of the molecule was shown previously in Figure . The molecule has VDW and electrostatic interactions for atoms further than 1-4 apart (OPLS force field). Figure shows the comparison of CBMC and MD. Note that for this molecule the energy landscape is already quite complex. The MD result for 16 and 1024 molecules is different. This is common and due to internal energy barriers between different conformations of molecules. For example, for n-alkanes, the experimental gauche–trans energy differences range from 0.5 to 1.0 kcal/mol.[Citation286] Alkenes and cycloalkanes have rigid double bonds that prevent rotation, giving rise to cis and trans isomers. These geometric isomers cannot interconvert without breaking and re-forming bonds. Using more molecules in MD helps because it is closer to the proper distribution. (CB-)MC inherently generates the proper distribution and is therefore the most appropriate tool to investigate conformation energy barriers.[Citation286] Another point to note is how different the dihedral distribution is without VDW and electrostatics. There are multiple torsions defined over the 4–7 bond, and the coupled–decoupled algorithm used here generates the correct internal energy distributions for the internal configurations. In the Supporting Information (Figures S1–S3) available via the article webpage, we have plotted all internal bond, bend and torsional distributions for CBMC and MD.
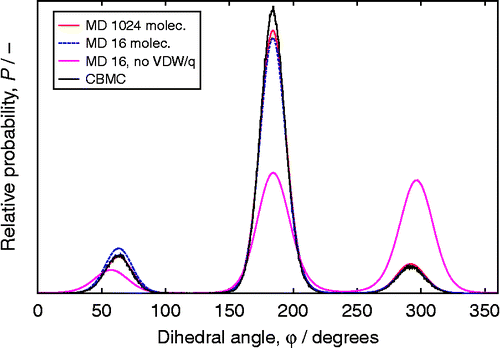
Martin and Siepmann validated their CBMC algorithms by performing some single-molecule calculations with a hit-and-miss integration program that creates individual molecule conformations from scratch by connecting n random vectors (where n is the number of bonds in that molecule).[Citation71] Properties are then calculated by simple Boltzmann averaging using the total energy (the sum of bond bending, torsional and intra-molecular Lennard-Jones interactions). Although extremely inefficient, this allows one to compare the angle and torsional distributions generated via CBMC simulations with an independent, non-biased generation of the same quantities.
Validating adsorption isotherms
It is possible to validate the adsorption results by comparing to other codes, but there are less than a handful of codes that can do CBMC. However, a very good check is to validate CBMC against CFMC. In CFMC, no configurational biasing is used, only energy evaluations. The approach differs so much that an agreement between these methods is a high indication of the correct implementation of both algorithms. Of course, one must keep in mind that CBMC is limited up to a certain density while CFMC can handle dense systems.
Table lists data for CO2 and CH4 in MFI-type zeolite. When run long enough both methods are statistical equivalent. Because of the diffusive nature of λ it can take quite some λ moves before an insertion or a deletion move is attempted. Equilibration therefore takes significantly longer at low loadings when compared to CBMC. However, for low temperatures and/or very high densities CFMC equilibrates much faster than CBMC. Figure shows several isotherms run up to high densities. At low temperatures the CFMC leads to higher values, while the CBMC results are hampered by very low insertion rates (e.g. propane in UiO-66 at 200K). Note, however, that acceptance probabilities are only an indication of performance and not a proof. If a molecule is inserted, but subsequently removed before the environment could adjust to the insertion, then the insertion effort was basically in vain. Nonetheless, it is clear that acceptance probabilities should be at least a few per mill, and insertion and deletion probabilities should be equal in equilibrium.
Table 2 Comparison of CBMC and CFMC for adsorption of CO2 and CH4 in MFI, as a function of fugacity f.
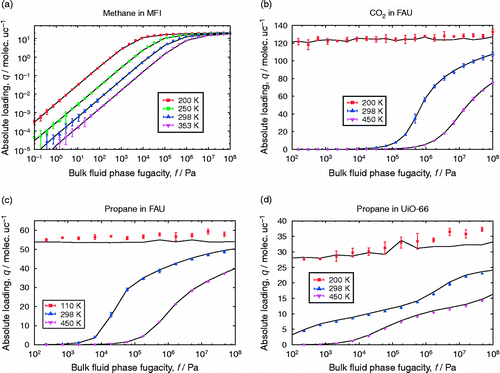
Validating Gibbs simulations
Similar to adsorption isotherms, also Gibbs CBCM simulations can be compared to CFMC. Numerical values for ethane and CO2 VLE are listed in Tables and , respectively. Again, excellent agreement is obtained. For strongly interacting fluids e.g. ionic liquids, methanol and water, the CFMC Gibbs is superior. Figure shows the VLE data for CBMC and CFMC, also compared to published simulation data of Martin and Siepmann (who developed the TraPPE model for alkanes based on reproducing experimental VLE data). The right figure shows typical behaviour of λ. Without λ biasing the histogram of λ is peaked near unity. For more strongly interacting fluids λ will ‘get stuck’. Using WL sampling, biasing factors can be obtained which lead to essentially flat λ histograms. By construction, the vapour and liquid distributions are mirror images of each other. In the limit of the λ distribution being completely flat, the difference between adjacent values of is the free energy difference between these adjacent states.[Citation181]
Table 3 Gibbs simulations of TraPPE ethane.
Table 4 Gibbs simulations of TraPPE CO2.
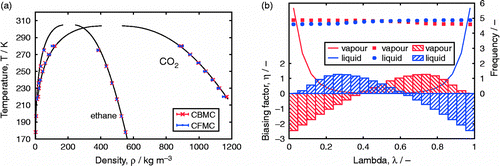
Mixture isotherms, IAST and identity change
Mixtures can also be validated by comparing CBMC to CFMC. At low and medium densities, the results with and without the identity switch move should be identical. Furthermore, it is known that Ideal Adsorption Solution Theory (IAST) works very well for homogeneous systems (i.e. no segregation). IAST predicts the mixture based on pure components,[Citation287] and can be used as a first guess where to expect the mixture results to be. MC simulation can be used to validate IAST, which then can be confidently applied to experimental isotherms (in experiments it is very difficult to measure mixture isotherms directly). Figure S4 in the Supporting Information available via the article webpage shows that IAST works qualitatively well for this system. In Table , we show that for a CO2/N2 mixture all the mixture simulations are in agreement with each other within the statistical error. The results with and without identity switch move are identical. Note that acceptance probabilities of the identity switch moves are high over the full loading range for molecules that are similar in size and shape.
Table 5 CBMC versus CFMC for an equimolar mixture of CO2/N2 in DMOF at 300 K, with and without the identity switch move.
Conclusions
We have reviewed modern MC algorithms and discussed some validation methodology. The basis of an MC code, the energy evaluations, can be compared to other codes or checked by hand for snapshots of molecules. Derivatives of energies can be easily checked numerically. To properly check more advanced MC algorithms, it is useful to implement two (or more) quite different techniques (e.g. CBMC and CFMC) that overlap in applicability for a large range of conditions. Comparison of MC and MD is feasible, but care must be taken so that the same property is measured.
