
Abstract
A new software package, RASPA, for simulating adsorption and diffusion of molecules in flexible nanoporous materials is presented. The code implements the latest state-of-the-art algorithms for molecular dynamics and Monte Carlo (MC) in various ensembles including symplectic/measure-preserving integrators, Ewald summation, configurational-bias MC, continuous fractional component MC, reactive MC and Baker's minimisation. We show example applications of RASPA in computing coexistence properties, adsorption isotherms for single and multiple components, self- and collective diffusivities, reaction systems and visualisation. The software is released under the GNU General Public License.
1. Introduction
Molecular sieves are selective, high-capacity adsorbents because of their high intracrystalline surface areas and strong interactions with adsorbates. Molecules of different size generally have different diffusion properties in a given molecular sieve, and molecules can be separated on the basis of their size and structure relative to the size and geometry of the apertures of the sieve. Much progress has been made in understanding the subtle interaction between molecules and the confinement, and much of this understanding comes from computer simulations that are able to analyse the chemistry and physics at the atomistic level.
The two main computational approaches to tackle these systems are (i) quantum mechanical calculations and (ii) force field-based simulations. The first approach is required for studying properties such as bond breakage and formation and is available in many excellent commercial and non-commercial packages. The second approach is useful for studying larger systems and for calculating a wide variety of thermodynamic and dynamic properties. Force field-based approaches include Monte Carlo (MC) simulations, molecular dynamics (MD) simulations and energy minimisations. We introduce here a code, RASPA, that focuses on MC, MD and minimisation of systems described by classical force fields.
The RASPA code was written as a collaboration among Northwestern University (USA), the University of Amsterdam (The Netherlands) and the University Pablo de Olavide (Spain), with recent contributions also from the University of Delft (The Netherlands). The code evolved initially from the post-doc project (2006–2009) of David Dubbeldam at Northwestern University, where the Snurr group had another MUltipurpose SImulation Code (MUSIC), which was written in object-oriented Fortran 90.[Citation1,Citation2] MUSIC provides functionality for performing MD and MC simulations in a number of different ensembles, minimisations and free energy calculations for bulk and adsorbed phases using a variety of force fields, but not for treating flexible adsorbent frameworks and hence RASPA was developed. Version 1.0 of this code has been used internally by the authors and a growing list of collaborators. In this paper, we present version 2.0 available for public use. Its main areas of utility are thermodynamic properties of liquids and gases, and adsorption/diffusion behaviour of adsorbates in crystalline nanoporous materials.
Examples of nanoporous materials are clays, carbon nanotubes, zeolites and metal-organic frameworks (MOFs). MOFs are a relatively new class of materials composed of metal nodes connected by organic linkers. MOFs possess almost unlimited structural variety because of the many combinations of building blocks that can be imagined. The building blocks self-assemble during synthesis into crystalline materials that, after evacuation of the structure, may find applications in adsorption separations, gas storage and catalysis.Citation3Citation4Citation5 MOFs have crystal structures that exhibit unusual flexibility. An extreme example is the ‘breathing MOF’ MIL-53 that expands or shrinks to admit guest molecules such as CO2 and water.[Citation6] For simulation of zeolites, it is common practice to keep the positions of the framework atoms fixed, but this assumption is not valid for many large-pore MOFs. New algorithms and a new code were developed to handle these systems.
RASPA is a serial code. A single point of an isotherm can be obtained within hours for a simple system and in days for more complicated systems. MC codes are ideally suited for task-farm parallelism. Here, simulations are independent and are run as batches of serial simulations that differ in temperature, pressure, etc. For example, (assuming no hysteresis), each point of an isotherm can be run independently. Memory requirements of MC codes are modest.
Programs can be written in various ways, but often it is true that the fastest codes are probably the hardest to read, while programs strictly based on readability lack efficiency. RASPA (being a ‘research’ code) chooses the middle-ground and is based on the following ideas:
Correctness and accuracy. For all techniques and algorithms available in RASPA, we have implemented the ‘best’ ones (in our opinion) available in the literature. For example, RASPA uses configurational-bias Monte-Carlo (CBMC) and continuous fractional component Monte Carlo (CFCMC); it uses the Ewald summation for electrostatics; MD is based on symplectic and measure-preserving integrators.