SUPPLEMENTAL
Download PDF (1.1 MB)Acknowledgements
The authors thank T.J.H. Vlugt for comments on the manuscript and detailed discussions on CBMC, coupled–decoupled CBMC and CFMC and Steven Nielsen for very helpful discussions regarding nested sampling. This material is supported by the Netherlands Research Council for Chemical Sciences (NWO/CW) through a VIDI grant (David Dubbeldam) and the National Science Foundation CAREER Award 0969261 and PECASE Award ARO W911-10-0079 (Krista S. Walton).
REFERENCES
- AllenMP, TildesleyDJ. Computer simulation of liquids. Oxford: Clarendon Press; 1987.
- RapaportDC. The art of molecular dynamics simulation. 2nd ed.Cambridge: Cambridge University Press; 2004.
- FrenkelD, SmitB. Understanding molecular simulation. 2nd ed.London: Academic Press; 2002.
- TuckermanM. Statistical mechanics: theory and molecular simulations. New York: Oxford University Press; 2010.
- TheodorouDN. Progress and outlook in Monte Carlo simulations. Ind Eng Chem Res. 2010;49(7):3047–3058.
- VitalisA, PappuRV. Methods for Monte Carlo simulations of biomacromolecules. Ann Rep Comput Chem. 2009;5(9):49–76.
- HillTLJ. On steric effects. J Chem Phys. 1946;14(7):465.
- WestheimerFH, MayerJE. The theory of the racemization of optically active derivatives of diphenyl. J Chem Phys. 1946;14(12):733–738.
- WestheimerFH. Calculation of the magnitud of steric effects. In: NewmanMS, editor. Steric effects in organic chemistry. New York: John Wiley & Sons; 1956.
- WalesDJ. Energy landscapes. University Press, Cambridge; 2003.
- DinurU, HaglerAT. New approaches to empirical force fields. In: LipkowitzKB, BoydDB, editors. Reviews in computational chemistry. Vol. 2. New Jersey: Wiley Online Library; 1991. p. 99–164.
- HillJR, FreemanCM, SubramanianL. Use of force fields in materials modeling. In: LipkowitzKB, BoydDB, editors. Reviews in computational chemistry. Vol. 16. New Jersey: Wiley Online Library; 2000. p. 141–216.
- BowenJP, AllingerNL. Molecular mechanics: the art and science of parameterization. In: LipkowitzKB, BoydDB, editors. Reviews in computational chemistry. Vol. 2. New Jersey: Wiley Online Library; 1991. p. 81–97.
- TafipolskyM, SchmidR. Systematic first principles parameterization of force fields for metal-organic frameworks using a genetic algorithm approach. J Phys Chem B. 2009;113(5):1341–1352.
- JudsonR. Genetic algorithms and their use in chemistry. In: LipkowitzKB, BoydDB, editors. Reviews in computational chemistry. Vol. 10. New Jersey: Wiley Online Library; 1997. p. 1–73.
- MomanyFA. Determination of partial atomic charges from ab initio molecular electrostatic potentials. Application to formamide, methanol, and formic acid. J Phys Chem. 1978;82(5):592–601.
- CoxSR, WilliamsDE. Representation of the molecular electrostatic potential by a net atomic charge model. J Comput Chem. 1981;2(3):304–323.
- FranclMM, ChirlianLE. The pluses and minuses of mapping atomic charges to electrostatic potentials. In: LipkowitzKB, BoydDB, editors. Reviews in computational chemistry. Vol. 14. New Jersey: Wiley Online Library; 2000. p. 1–31.
- CampanaC, MussardB, WooTK. Electrostatic potential derived atomic charges for periodic systems using a modified error functional. J Chem Theory Comput. 2009;5(10):2866–2878.
- WatanabeT, ManzTA, ShollDS. Accurate treatment of electrostatics during molecular adsorption in nanoporous crystals without assigning point charges to framework atoms. J Phys Chem C. 2011;115(11):4824–4836.
- GasteigerJ, MarsiliM. Iterative partial equalization of orbital electronegativity – a rapid access to atomic charges. Tetrahedron. 1980;36:3219–3228.
- RappeAK, GoddardWA. Charge equilibration for molecular dynamics simulations. J Phys Chem. 1991;95(8):3358–3363.
- WilmerCE, SnurrRQ. Towards rapid computational screening of metal-organic frameworks for carbon dioxide capture: calculation of framework charges via charge equilibration. Chem Eng J. 2011;171(3):775–781.
- WilmerCE, KimKC, SnurrRQ. An extended charge equilibration method. J Phys Chem Lett. 2012;3(17):2506–2511.
- DubbeldamD, CaleroS, VlugtTJH, KrishnaR, MaesenTLM, BeerdsenE, SmitB. Force field parametrization through fitting on inflection points in isotherms. Phys Rev Lett. 2004;93:088302-1.
- DubbeldamD, CaleroS, VlugtTJH, KrishnaR, MaesenTLM, SmitB. United atom force field for alkanes in nanoporous materials. J Phys Chem B. 2004;108(33):12301–12313.
- CornellWD, CieplakP, BaylyCI, GouldIR, MerzKMJr, FergusonDM, SpellmeyerDC, FoxT, CaldwellJW, KollmanPA. A second generation force field for the simulation of proteins, nucleic acids and organic molecules. J Am Chem Soc. 1992;117(3):5179–5197.
- PearlmanDA, CaseDA, CaldwellJC, SeibelGL, SinghUC, WeinerP, KollmanPA. AMBER 4.0. San Francisco, CA: University of California Press; 1991.
- WeinerPK, KollmanPA. AMBER: assisted model building with energy refinement. A general program for modeling molecules and their interactions. J Comp Chem. 1981;2(3):287–303.
- WeinerSJ, KollmanPA, CaseDA, SinghUC, GhioC, AlagonaG, ProfetaSJr, WeinerPK. A new force field for molecular mechanical simulation of nucleic acids and proteins. J Am Chem Soc. 1984;106(3):765–784.
- WeinerSJ, KollmanPA, NguyenDT, CaseDA. An all atom force field for simulations of proteins and nucleic acids. J Comp Chem. 1986;7(2):230–252.
- JorgensenWL, MaduraJD, SwensonCJ. Optimized intermolecular potential functions for liquid hydrocarbons. J Am Chem Soc. 1984;106(22):6638–6646.
- JorgensenWL, SwensonCJ. Optimized intermolecular potential functions for amides and peptides structure and properties of liquid amides. J Am Chem Soc. 1985;107(3):569–578.
- JorgensenWL. Intermolecular potential functions and Monte Carlo simulations for liquid sulfur compounds. J Phys Chem. 1986;90(23):6379–6388.
- JorgensenWL. Optimized intermolecular potential functions for liquid alcohols. J Phys Chem. 1986;90(7):1276–1284.
- DammW, FronteraA, Tirado-RivesJ, JorgensenWL. OPLS all-atom force field for carbohydrates. J Comp Chem. 1997;18(16):1955–1970.
- JorgensenWL, MaxwellDS, Tirado-RivesJ. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J Am Chem Soc. 1996;118(45):11225–11236.
- JorgensenWL, Tirado-RivesJ. The OPLS potential functions for proteins. Energy minimization for crystals of cyclic peptides and crambin. J Am Chem Soc. 1988;110(6):1657–1666.
- KaminskiG, DuffyEM, MatsuiT, JorgensenWL. Free-energies of hydration and pure liquid properties of hydrocarbons from the OPLS all-atom model. J Phys Chem. 1994;98(49):13077–13082.