Functional design. Examining the source code, one can notice that there are not a large number of files. The program is split up according to its function: ‘grid.c’ contains the code to make and use an energy/force grid of a framework, ‘ewald.c’ handles all the electrostatic calculations, ‘mc_moves.c’ contains all the moves to be used in MC, ‘potentials.c’ contains all the van der Waals potentials, etc.
Input made easy. The requirements for the input files are kept as minimal as possible. Only for more advanced options are extra commands in the input file needed. Also the format of the input is straightforward. Fugacity coefficients and excess adsorption are automatically computed.
Integrated simulation environment. The code is built up of many functions and routines which can be easily combined. MD can be used in MC and vice versa. Extension and modification of the code is relatively straightforward.
This article provides an overview of the application areas of the code. For a detailed description of the algorithms themselves and the inner working of MC and MD codes, we refer to Refs.[Citation7,Citation8]
2. Units, input and conventions
A small set of internal units needs to be chosen. A convenient set, which is chosen in DLPOLY [Citation9], RASPA, and many other codes, is the following:
The unit of length l0 is chosen as the Ångstrom, i.e.
m
The unit of time
The unit of mass
The unit of charge
All other units follow from this choice. For example, one Pascal is
in internal units. A pressure input of 10 Pa in the input file is converted to ‘internal units’ by dividing by
. Similarly, at output the pressure in internal units is converted to Pa by multiplying by
.
RASPA used three generic ‘types’ or ‘groups’ for the particles: (1) ‘framework atoms’, (2) ‘adsorbates’ and (3) ‘cations’. (The classification was done with respect to porous materials, for pure fluids the meaning of ‘adsorbates’ reduces to ‘molecules’.) The advantage is that the different components of the total energy are available and the interactions can be examined (also the energies in the Ewald Fourier part are split [Citation10]). Cations are considered as part of the framework (they are included in the total mass of the framework). Another example is when using thermostats, e.g. in LTA5A, a different thermostat can operate on the framework atoms, the adsorbates and the cations (these all vibrate/move at different length- and time scales). There are no restrictions on the number of molecules or the number of components. This allows for example an adsorption simulation of a mixture of and
in LTA5A with Na and Ca ions. In this example, there are four components: two adsorbate components, CO2 and N2, and two cation components, Na and Ca. For each component the MC move types and relative attempt frequency can be specified. In this case, CO2 and N2 can undergo particle insertion and deletion moves, while Na and Ca only use translation (the cations can be simulated as mobile). A typical input for such a simulation at 298 K, 1 bar (1:3 mixture of CO2/N2) looks like:
SimulationType MonteCarlo
NumberOfCycles 250000
NumberOfInitialisationCycles 100000
PrintEvery 1000
Forcefield GenericZeolites
ModifyOxgensConnectedToAluminium yes
Framework 0
FrameworkName LTA5A
RemoveAtomNumberCodeFromLabel yes
UnitCells 1 1 1
ExternalTemperature 298.0
ExternalPressure 10000.0
Component 0 MoleculeName sodium
MoleculeDefinition Cations
TranslationProbability 1.0
RandomTranslationProbability 1.0
ExtraFrameworkMolecule yes
CreateNumberOfMolecules 32
Component 1 MoleculeName calcium
MoleculeDefinition Cations
TranslationProbability 1.0
RandomTranslationProbability 1.0
ExtraFrameworkMolecule yes
CreateNumberOfMolecules 32
Component 2 MoleculeName CO2
MoleculeDefinition TraPPE
MolFraction 0.25
BlockPockets yes
BlockPocketsFilename LTA
TranslationProbability 1.0
RotationProbability 1.0
ReinsertionProbability 1.0
SwapProbability 2.0
ExtraFrameworkMolecule no
CreateNumberOfMolecules 0
Component 3 MoleculeName N2
MoleculeDefinition TraPPE
MolFraction 0.75
BlockPockets yes
BlockPocketsFilename LTA
TranslationProbability 1.0
RotationProbability 1.0
ReinsertionProbability 1.0
SwapProbability 2.0
ExtraFrameworkMolecule no
CreateNumberOfMolecules 0
Numbering of frameworks, components, etc., is based on the C-convention, i.e. starting from zero. The ‘SimulationType’ line sets the simulation type (e.g. MC, MD, transition state theory (TST), minimisation). RASPA ‘cycles’ for MD is the number of integration steps. For MC a cycle is max(20, N)-move-attempts with N being the number of molecules. In one cycle each molecule experiences on average one MC move attempt (either accepted or rejected). More molecules require more MC moves and the use of a ‘cycle’ allows for a specification of the simulation length that is relatively insensitive to the number of molecules. Once a line like ‘Framework 0’ has been read, the lines below it refer to that framework until another line like ‘Framework 1’ is encountered (it is possible to define multiple frameworks per system). Similarly, the MC-move probabilities are set for a specific component (set with the ‘Component [number] MoleculeName [string]’ line). For each component you can specify mole fraction, fugacity coefficient, MC moves, whether the component is an adsorbate or a cation, the initial number of molecules, etc. The MC-move probabilities are appropriately normalised by the code and therefore only have to be given relative to each other. In this example, twice the number of ‘swap’-moves (i.e. insertion/deletion) will be used compared to translation, rotation and reinsertion. If no fugacity coefficients are given in the input, then the Peng–Robinson equation of state will be used to convert pressure to fugacity. Therefore, if a fugacity coefficient of unity is specified, then adsorption will be computed as a function of fugacity instead of pressure. Any specified initial number of molecules will be created at start up using the CBMC algorithm. This avoids the need to create the initial positions by hand.
In addition to this input file, force-field files need to be created. If one uses a generic force field, then a simple ‘Forcefield GenericZeolites’ is sufficient. If one creates force field files in the current directory, then these files are read instead of the generic files. This is convenient for force field fitting where one needs to change the parameters frequently. The first two force field files are ‘force_field_mixing_rules.def’ and ‘force_field.def’. The first is read to construct an initial force field based on the parameters for each atom-type and using mixing rules. The second file allows you to overwrite an interaction pair directly. Both ways of specifying a force field occur in the zeolite and MOF literature. A third file making up the force field is ‘pseudo_atoms.def’ which defines atom properties such as the name, atomic weight, charge and so on.
Next, for each component a definition must be provided. Many molecules have been already been created for general use and for these one can simply specify, e.g. ‘MoleculeDefinition TraPPE’. The atom names from this file are also those used in the files for defining the molecules. These list all the bond, bend and torsion interactions. For defining a flexible framework, the bond, bend and torsions are specified by type and an algorithm searches automatically for all occurrences of these. Details and examples are given in the manual accompanying the source code.
3. Vapour–liquid coexistence
RASPA was initially developed to simulate porous materials. However, it can also be used to model vapour–liquid equilibrium (VLE) as illustrated in this section.
3.1 Coexistence properties
The enthalpy of vapourisation (or heat of vapourisation) is the enthalpy change required to transform a given quantity of a substance from a liquid into a gas at a given pressure. The enthalpy of vapourisation is given without approximation by
3.2 NVT Gibbs ensemble for vapour–liquid equilibrium
The Gibbs ensemble MC simulation technique allows direct simulation of phase equilibria in fluids.[Citation16,Citation17] NVT (also called ‘canonical’) Gibbs ensemble simulations are performed in two separate microscopic regions, each with periodic boundary conditions. The temperature T is held constant, as a well as the total volume V and the total number of particles N. The equilibrium conditions are (i) equal temperature, (ii) equal pressure and (iii) equal chemical potential for each species in the two boxes. The equal temperature in both boxes is imposed via the MC scheme for configurational equilibration (MC moves like translation and rotation). Condition (ii) is enforced by a volume move, and condition (iii) by a particle transfer move. The volume move makes one box larger and the other box smaller, which leads to pressure equilibration. The transfer of particles between the boxes leads to equal chemical potential.
Van der Waals parameters are very difficult to obtain from experiment or from ab initio calculations. However, the VLE curves are very sensitive to, e.g. the strength parameter and size parameter
of the Lennard-Jones potentials. Martin and Siepmann developed the transferable potentials for phase equilibria (TraPPE) force field for a large variety of molecules. It includes (and historically started with) united-atom linear and branched alkanes.[Citation12,Citation13] Figure shows simulation data calibrated to experimentally available VLE data. The simulation results of RASPA agree very well with the simulation data of Martin and Siepmann. By fitting sequentially to methane (CH4), ethane (CH3), propane (CH2), isobutane (CH) and neopentane (C), the five atom types in the model can be uniquely fitted. Other data for alkanes can then be used to validate the force field. These types of simulations are also useful for re-optimising force fields for a different cut-off or change from tail-correction to a shifted potential type.[Citation18,Citation19] The chief advantage of VLE-fitted force fields is that over a large range of pressures and temperatures, the density of the fluid is accurately reproduced.