- MacKerellAD, BashfordD, BellottM, DunbrackRL, EvanseckJD, FieldMJ, FischerS, GaoJ, GuoH, HaS, Joseph-McCarthyD, KuchnirL, KuczeraK, LauFTK, MattosC, MichnickS, NgoT, NguyenDT, ProdhomB, ReiherWE, RouxB, SchlenkrichM, SmithJC, StoteR, StraubJ, WatanabeM, Wiorkiewicz-KuczeraJ, YinD, KarplusM. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J Phys Chem B. 1998;102(18):3586–3617.
- MomanyFA, RoneR. Validation of the general purpose QUANTA 3.2/CHARMM force field. J Comp Chem. 1992;13(7):888–900.
- PavelitesJJ, GaoJ, BashPA, MackerellADJr. A molecular mechanics force field for NAD+, NADH, and the pyrophosphate groups of nucleotides. J Comp Chem. 1997;18(2):221–239.
- HermansJ, BerendsenHJC, van GunsterenWF, PostmaJPM. A consistent empirical potential for water-protein interactions. Biopolymers. 1984;23(8):1513–1518.
- OttK-H, MeyerB. Parametrization of GROMOS force field for oligosaccharides and assessment of efficiency of molecular dynamics simulations. J Comp Chem. 1996;17(8):1068–1084.
- van GunsterenWF, DauraX, MarkAE. The GROMOS force field. ECC. 1997;17.
- Dauber-OsguthorpeP, RobertsVA, OsguthorpeDJ, WolffJ, GenestM, HaglerAT. Structure and energetics of ligand binding to proteins: E. coli dihydrofolate reductase-trimethoprim, a drug-receptor system. Prot: Struct Funct Genet. 1988;4(31–47):31–47.
- HaglerAT, EwigCS. On the use of quantum energy surfaces in the derivation of molecular force fields. Comp Phys Commun. 1994;84(1–3):131–155.
- HwangM-J, StockfischTP, HaglerAT. Derivation of class II force fields. 2. Derivation and characterization of a class II force field, CFF93, for the alkyl functional group and alkane molecules. J Am Chem Soc. 1994;116(6):195–231.
- MapleJR, HwangM-J, StockfischTP, HaglerAT. Derivation of class II force fields. 3. Characterization of a quantum force field for the alkanes. Israel J Chem. 1994;34(2):195–231.
- AllingerNL. Conformational analysis 130. MM2. A hydrocarbon force field utilizing V1 and V2 torsional terms. J Am Chem Soc. 1977;99(25):8127–8134.
- AllingerNL, KokR, ImamMR. Hydrogen bonding in MM2. J Comp Chem. 1988;9(6):591–595.
- LiiJ-H, GallionS, BenderC, WikstromH, AllingerNL, FlurchickKM, TeeterMM. Molecular mechanics (MM2) calculations on peptides and on the protein crambin using the cyber 205. J Comp Chem. 1989;10(4):503–513.
- LiiJ-H, AllingerNL. The MM3 force field for amides, polypeptides and proteins. J Comp Chem. 1991;12(2):186–199.
- LiiJ-H, AllingerNL. Directional hydrogen bonding in the MM3 force field. II. J Comp Chem. 1998;19(9):1001–1016.
- AllingerNL, ChenK, LiiJ-H. An improved force field (MM4) for saturated hydrocarbons. J Comp Chem. 1996;17(5–6):642–668.
- AllingerNL, ChenK, KatzenellenbogenJA, WilsonSR, AnsteadGM. Hyperconjugative effects on carbon-carbon bond lengths in molecular mechanics (MM4). J Comp Chem. 1996;17(5–6):747–755.
- AllingerNL, FanY. Molecular mechanics studies (MM4) of sulfides and mercaptans. J Comp Chem. 1997;18(15):1827–1847.
- NevensN, ChenK, AllingerNL. Molecular mechanics (MM4) calculations on alkenes. J Comp Chem. 1996;17(5–6):669–694.
- NevensN, LiiJ-H, AllingerNL. Molecular mechanics (MM4) calculations on conjugated hydrocarbons. J Comp Chem. 1996;17(5–6):695–729.
- NevensN, AllingerNL. Molecular mechanics (MM4) vibrational frequency calculations for alkenes an conjugated hydrocarbons. J Comp Chem. 1996;17(5–6):730–746.
- HalgrenTA. The representation of the van der Waals (vdW) interactions in molecular mechanics force fields: potential form, combination rules, and vdW parameters. J Am Chem Soc. 1992;114(20):7827–7843.
- HalgrenTA. Merck molecular force field. I. Basis, form, scope, parameterization and performance of MMFF94. J Comp Chem. 1996;17(5–6):490–519.
- HalgrenTA. Merck molecular force field. II. MMFF94 van der Waals and electrostatic parameters for intermolecular interactions. J Comp Chem. 1996;17(5–6):520–552.
- HalgrenTA. Merck molecular force field. III. Molecular geometrics and vibrational frequencies for MMFF94. J Comp Chem. 1996;17(5–6):553–586.
- HalgrenTA, NachbarRB. Merck molecular force field. IV. Conformational energies and geometries. J Comp Chem. 1996;17(5–6):587–615.
- HalgrenTA, NachbarRB. Merck molecular force field. V. Extension of MMFF94 using experimental data, additional computational data and empirical rules. J Comp Chem. 1996;17(5–6):616–641.
- MayoSL, OlafsonBD, GoddardWA. DREIDING: a generic force field for molecular simulations. J Phys Chem. 1990;94(26):8897–8909.
- SunH, RenP, FriedJR. The COMPASS force field: parameterization and validation for phosphazenes. Comput Theor Polym Sci. 1998;8(1–2):229–246.
- RappéAK, CasewitCJ, ColwellKS, GoddardWA, SkiffWMJ. UFF, a full periodic table force field for molecular mechanics and molecular dynamics simulations. J Am Chem Soc. 1992;114(25):10024–10035.
- MartinMG, SiepmannJI. Transferable potentials for phase equilibria. 1. United-atom description of n-alkanes. J Phys Chem B. 1998;102(14):2569–2577.
- MartinMG, SiepmannJI. Novel configurational-bias Monte Carlo method for branched molecules. Transferable potentials for phase equilibria. 2. United-atom description of branched alkanes. J Phys Chem B. 1999;103(21):4508–4517.
- RaiN, SiepmannJI. Transferable potentials for phase equilibria. 9. Explicit hydrogen description of benzene and five-membered and six-membered heterocyclic aromatic compounds. J Phys Chem B. 2007;111(36):10790–10799.
- LubnaN, KamathG, PotoffJJ, RaiN, SiepmannJI. Transferable potentials for phase equilibria. 8. United-atom description for thiols, sulfides, disulfides, and thiophene. J Phys Chem B. 2005;109(50):24100–24107.
- StubbsJM, PotoffJJ, SiepmannJI. Transferable potentials for phase equilibria. 6. United-atom description for ethers, glycols, ketones, and aldehydes. J Phys Chem B. 2004;108(45):17596–17605.
- WickCD, StubbsJM, RaiN, SiepmannJI. Transferable potentials for phase equilibria. 7. Primary, secondary, and tertiary amines, nitroalkanes and nitrobenzene, nitriles, amides, pyridine, and pyrimidine. J Phys Chem B. 2005;109(40):18974–18982.
- ChenB, SiepmannJI. Transferable potentials for phase equilibria. 3. Explicit-hydrogen description of normal alkanes. J Phys Chem B. 1999;103(25):5370–5379.
- ToxvaerdS. Molecular-dynamics calculation of the equation of state of alkanes. J Chem Phys. 1990;93(6):4290–4295.
- UngererP, BeauvaisC, DelhommelleJ, BoutinA, RousseauB, FuchsAH. Optimization of the anisotropic united atoms intermolecular potential for n-alkanes. J Chem Phys. 2000;112(12):5499–5510.
- van WestenT, VlugtTJH, GrossJ. Determining force field parameters using a physically based equation of state. J Phys Chem B. 2011;115(24):7872–7880.
- GrossJ, SadowskiG. Perturbed-chain SAFT: an equation of state based on a perturbation theory for chain molecules. Ind Eng Chem Res. 2001;40(4):1244–1260.
- TrachenkoK, TodorovIT, SmithW, DoveMT. DLPOLY: 3 new dimensions in molecular dynamics simulations via massive parallelism. J Mater Chem. 2006;16:1911–1918.
- Ponder JW. TINKER software tools for molecular design. http://dasher.wustl.edu/tinker.
- FieldMJ, AlbeM, BretC, Proust-De MartinF, ThomasA. The dynamo library for molecular simulations using hybrid quantum mechanical and molecular mechanical potentials. J Comput Chem. 2000;21(12):1088–1100.
- FieldMJ. A practical introduction to the simulation of molecular systems. New York: Cambridge University Press; 1999.
- GaleJD. GULP – a computer program for the symmetry adapted simulation of solids. JCS Faraday Trans. 1997;93(629)
- Accelrys. © 2001-2007 Accelrys Software Inc., Materials Studio. http://accelrys.com/products/materials-studio/polymers-and-classical-simulation-software.html.
- Accelrys. ; 2013.
- PlimptonS. Fast parallel algorithms for short-range molecular dynamics. J Comput Phys. 1995;117(1):1–19.
- van der SpoelD, BerendsenHJC, van DrunenR. GROMACS: a message-passing parallel molecular dynamics implementation. Comp Phys Commun. 1995;91:43–56.
- LindahlBHE, van der SpoelD. GROMACS 3.0: a package for molecular simulation and trajectory analysis. J Mol Mod. 2001;7:306–317.
- JorgensenWL. Encyclopedia of computational chemistry. Vol. 5. New York: P. v.R. Schleyer,Wiley; 1998.
- Yale University W.L. Jorgensen Research Group. MCPRO. http://www.cemcomco.com/BOSS_and_MCPRO_Distribution125.html.
- BucherHF, SchultzAJ, KofkeDA. An eclipse-based environment for molecular simulation. Proceedings of the 2005 OOPSLA Workshop on Eclipse Technology eXchange, eclipse 2005. New York, NY: ACM; 2005. p. 130–134.
- Martin MG. Towhee. http://towhee.sourceforge.net.
- MedeA Gibbs. http://www.materialsdesign.com/medea/medea-gibbs-thermodynamics-fluids-adsorption.
- Vlugt TJH, Smit B. The BIGMAC: a configurational bias Monte Carlo program. http://molsim.chem.uva.nl/bigmac/, 1998.
- Vlugt TJH. Adsorption and diffusion in zeolites: a computational study [PhD thesis]. University of Amsterdam, Amsterdam ; 2000.
- Cassandra. http://www3.nd.edu/ ed/research/cassandra.html.
- GuptaA, ChempathS, SanbornMJ, ClarkLA, SnurrRQ. Object-oriented programming paradigms for molecular modeling. Mol Sim. 2003;29:29–46.
- Sarkisov L, Duren T, Chempath S, Snurr RQ, 2013.
- Dubbeldam D, Calero S, Ellis DE, Snurr RQ. RASPA 1.0: molecular software package for adsorption and diffusion in flexible nanoporous materials, 2013.
- HeffelfingerGS. Parallel atomistic simulations. Comp Phys Commun. 2000;128(1–2):219–237.
- UhlherrA. Parallel Monte Carlo simulations by asynchronous domain decomposition. Comp Phys Commun. 2003;155(1):31–41.
- EsselinkK, LoyensLDJC, SmitB. Parallel Monte Carlo simulations. Phys Rev E. 1995;51(2):1560–1568.
- LoyensLDJC, SmitB, EsselinkK. Parallel Gibbs-ensemble simulations. Mol Phys. 1995;86(2):171–183.
- VlugtTJH. Efficiency of parallel CBMC simulations. Mol Simul. 1999;23(1):63–78.
- AlbertyRA. Legendre transforms in chemical thermodynamics. J Chem Thermo. 1997;29(5):501–516.
- ZemanskyMW, DittmanRH. Heat and thermodynamics. 7th ed.New York: McGraw-Hill; 1997.
- RayJ. Ensembles and computer simulation calculation of response functions. In: YipS, editor. Handbook of materials modeling, chapter 12. Netherlands: Springer; 2005. p. 729–743.
- FayPJ, RayJR. Monte-Carlo simulations in the isoenthalpic-isotension-isobaric ensemble. Phys Rev A. 1992;46(8):4645–4649.
- RayJR, WolfRJ. Monte-Carlo simulations at constant chemical-potential and pressure. J Chem Phys. 1993;98(3):2263–2267.
- BornM, von KarmanTh. Uber schwingungen in raumgittern. Physik Z. 1912;13:297–309.
- SiepmannIJ. Monte Carlo methods for simulating phase equilibria of complex fluids. In: FergusonDM, SiepmannJI, TruhlarDG, editors. Advances in chemical physics: Monte Carlo methods in chemical physics. Vol. 105. New York: John Wiley & Sons; 1999. p. 433–460.
- HillTL. Thermodynamics of small systems. New York: W.A. Benjamin, Inc.; 1963.
- SchnellSK, LiuX, SimonJM, BardowA, BedeauxD, VlugtTJH, KjelstrupS. Calculating thermodynamic properties from fluctuations at small scales. J Phys Chem B. 2011;115(37):10911–10918.
- SchnellSK, VlugtTJH, SimonJM, BedeauxB, KjelstrupS. Thermodynamics of small systems embedded in a reservoir: a detailed analysis of finite size effects. Mol Phys. 2012;110(11–12):1069–1079.
- KrishnaR, van BatenJM. Comment on comparative molecular simulation study of CO2/N2 and CH4/N2 separation in zeolites and metal-organic frameworks. Langmuir. 2010;26(4):2975–2978.
- KimJ, MartinRL, RübelO, HaranczykM, SmitB. High-throughput characterization of porous materials using graphics processing units. J Chem Theory Comput. 2012;8(5):1684–1693.
- SchnabelT, VrabecJ, HasseH. Unlike Lennard-Jones parameters for vapor-liquid equilibria. J Mol Liq. 2007;135(1–3):170–178.
- AdamsDJ, AdamsEM, HillsGJ. Computer simulation of polar liquids. Mol Phys. 1979;38(2):387–400.