![Figure 1 (Colour online) Vapour–liquid coexistence curves for methane, ethane, propane, isobutane and neopentane computed in the Gibbs ensemble. Line, experimental data taken from the NIST database [Citation11]; closed symbols, previous simulation data of Martin and Siepmann [Citation12,Citation13] using the Towhee code [Citation14], open symbols, this work using RASPA. The order of the data from top-to-bottom is the same as the order in the legend.](/cms/asset/65a65b7f-dad7-4918-a0ef-d53ae6ed9ead/gmos_a_1010082_f0001_oc.gif)
For pure component systems, the Gibbs phase rule requires that only one intensive variable (usually the temperature) can be independently specified when two phases coexist. The density and pressure are obtained from the simulation. By contrast, for multi-component systems, pressure can be specified in advance, with the total system being considered at constant NpT. The only change necessary is that the volume changes in the two regions are now independent.
4. Adsorption
4.1 Adsorption in the NpT Gibbs ensemble
The fundamental concept in adsorption science is the adsorption isotherm. It is the equilibrium relationship between the quantity of the molecules adsorbed and the pressure or concentration in the bulk fluid phase at constant temperature.[Citation21] The Gibbs ensemble method can be used to compute adsorption isotherms in nanoporous materials.[Citation16,Citation22] One of the boxes contains the framework, while the other box contains the fluid phase (either gas or liquid) that is in equilibrium with the adsorbed phase. For adsorption of a system of n components, the Gibbs phase rule requires that n+1 intensive variables be set, if you consider the adsorbent as an additional component. These n+1 variables are conveniently taken as the temperature, the pressure of the fluid phase, and n–1 mole fractions in the fluid phase. The system is then simulated using the NpT Gibbs ensemble. The fluid-phase box is maintained at constant pressure (and temperature) by applying volume moves. For adsorption in a flexible framework, the adsorbed-phase box is also maintained at constant pressure, but volume moves in a multi component Gibbs ensemble are performed independently for each box. For the simulation of adsorption in a rigid framework, the volume moves on the adsorbed-phase box are switched off; there is no requirement for mechanical equilibrium.[Citation16] The equilibrium constraints are equal temperature in both systems and equal chemical potentials in the bulk and in the interior of the framework (similar to the VLE, the chemical potential equilibrium is enforced by particle swap moves between the boxes).
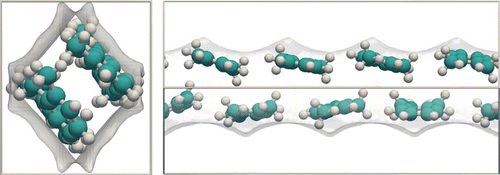
Figure (a) shows adsorption isotherms of xylenes in MIL-47 using the NpT-Gibbs ensemble. Although we use a slightly different model (all-atom OPLS/DREIDING/UFF model,[Citation23]) the results generally agree with the previous simulation data of Castillo et al. [Citation24]. Note that experiments measure ‘excess adsorption’ while simulations calculate ‘absolute adsorption’. Excess adsorption is the number of molecules in the nanopores in excess of the amount that would be present in the pore volume at the equilibrium density of the bulk gas.[Citation25,Citation26] RASPA calculates absolute adsorption and, if the pore-volume fraction is given (can be computed separately), it also calculates the excess adsorption for convenience. With this type of modelling, the experimental results are well reproduced. The snapshots from the simulation (Figure ) explain the ortho-selectivity: because ortho-xylene is commensurate with the size of the channel, it forms two layers of molecules that stack very efficiently.[Citation24,Citation23] The advantage of the Gibbs adsorption method is that the reservoir is explicitly simulated and hence the conversion from pressure to fugacity is consistently computed with the same force field. This avoids having to find and use an accurate equation of state for the adsorbates. Downsides include having to explicitly simulate the fluid phase (which can be expensive, especially in the liquid phase), and also the computed fugacity coefficient depends on the quality of the chosen force field and representation of the adsorbates.
![Figure 2 (Colour online) Pure component isotherms of o-, m- and p-xylene in MIL-47 at 423 K: closed symbols represent experimental data [Citation20], open symbols represent simulation data.](/cms/asset/a7293aa2-2203-4163-8dd1-5fe80122a14a/gmos_a_1010082_f0002_oc.gif)
4.2 Adsorption in the grand-canonical ensemble
In the limit of low pressure, fugacity and pressure are equal (i.e. the fugacity coefficient is unity). There is, therefore, no need to explicitly simulate the fluid phase. But also if an accurate equation of state is available, or if the fugacity coefficient is known experimentally, or if one is simply interested in adsorption as a function of fugacity, then the reservoir computation is not necessary. In the grand-canonical (GC) ensemble ( ensemble), the chemical potential is imposed at fixed temperature in a fixed volume (determined in this case by the crystallographic definition of the host framework). Insertion and deletion moves are used in the
ensemble to equilibrate the system at the fixed value of the chemical potential which is directly related to the fugacity
by
(where
is the reference chemical potential).
Figure (b) shows the results for the xylene-MIL-47 system using the GC ensemble. The results of the Gibbs ensemble and grand canonical Monte Carlo (GCMC) simulations agree very well. In the GCMC simulations, the pressure was converted to fugacity using the Peng–Robinson equation of state, which uses the critical temperature, critical pressure and the ‘acentric factor’ that has been tabulated for many compounds. As output it gives whether under these conditions the fluid is a gas or liquid (or metastable), as well as properties such as the fugacity coefficient, the compressibility and the density of the bulk fluid phase. The density is needed to convert absolute adsorption to excess adsorption and vice versa.
In theory and simulation, it is common to plot loading as a function of fugacity because these plots are unaffected by the gas–liquid transition. For validation of the GCMC capabilities of RASPA, we show in Figure adsorption results for benzene in MFI-type zeolite compared to the previous simulation results of Hansen [Citation27] using the BIGMAC code.[Citation28] Both simulations use fugacity and absolute loadings, and the agreement is excellent.
![Figure 4 (Colour online) Pure component adsorption isotherms of benzene in MFI at 603 and 703 K. Closed symbols, previous simulation result of Hansen [Citation27] using BIGMAC [Citation28]; open symbols, this work using RASPA.](/cms/asset/3a4d74ea-2417-477f-895c-a739b406c37e/gmos_a_1010082_f0004_oc.gif)
Simulations of mixture adsorption require the specification of chemical potentials for each component. For gas phase adsorption, an empirical equation of state can be employed. The same treatment is often not readily generalisable to liquid mixtures because of the lack of accurate activity models.[Citation29] A convenient simulation set-up for such systems is to use NpT Gibbs-ensemble simulations using three boxes: (i) the adsorbed phase with the host framework, (ii) the solution phase and (iii) a vapour phase transfer medium. The molecules are not swapped directly between the adsorbed phase and the liquid phase but instead rely on the vapour phase as an intermediate transfer medium.[Citation29]
Measuring mixed-gas adsorption experimentally is difficult. The ideal adsorption solution theory (IAST) of Myers and Prausnitz [Citation30] is often used to estimate the mixture loading from the pure component isotherms. The validity of IAST can be checked using simulations. Figure shows single component isotherms, and results for an equimolar four-component mixture of para-, meta-, ortho-xylene and ethylbenzene in MAF-x8 at 433 K.[Citation23] The IAST prediction is validated with explicit mixture simulations and for this system the IAST is applicable. Another reason to validate IAST is because it is convenient to use IAST as the input for breakthrough simulations.[Citation31]
![Figure 5 (Colour online) Xylene separation at 433 K using MAF-x8,[Citation23] (a) single components fitted with dual-site Langmuir–Freundlich isotherms, (b) equimolar mixture simulations and IAST prediction based on single component isotherms.](/cms/asset/6624ee34-a3df-46ba-b324-56e37f505300/gmos_a_1010082_f0005_oc.gif)
4.3 Adsorption in the ensemble (flexible frameworks)
The ensemble [Citation35,Citation36] is the natural ensemble to compute adsorption for flexible frameworks. The system is considered as two components, where the chemical potential of component 1 (the guest species) is kept constant (and has variable particle number), while component 2 (the framework) has constant particle number. As in GCMC, only the adsorbed phase is simulated, but now the volume moves are included to hold the pressure constant. For a single component system, it is not possible to vary three intensive variables independently because of the Gibbs–Duhem relation (from which Gibbs' phase rule follows) which relates them. However, for two (or more) species systems, it is possible to derive, rigorously, a statistical ensemble in which
,
and
and
are held fixed. For this ensemble,
is the chemical potential of the adsorbate and
is the fixed number of atoms of the framework (host). This is a hybrid statistical ensemble which has some properties similar to the single species (
) and (
) ensembles. In (
) MC simulation, one carries out (at least) three distinct types of trial procedures [Citation35,Citation36] (i) the conventional configuration change moves, (ii) the change of volume and/or size of the system and (iii) a creation or deletion move.