- AndreaTA, SwopeWC, AndersenHC. The role of long ranged forces in determining the structure and properties of liquid water. J Chem Phys. 1983;79(9):4576–4584.
- SteinbachPJ, BrooksBR. New spherical-cutoff methods for lone-range forces in macromolecular simulation. J Comput Chem. 1994;15(7):667–683.
- MacedoniaMD, MaginnEJ. A biased grand canonical Monte Carlo method for simulating adsorption using all-atom and branched united atom models. Mol Phys. 1999;96:1375–1390.
- EwaldPP. Die berechnung optischer und elektrostatischer Gitterpotentiale. Ann Phys. 1921;369:253–287.
- de LeeuwSW, PerramJW, SmithER. Simulation of electrostatic systems in periodic boundary-conditions. 1. Lattice sums and dielectric-constants. P Roy Soc Lond A Mat. 1980;373(1752):27–56.
- AdamsDJ. On the use of the Ewald summation in computer-simulation. J Chem Phys. 1983;78(5):2585–2590.
- NymandTW, LinseP. Ewald summation and reaction field methods for potentials with atomic charges, dipoles, and polarizabilities. J Chem Phys. 2000;112(14):6152–6160.
- AguadoA, MaddenPA. Ewald summation of electrostatic multipole interactions up to the quadrupolar level. J Chem Phys. 2003;119(14):7471–7483.
- BoguszS, CheathamTE, BrooksBR. Removal of pressure and free energy artifacts in charged periodic systems via net charge corrections to the Ewald potential. J Chem Phys. 1998;108(17):7070–7084.
- VlugtTJH, Garcia-PerezE, DubbeldamD, BanS, CaleroS. Computing the heat of adsorption using molecular simulations: the effect of strong coulombic interactions. J Chem Theory Comput. 2008;4(7):1107–1118.
- WolfD, KeblinskiP, PhillpotSR, EggebrechtJ. Exact method for the simulation of coulombic systems by spherically truncated, pairwise r( − 1) summation. J Chem Phys. 1999;110(17):8254–8282.
- AgarwalBK, EisnerM. Statistical mechanics. 2nd ed.New Dehli: New Age International Ltd; 1998.
- MatsumotoM, NishimuraT. Mersenne Twister: a 623-dimensionally equidistributed uniform pseudorandom number generator. ACM Trans Model Comput Simul. 1998;8(1):3–30.
- NishimuraT. Tables of 64-bit Mersenne twisters. ACM Trans Model Comput Simul. 2000;10:348–357.
- MetropolisN, RosenbluthAW, RosenbluthMN, TellerAH, TellerE. Equations of state calculations by fast computing machines. J Chem Phys. 1953;21(6):1087–1092.
- ValleauJP, WhittingtonSG. A guide to Monte Carlo for statistical mechanics. 1. Highways. In: BerneBJ, editor. Statistical mechanics A. modern theoretical chemistry. Vol. 5. New York: Plenum Press; 1977. p. 137–168.
- McQuarrieDA. Statistical mechanics. Mill Valley, CA: University Science Books; 2000.
- FrenkelD. Monte Carlo simulations. In: CatlowCRA, ParkerSC, AllenMP, editors. Computer modelling of fluids polymers and solids, chapter 4. Netherlands: Springer; 1988. p. 83–124.
- FowlerRH. Statistical mechanics. The theory of the properties of matter in equilibrium. Cambridge: University Press; 1936.
- FowlerRH, GuggenheimEA. Statistical mechanics. 2d ed. New York: Macmillian; 1939.
- HillTR. An introduction to statistical thermodynamics. 2nd ed.New York: Dover Publications; 1968.
- WoodWW. Monte Carlo calculations for hard disks in the isothermal-isobaric ensemble. J Chem Phys. 1968;48(1):415–434.
- WoodWW. NpT ensemble Monte Carlo calculations for the hard disk fluid. J Chem Phys. 1970;52(2):729–741.
- GuggenheimEA. Grand partition functions and so-called ‘thermodynamic probability’. J Chem Phys. 1939;7(2):103–107.
- HansenJ-P, McDonaldIR. Theory of simple liquids. 3th ed.Amsterdam: Elsevier; 2006.
- AttardP. On the density of volume states in the isobaric ensemble. J Chem Phys. 1995;103(22):9884–9885.
- KoperGJM, ReissH. Length scale for the constant pressure ensemble: application to small systems and relation to Einstein fluctuation theory. J Phys Chem. 1996;100(1):422–432.
- CortiDS, Soto-CampusG. Deriving the isothermal-isobaric ensemble: the requirement of a ‘shell’ molecule and applicability to small systems. J Chem Phys. 1998;108(19):7959–7966.
- HanK-K, SonHS. On the isothermal-isobaric ensemble partition function. J Chem Phys. 2001;115(16):7793–7794.
- PanagiotopoulosAZ. Direct determination of the phase coexistence properties of fluids by Monte Carlo simulation in a new ensemble. Mol Phys. 1987;61(4):813–826.
- PanagiotopoulosAZ, QuirkeN, StapeltonNM, TildesleyDJ. Phase-equilibria by simulation in the Gibbs ensemble-alternative derivation, generalization and application to the mixture and membrane equilibria. Mol Phys. 1988;63(4):527–545.
- PanagiotopoulosAZ. Gibbs ensemble techniques. In: RullLR, BausM, RyckaertJP, editors. Observation, prediction and simulation of phase transitions in complex fluids, volume NATO ASI Series C,460. Netherlands: Kluwer Academic; 1995. p. 463–501.
- NicholsonD, ParsonageNG. Computer simulation and the statistical mechanics of adsorption. New York: Academic Press; 1988.
- NormanGE, FilinovVS. Investigations of phase transitions by a Monte Carlo method. High Temp USSR. 1969;7(7):216–222.
- AdamsDJ. Chemical potential of hard-sphere fluids by Monte Carlo methods. Mol Phys. 1974;28(5):1241–1252.
- SmithJM, Van NessHC, AbbottMM. Introduction to chemical engineering thermodynamics. 7th ed.New York: Mcgraw-Hill; 2005.
- VorosNG, TassiosDP. Vapour-liquid equilibria in nonpolar/weakly polar systems with different types of mixing rules. Fluid Phase Equilib. 1993;91(1):1–29.
- WolfRJ, LeeMW, DavisRC, FayPJ, RayJR. Pressure-composition isotherms for palladium hydride. Phys Rev B. 1993;48(17):12415–12418.
- BrennanJK, MaddenWG. Phase coexistence curves for off-lattice polymersolvent mixtures: Gibbs-ensemble simulations. Macromolecules. 2002;35(7):2827–2834.
- BanaszakBJ, FallerR, de PabloJJ. Simulation of the effects of chain architecture on the sorption of ethylene in polyethylene. J Chem Phys. 2004;120(23):11304–11315.
- ZangJ, NairS, ShollDS. Osmotic ensemble methods for predicting adsorption-induced structural transitions in nanoporous materials using molecular simulations. J Chem Phys. 2011;134(18):184103.
- RosenbluthMN, RosenbluthAW. Monte Carlo simulations of the average extension of molecular chains. J Chem Phys. 1955;23:356–359.
- HarrisJ, RiceSA. A lattice model of a supported monolayer of amphiphile molecules: Monte Carlo simulations. J Chem Phys. 1988;88(2):1298–1307.
- SiepmannJI. A method for the direct calculation of chemical-potentials for dense chain systems. Mol Phys. 1990;70(6):1145–1158.
- SiepmannJI, FrenkelD. Configurational bias Monte-Carlo – A new sampling scheme for flexible chains. Mol Phys. 1992;75(1):59–70.
- FrenkelD, MooijGCAM, SmitB. Novel scheme to study structural and thermal-properties of continuously deformable molecules. J Phys: Condens Matter. 1992;4(12):3053–3076.
- de PabloJJ, SuterM, SuterUW. Simulation of polyethylene above and below the melting point. J Chem Phys. 1992;96(3):2395–2403.
- LasoM, de PabloJJ, SuterUW. Simulation of phase-equilibria for chain molecules. J Phys: Condens Matter. 1992;97(4):2817–2819.
- SiepmannJI, McDonaldIR. Monte-Carlo simulation of mixed monolayers. Mol Phys. 1992;75(2):255–259.
- VlugtTJH, KrishnaR, SmitB. Molecular simulations of adsorption isotherms for linear and branched alkanes and their mixtures in silicalite. J Phys Chem B. 1999;103:1102–1118.
- MartinMG, ThompsonAP. Industrial property prediction using Towhee and LAMMPS. Fluid Phase Equilib. 2004;217(1):105–110.
- DeemMW, BaderJS. A Configurational Bias Monte Carlo method for linear and cyclic peptides. Mol Phys. 1996;87:1245–1260.
- ChenZ, EscobedoFA. A configurational-bias approach for the simulation of linear and cyclic molecules. J Chem Phys. 2000;113:11382–11392.
- WickCD, SiepmannJI. Self-adapting fixed-end-point configurational-bias Monte Carlo method for the regrowth of interior segments of chain molecules with strong intramolecular interactions. Macromolecules. 2000;33(19):7207–7218.
- UhlherrA. Monte Carlo conformational sampling of the internal degrees of freedom of chain molecules. Macromolecules. 2000;33(4):1351–1360.
- ShahJK, MaginnEJ. A general and efficient Monte Carlo method for sampling intramolecular degrees of freedom of branched and cyclic molecules. J Chem Phys. 2011;135(13):134121.
- JakobtorweihenS, HansenN, KeilFJ. Combining reactive and configurational-bias Monte Carlo: confinement influence on the propene mathesis reaction system in various zeolites. J Chem Phys. 2006;125:224709/1–2247099.
- VlugtTJH, MartinMG, SmitB, SiepmannJI, KrishnaR. Improving the efficiency of the configurational-bias Monte Carlo algorithm. Mol Phys. 1998;94(4):727–733.
- SmitB, KaraborniS, SiepmannJI. Computer-simulations of vapor-liquid phase-equilibria of n-alkanes. J Chem Phys. 1995;102(5):2126–2140.
- ShiW, MaginnEJ. Continuous fractional component Monte Carlo: an adaptive biasing method for open system atomistic simulations. J Chem Theory Comput. 2007;3(4):1451–1463.
- ShiW, MaginnEJ. Improvement in molecule exchange efficiency in Gibbs ensemble Monte Carlo: development and implementation of the continuous fractional component move. J Comput Chem. 2008;29(15):2520–2530.
- RoschTW, MaginnEJ. Reaction ensemble Monte Carlo simulation of complex molecular systems. J Chem Theory Comput. 2011;7(2):269–279.
- JohnsonJK. Reactive canonical monte carlo. In: JohnsonDM, SiepmannJI, TruhlarDG, editors. Advances in chemical physics: Monte Carlo methods in chemical physics. Vol. 105. New York: John Wiley & Sons; 1999. p. 461–481.
- TurnerCH, BrennanJK, LisalM, SmithWR, JohnsonJK, GubbinsKE. Simulation of chemical reaction equilibria by the reaction ensemble Monte Carlo method: a review. Mol Simul. 2008;34(2):119–146.
- SmithWR, TriskaB. The reaction ensemble method for the computer simulation of chemical and phase equilibria. i. Theory and basic examples. J Chem Phys. 1994;100:3019.
- JohnsonJK, PanagiotopoulosAZ, GubbinsKE. Reactive canonical Monte Carlo: a new simulation technique for reacting or associating fluids. Mol Phys. 1994;81(3):717–733.
- HansenN, JakobtorweihenS, KeilFJ. Reactive Monte Carlo and grand-canonical Monte Carlo simulations of the propene metathesis reaction system. J Chem Phys. 2005;122:164705.
- LísalM, BrennanJK, SmithWR. Chemical reaction equilibrium in nanoporous materials: no dimerization reaction in carbon slit nanopores. J Chem Phys. 2006;124(6):064712.
- TurnerCH, JohnsonJK, GubbinsKE. Effect of confinement on chemical reaction equilibria: the reactions 2NO(NO) and N+ 3H 2NH in carbon micropores. J Chem Phys. 1851;114(4):2001.
- TurnerCH, PikunicJ, GubbinsKE. Influence of chemical and physical surface heterogeneity on chemical reaction equilibria in carbon micropores. Mol Phys. 2001;99(24):1991–2001.
- LísalM, CosoliP, SmithWR, JainSK, GubbinsKE. Fluid phase equilibria molecular-level simulations of chemical reaction equilibrium for nitric oxide dimerization reaction in disordered nanoporous carbons. Fluid Phase Equilib. 2008;272:18–31.
- LísalM, NezbedaI, SmithWR. The reaction ensemble method for the computer simulation of chemical and phase equilibria. ii. The Br+ Cl+ BrCl system. J Chem Phys. 1999;110(17):8597.
- TurnerCH, BrennanJK, LísalM. Replica exchange for reactive Monte Carlo simulations. J Phys Chem C. 2007;111(43):15706–15715.
- BrennanJK, LśalM, GubbinsKE, RiceBM. Reaction ensemble molecular dynamics: direct simulation of the dynamic equilibrium properties of chemically reacting mixtures. Phys Rev E. 2004;70(6):061103.
- PanagiotopoulosAZ. Exact calculations of fluid-phase equilibria by Monte-Carlo simulation in a new statistical ensemble. Int J Thermophys. 1989;10(2):447–457.
- de PabloJJ, PrausnitzJM. Phase equilbria for fluid mixtures from Monte-Carlo simulation. Fluid Phase Equilib. 1989;53(0):177–189.
- MartinMG, SiepmannJI. Predicting multicomponent phase equilibria and free energies of transfer for alkanes by molecular simulation. J Am Chem Soc. 1997;119(38):8921–8924.
- KofkeDA, GlandtED. Monte Carlo simulation of multicomponent equilibria in a semigrand canonical ensemble. Mol Phys. 1988;64(6):1105–1131.
- DoddLR, BooneTD, TheodorouDN. A concerted rotation algorithm for atomistic Monte Carlo simulation of polymer melts and glasses. Mol Phys. 1993;78:961–996.
- PantPVK, TheodorouDN. Variable connectivity method for the atomistic Monte Carlo simulation of polydisperse polymer melts. Macromolecules. 1995;28(21):7224–7234.
- MavrantzasVG, BooneTD, ZervopoulouE, TheodorouDN. End-bridging Monte Carlo: a fast algorithm for atomistic simulation of condensed phases of long polymer chains. Macromolecules. 1999;32(15):5072–5096.