Figure shows simulated adsorption results of CO2 in a flexible IRMOF-1 compared to simulations using a rigid structure (the energy-minimised structure with the same force field), and also compared to experimental data. The results for the rigid and flexible model are very similar for this system and in excellent agreement with experimental data. The computation of adsorption in the flexible structure was feasible because the IRMOF-1 structure stays relatively close to its equilibrium structure. The framework motions are efficiently sampled using the MC/MD-hybrid move.[Citation37,Citation38]
![Figure 6 (Colour online) CO2 adsorption in IRMOF-1 showing step-like isotherms [Citation32]. Lines, experimental data [Citation33];, filled coloured points, rigid framework model [Citation34];, open symbols, flexible framework model.[Citation34]](/cms/asset/5f552b46-a99a-4681-95e4-5b534826681e/gmos_a_1010082_f0006_oc.gif)
4.4 Efficient algorithms for open ensembles
A system where the number of molecules varies is called an open system. All open-ensemble methods suffer from a major drawback: the insertion and deletion probabilities become vanishingly low at high densities. This problem is particularly severe for long chain molecules. For adsorption simulations, the fraction of successful insertions into the pores becomes too low. To increase the number of successfully inserted molecules, the CBMC technique was developed in the early 1990s.Citation7Citation39Citation40Citation41 Instead of generating ideal gas configurations and trying to insert the molecule as a whole, the CBMC method inserts chains part by part, biasing the growth process towards energetically favourable configurations, and therefore significantly reduces overlap with the framework and other particles.
An alternative scheme to remedy the insertion problem is the recently developed CFCMC method of Shi and Maginn Citation42Citation43Citation44. The system is expanded with an additional particle whose interactions with the other atoms in the system are scaled by a parameter , where
. Note that only the inter-molecular energy is scaled (not the intra-molecular energy). Many variations on the algorithm are possible. For example
can be changed per molecule or per atom. Both methods slowly ‘inflate’ and ‘deflate’ the molecule like a balloon but differently.
CFCMC uses conventional NVT MC for thermalisation (such as translation, rotation and/or MC–MD hybrid moves), but in addition attempts to change of the fractional molecule using
.
is chosen uniformly between
and
and scaled to achieve around 50% acceptance. However, many systems show a behaviour where
-changes are hard. An additional bias
on
can be used. This bias will be removed by the acceptance rules. A careful calibration of
can make
histograms flat and hence can avoid that the system gets stuck in a certain range of
. There are three possible outcomes of a change of
to
: (i)
remains between 0 and 1; there is no change in the number of particles, nor in the positions, nor in the intra-molecular energies. Only
and the inter-molecular energy have changed. (ii)
becomes larger than 1; when
exceeds unity,
, the current fractional molecule is made fully present (
), and a new fractional molecule is randomly inserted with
. Shi and Maginn used a methodology where a rigid conformation is chosen from a ‘reservoir’ of ideal gas molecules generated before the simulation. (iii)
becomes smaller than 0: when
falls below 0,
, the current fractional molecule is removed from the system
, and a new fractional molecule is chosen with
.
RASPA implements both CBMC and CFCMC, but also a combination (named CB/CFCMC) of the two developed by Torres-Knoop et al. [Citation45]. The basic CFCMC algorithm is used with -biasing, but the insertion and deletion moves are performed using configurational biasing. Figure (c) shows that smoother curves are obtained by using this method. The method leads to very reliable results. Other implemented methods in RASPA to improve the efficiency of MC simulations are parallel-tempering and mole fraction replica exchange.[Citation46,Citation47]
5. Screening
Continued research and investments in high-performance computing have produced computing platforms that are now fast enough to permit predictive simulations and large-scale screening studies. Simulation (virtual) screening is significantly cheaper than experimental screening and can be used to increase the successful hit rate. A hierarchical or step-wise approach is often used.