- TheodorouDN. Variable-connectivity Monte Carlo algorithms for the atomistic simulation of long-chain polymer systems. In: NielabaP, MareschalM, CiccottiG, editors. Bridging time scales: molecular simulations for the next decade. Berlin: Springer-Verlag; 2002. p. 69–128.
- DuaneS, KennedyAD, PendletonBJ, RowethD. Hybrid Monte Carlo. Phys Lett B. 1987;195:216–222.
- ChempathS, ClarkLA, SnurrRQ. Two general methods for grand canonical ensemble simulation of molecules with internal flexibility. J Chem Phys. 2003;118(16):7635–7643.
- FallerR, de PabloJJ. Constant pressure hybrid molecular dynamics-Monte Carlo simulations. J Chem Phys. 2002;116(1):55–59.
- ShinodaW, ShigaM, MikamiM. Rapid estimation of elastic constants by molecular dynamics simulation under constant stress. Phys Rev B. 2004;69:134103.
- AkhmatskayaE, ReichS. GSHMC: an efficient method for molecular simulation. J Comput Phys. 2008;227(10):4934–4954.
- van ErpTS, CaremansTP, DubbeldamD, Martin-CalvoA, CaleroS, MartensJA. Enantioselective adsorption in achiral zeolites. Angew Chem Int Ed. 2010;49(17):3010–3013.
- van ErpTS, DubbeldamD, CaremansTP, CaleroS, MartensJA. Effective Monte Carlo scheme for multicomponent gas adsorption and enantioselectivity in nanoporous materials. J Phys Chem Lett. 2010;1(14):2154–2158.
- UhlherrA, TheodorouDN. Accelerating molecular simulations by reversible mapping between local minima. J Chem Phys. 2006;125:084107.
- SnurrRQ, BellAT, TheodorouDN. Prediction of adsorption of aromatic-hydrocarbons in silicalite from grand canonical Monte-Carlo simulations with biased insertions. J Phys Chem. 1993;97(51):13742–13752.
- WallFT, MandelF. Macromolecular dimensions obtained by an efficient Monte Carlo method without sample attrition. J Chem Phys. 1975;63:4592–4595.
- ChenB, SiepmannJI. Improving the efficiency of the aggregation-volume-bias Monte Carlo algorithm. J Phys Chem B. 2001;105(45):11275–11282.
- ConstaS, WildingNB, FrenkelD, AlexandrowiczZ. Recoil growth: an efficient simulation method for multi-polymer systems. J Chem Phys. 1999;110(6):3220–3228.
- ConstaS, VlugtTJH, Wichers HoethJ, SmitB, FrenkelD. Recoil growth algorithm for chain molecules with continuous interactions. Mol Phys. 1999;97(12):1243–1254.
- McGreevyRL, PusztaiL. Reverse Monte Carlo simulation: a new technique for the determination of disordered structures. Mol Simul. 1988;1:359–367.
- BruceAD, WildingNB, AcklandGJ. Free energy of crystalline solids: a lattice-switch Monte Carlo method. Phys Rev Lett. 1997;79(16):3002–3005.
- WildingNB, BruceAD. Freezing by Monte Carlo phase switch. Phys Rev Lett. 2000;85:5138–5141.
- BruceAD, WildingNB. Computational strategies for mapping equilibrium phase diagrams. Adv Chem Phys. 2002;127:1–64.
- YoungWM, ElcockEW. Monte Carlo studies of vacancy migration in binary ordered alloys: I. Proc Phys Soc. 1966;89:735–746.
- VoterAF. Introduction to the kinetic Monte Carlo method. Rad Effects Solids (NATO Sci Ser). 2007;235:1–23.
- ChatterjeeA, VlachosDG. An overview of spatial microscopic and accelerated Kinetic Monte Carlo methods. J Comput Mater Design. 2007;14:253–308.
- HoneycuttJD. A general simulation method for computing conformational properties of single polymer chains. Comput Theor Polym Sci. 1998;8(1–2):1–8.
- WalesDJ, DoyeJPK. Global optimization by basin-hopping and the lowest energy structures of Lennard-Jones clusters containing up to 110 atoms. J Phys Chem. 1997;101(28):5111–5116.
- FernandezLA, Martin-MayorV, YllanesD. Tethered Monte Carlo: computing the effective potential without critical slowing down. Nucl Phys B. 2009;807:424–454.
- ZhouR, BerneBJ. Smart walking: a new method for Boltzmann sampling of protein conformations. J Chem Phys. 1997;107:9185–9196.
- SweatmanMB, QuirkeN. Simulating fluid-solid equilibrium with the Gibbs ensemble. Mol Simul. 2004;30(1):23–28.
- SweatmanMB. Self-referential Monte Carlo method for calculating the free energy of crystalline solids. Phys Rev E. 2005;72:016711.
- SweatmanMB. New techniques for simulating crystals. Mol Simul. 2009;35:897–909.
- EscobedoFA, de PabloJJ. Monte-Carlo simulation of the chemical potential of polymers in an expanded ensemble. J Chem Phys. 1995;103(7):2703–2710.
- EscobedoFA, de PabloJJ. Expanded grand canonical and Gibbs ensemble Monte Carlo simulation of polymers. J Chem Phys. 1996;105(10):4391–4394.
- ZhangJ, KouSC, LiuJS. Biopolymer structure simulation and optimization via fragment regrowth Monte Carlo. J Chem Phys. 2007;126:225101.
- FrenkelD. Speed-up of Monte Carlo simulations by sampling of rejected states. Proc Natl Acad Sci USA. 2004;101(51):17571–17575.
- FrenkelD. Waste-recyling Monte Carlo. Lecture Notes in Phys. 2006;703:127–137.
- SwendsenRH, WangJS. Replica Monte Carlo simulation of spin glasses. Phys Rev Lett. 1986;57:2607–2609.
- GeyerCJ. Proceedings of the 23rd symposium on the interface. Computing science and statistics. New York: American Statistical Association; 1991. p. 156–163.
- SugitaY, OkamotoY. Replica-exchange molecular dynamics method for protein folding. Chem Phys Lett. 1999;314(1–2):141–151.
- FallerR, YanQL, de PabloJJ. Multicanonical parallel tempering. J Chem Phys. 2002;116(13):5419–5423.
- EarlDJ, DeemMW. Parallel tempering: theory, applications, and new perspectives. Phys Chem Chem Phys. 2005;7:3910–3916.
- YanQL, de PabloJJ. Hyperparallel tempering Monte Carlo simulation of polymeric systems. J Chem Phys. 2000;113(3):1276–1282.
- PerssonRAX. Perturbation method to calculate the density of states. Phys Rev E. 2012;86:066708.
- MingL, NordholmS, SchranzHW. On the estimation of anharmonic densities of states of molecules. Chem Phys Lett. 1996;248(3–4):228–236.
- LabastieP, WhettenRL. Statistical thermodynamics of the cluster solid-liquid transition. Phys Rev Lett. 1990;65(13):1567–1570.
- ChengH-P, LiX, WhettenRL, BerryRS. Complete statistical thermodynamics of the cluster solid-liquid transition. Phys Rev A. 1992;46(2):791–800.
- PoteauR, SpiegelmannF, LabastieP. Isomerisation and phase transitions in small sodium clusters. Z Phys D: At, Mol Cluster. 1994;30(1):57–68.
- CalvoF, LabastieP. Configurational density of states from molecular dynamics simulations. Chem Phys Lett. 1995;247(4–6):395–400.
- FerrenbergAM, SwendsenRH. New Monte Carlo technique for studying phase transitions. Phys Rev Lett. 1988;61(23):2635–2638.
- FerrenbergAM, SwendsenRH. Optimized monte carlo data analysis. Phys Rev Lett. 1988;63(12):1195–1198.
- WangF, LandauDP. Determining the density of states for classical statistical models: a random walk algorithm to produce a flat histogram. Phys Rev E. 2001;64:056101.
- WangF, LandauDP. Efficient, Multiple-Range Random Walk Algorithm to Calculate the Density of States. Phys Rev Lett. 2001;86(10):2050–2053.
- BergBA, NeuhausT. Multicanonical algorithms for first order phase transitions. Phys Lett B. 1991;267(2):249–253.
- LeeJ. New Monte Carlo algorithm: entropic sampling. Phys Rev Lett. 1993;71(2):211–214.
- GeyerCJ, ThompsonEA. Annealing markov chain monte carlo with applications to ancestral inference. J Am Stat Assoc. 1995;90(431):909–920.
- WangJ-S, TayTK, SwendsenRH. Transition matrix Monte Carlo reweighting and dynamics. Phys Rev Lett. 1999;82(3):476–479.
- HeilmannF, HoffmannKH. ParQ-high-precision calculation of the density of states. Europhys Lett. 2005;70(2):155–161.
- PárayLB, BartókAP, CsányiG. Efficient sampling of atomic configurational spaces. J Phys Chem B. 2010;114(32):10502–10512.
- BrewerBJ, PártayLB, CsányiG. Diffusive nested sampling. Stat Comput. 2011;21(4):649–656.
- DoH, HirstJD, WheatleyRJ. Rapid calculation of partition functions and free energies of fluids. J Chem Phys. 2011;135(17):174105.
- DoH, HirstJD, WheatleyRJ. Calculation of partition functions and free energies of a binary mixture using the energy partitioning method: application to carbon dioxide and methane. J Phys Chem B. 2012;116(15):4535–4542.
- KofkeDA. Free energy methods in molecular simulation. Fluid Phase Equilib. 2005;228–229(0):41–48.
- WildingNB. Critical-point and coexistence-curve properties of the Lennard-Jones fluid: a finite-size scaling study. Phys Rev E. 1995;52:602–611.
- KiyoharaK, GubbinsKE, PanagiotopoulosAZ. Phase coexistence properties of polarizable Stockmayer fluids. J Chem Phys. 1997;106(8):3338.
- FenwickMK. A direct multiple histogram reweighting method for optimal computation of the density of states. J Chem Phys. 2008;129(12):125106.
- ConradPB, de PabloJJ. Phase coexistence properties of polarizable Stockmayer fluids. Fluid Phase Equilib. 1998;150–151:51–61.
- de PabloJJ, YanQ, EsobedoFA. Simulation of phase transitions in fluids. Ann Rev Phys Chem. 1999;50:377–411.
- PanagiotopoulosAZ. Monte Carlo methods for phase equilibria of fluids. J Phys-cond Mat. 2000;12(3):25–52.
- WildingNB. Computer simulation of fluid phase transitions. Am J Phys. 2001;69(11):1147–1155.
- PotoffJJ, PanagiotopoulosAZ. Critical point and phase behavior of the pure fluid and a Lennard-Jones mixture. J Chem Phys. 1998;109(24):10914–10920.
- WildingNB. Critical end point behavior in a binary mixture. Phys Rev E. 1997;55:6624–6631.
- ShellMS, DebenedettiPG, PanagiotopoulosAZ. Generalization of the Wang-Landau method for off-lattice simulations. Phys Rev E. 2002;66:056703.
- SchulzBJ, BinderK, MullerM, LandauDP. Avoiding boundary effects in Wang-Landau sampling. Phys Rev E. 2003;67:067102.
- LandauDP, TsaiS-H, ExlerM. A new approach to Monte Carlo simulations in statistical physics: Wang-Landau sampling. Am J Phys. 2004;72(10):1294–1302.
- Cunha-NettoAG, CaparicaAA, TsaiS-H, DickmanR, LandauDP. Improving Wang-Landau sampling with adaptive windows. Phys Rev E. 2008;78(5):055701.
- NielsenSO. Nested sampling in the canonical ensemble: direct calculation of the partition function from NVT trajectories. J Chem Phys, in preparation2013.
- SkillingJ. Nested sampling. Bayesian Inference and Maximum Entropy Methods in Science and Engineering: 24th International Workshop on Bayesian Inference and Maximum Entropy Methods in Science and Engineering, Garching, Germany; 2004 p. 395–405.
- SkillingJ. Nested sampling for general Bayesian computation. Bayesian Anal. 2006;1(4):833–859.
- De LangeMF, Gutierrez-SevillanoJ-J, HamadS, VlugtTJH, CaleroS, GasconJ, KapteijnF. Understanding adsorption of highly polar vapors on mesoporous MIL-100(Cr) and MIL-101(Cr): experiments and molecular simulations. J Phys Chem. 2013.
- MartynaGJ, TuckermanM, TobiasDJ, KleinML. Explicit reversible integrators for extended systems dynamics. Mol Phys. 1996;87:1117–1157.
- BondSD, LeimkuhlerBJ, LairdBB. The Nosé Poincare method for constant temperature molecular dynamics. J Comp Phys. 1999;151(1):114–134.
- MillerTF, EleftheriouM, PattnaikP, NdirangoA, NewnsD, MartynaGJ. Symplectic quaternion scheme for biophysical molecular dynamics. J Chem Phys. 2002;116(20):8649–8659.
- LeimkuhlerBJ, SweetCR. The canonical ensemble via symplectic integrators using Nosé and Nosé Poincare chains. J Chem Phys. 2004;121(1):108–116.
- LeimkuhlerBJ, SweetC. A hamiltonian formulation for recursive multiple thermostats in a common timescale. SIAM J Appl Dyn Syst. 2005;4(1):187–216.
- TuckermanME, AlejandreJ, Lopez-RendonR, JochimAL, MartynaGJ. A Liouville-operator derived. measure-preserving integrator for molecular dynamics simulations in the isothermal-isobaric ensemble. J Phys A. 2006;39:5629–5651.
- YuT-Q, AlejandreJ, Lopez-RendonR, MartynaGJ, TuckermanME. Measure-preserving integrators for molecular dynamics in the isothermal-isobaric ensemble derived from the Liouville operator. Chem Phys. 2010;370:294–305.
- KaraviasF, MyersAL. Isosteric heats of multicomponent adsorption – thermodynamics and computer-simulations. Langmuir. 1991;7(12):3118–3126.
- ThomasLL, ChristakisTJ, JorgensenWL. Conformation of alkanes in the gas phase and pure liquids. J Phys Chem B. 2006;110(42):21198–21204.
- MyersAL, PrausnitzJM. Thermodynamics of mixed-gas adsorption. AIChE J. 1965;11(1):121.