Initial screening (millions of structures). Screening on the basis of properties that can be computed very quickly. Example properties are pore volume, surface area, pore-size distribution, Henry coefficients and heats of adsorption at infinite dilution.
High throughput screening (hundreds of thousands of structures). In the pressure range of practical interest, the heat of adsorption and loadings are simulated (usually using relatively short runs). This allows a comparison of structures and an elucidation of structure–property relationships.Citation49Citation50Citation51Citation52Citation48
Detailed analysis (tens or hundreds of structures). Detailed analysis of the most promising structures could include simulations of single-component and mixture isotherms, diffusivities and efficiency estimates of the performance in a fixed-bed adsorber using breakthrough simulations.[Citation23,Citation53]
Figure shows two examples of screening. Gómez-Gualdrón et al. [Citation48] investigated physical limits for methane storage and delivery in nanoporous materials, with a focus on whether it is possible to reach a methane deliverable capacity of 315 cm3(STP)/cm3 in line with the adsorption target established by the ARPA-E agency. Using GCMC simulations, methane adsorption and delivery properties were studied in a population of 122,835 hypothetical pcu MOFs and 39 idealised carbon-based porous materials. From the simulation results, an analytical equation was obtained that delimits the necessary material properties to reach specific methane deliverable capacity targets. This high-throughput analysis elucidates how both geometric and chemical properties, such as void fraction, volumetric surface area and heat of adsorption, impact methane deliverable capacity.
![Figure 7 (Colour online) Screening results for (a) physical limits for methane storage and delivery in about hundred thousand nanoporous materials,[Citation48] (b) fixed bed performance of para-selective MOFs.[Citation23] Figure (a) courtesy of Diego A. Gómez-Gualdrón.](/cms/asset/ce96a5c6-86b4-456d-8d2c-88a13e876116/gmos_a_1010082_f0007_oc.gif)
The second example is a detailed analysis by Torres-Knoop et al. on separation of benzene, toluene, meta-, ortho-, para-xylene and ethylbenzene (BTEX process).[Citation23] Many ortho-xylene selective structures have been found, but finding para-selective structures is much harder. Small pore structures are able to separate para-xylene using ‘sieving’ (the smaller molecules fit into the structure, but the larger molecules are excluded), but these are unable to separate para-xylene from ethylbenzene (same smallest dimension) and are usually diffusion limited. Torres-Knoop et al. studied about 30 structures in full detail and elucidated why some structures are ortho-selective and others are para-selective. Using snapshots, the reason for a selectivity was explained. Snapshots in Figure show that strong ortho-selectively can be obtained by a two-layer molecular stacking in the MIL-47 structure. The ortho-xylene fits in perfectly, while meta-xylene fits less well. Para-xylene and ethylbenzene are too long and are forced to align obliquely. In their work, a para-selective structure was sought. Using the same mechanism, it is then required that the channel dimension are perfectly commensurate with the para-xylene dimensions. The screening found a strongly para-selective structure (MAF-x8). The single component isotherms were computed, the IAST prediction was validated with mixture simulations and the IAST solution was the input for simulating breakthrough curves. These breakthrough simulations have the ‘cycle-time’ as output, i.e. the time needed before needing to start the expensive desorption process. As can be seen from Figure , the MAF-x8 would be better than the currently used technology (BaX). The study also revealed that other para-xylene-selective structures, such as MIL-125 and JUC-77, would be diffusion limited (decreasing their performance).
RASPA provides perl scripts to submit jobs for screening purposes. One specifies the list of adsorbates, structures, temperatures, pressure range and number of pressure points, whether fugacity or pressure is used, whether the points are equally spaced in normal or log-scale, etc. The script then generates all the necessary input files and has as output the job scripts needed to submit it to a cluster (with a single command).
6. Reactive Monte Carlo
The RxMC method allows computation of equilibrium properties for chemically reacting or associating fluids.[Citation54,Citation55] The method samples forward and backward reactions using MC moves directly (without going through the transition states). No chemical potentials or chemical potential differences need to be specified for the reaction steps, just the stoichiometry of the reactions. Essentially, the method enhances the GCMC with a ‘forward’ and ‘backward’ reaction step, which ensures that the chemical reaction equilibria between the reactants and the products are maintained.
As an example, the industrially important propene metathesis is described by three equilibrium reactions [Citation57]
2C3H6 ↔ C2H4 + trans-C4H8
2C3H6 ↔ C2H4 + cis-C4H8
cis-C3H6 ↔ trans-C4H8
Only two reactions are independent and need to be included. In addition to the MC moves associated with simulating a chosen ensemble, also ‘reaction’ moves are performed:
randomly choose a reaction,
randomly choose whether to do a forward or backward reaction (this determines the ‘reactant’ and ‘product’ molecule types),
randomly select the reactant molecules and remove them from the system,
insert the product molecules at random positions,
accept or reject the reaction step with the appropriate acceptance probability.
Inserting molecules at high densities is difficult and even more so when one needs to insert several molecules at the same time. To overcome these difficulties, Hansen et al. [56] and Jakobtorweihen et al. . Recently, Rosch and Maginn combined the CFCMC method with the RxMC method.[Citation44] For the propene metathesis reactions simulated with reactions 2 and 3, six fractional reaction molecules are added to the system. Each reaction has an associated reaction (between 0 and 1) and MC moves are performed trying to change
to
. When
a forward reaction is performed and, if accepted,
is set to
. If
a backward reaction is performed and, if accepted,
is set to
. The insertions and deletions are biased in
which allows the method to efficiently overcome insertion and deletion difficulties.
Figure shows the results from RASPA using the CFCMC–RxMC method compared to the CBMC–RxMC simulation results of Hansen et al. [Citation56]. The RASPA results are in excellent agreement. Previously, Rosch and Maginn [Citation44] validated their implementation with the results of Hansen et al. and also found excellent agreement.
![Figure 8 (Colour online) Selectivity of the propene metathesis reaction system in the temperature range between 300 and 600 K. Closed symbols, previous simulation result of Hansen et al. [Citation56]; open symbols, this work using RASPA.](/cms/asset/7f6b7dc6-471f-4510-960c-e52c9618e7ae/gmos_a_1010082_f0008_oc.gif)
7. Diffusion
7.1 Molecular dynamics
In many applications of nanoporous materials, the rate of molecular transport inside the pores plays a key role in the overall process. The size of the pores is usually of the same order as the size of the adsorbates. Diffusion properties of guest molecules in nanoporous materials can therefore be quite sensitive to small differences between different host materials. Molecular-level modelling has become a useful tool for gaining a better understanding of diffusion in nanoporous materials. Many different diffusion coefficients can be defined for guest molecules in nanoporous materials, but it is useful to put them into two general classes: transport diffusivities and self-diffusivities. The transport (or Fickian) diffusivity describes the transport of mass and the decay of density fluctuations in the system, while self-diffusion describes the diffusive motion of a single particle. By omitting the thermodynamic contribution in the transport diffusion, the so-called ‘corrected diffusivity’ (also called ‘collective diffusivity’) is obtained. The self-, corrected and transport diffusivities are equal only in the limit of zero loading.
In an equilibrium molecular dynamics simulation, the self-diffusion coefficient of component
is computed by taking the slope of the mean-squared displacement (MSD) at long times
For a single adsorbed component, the transport diffusion coefficient is given by
Note that here first the distances are summed and then squared. Hence collective diffusion can be considered a ‘centre-of-mass’ diffusion. The thermodynamic factor is
If the framework is kept fixed, the potential energy surface induced by the framework can be pre-computed.[Citation64,Citation65] Instead of looping over all framework atoms in order to compute the host-adsorbate energy at each time step, one can construct a 3D grid before the simulation and then obtain the energy by interpolation during the simulation. The more points in the grid the higher the accuracy. RASPA implements the triclinic grid interpolation scheme in three dimensions of Lekien and Marsden [Citation45,Citation66]. The algorithm is based on a specific matrix that provides the relationship between the derivatives at the corners of the elements and the coefficients of the tricubic interpolant for this element. The cubic interpolant and its first three derivatives are continuous and consistent. The same grids can, therefore, be used for both MC and MD with no additional energy drift besides the drift due to the integration scheme, i.e. the energy gradients are the exact derivatives of the energy at each point in the element.
RASPA uses symplectic or measure-preserving and reversible integrators. Symplectic integrators tend to preserve qualitative properties of phase space trajectories: trajectories do not cross, and although energy is not exactly conserved, energy fluctuations are bounded. Since symplectic integrators preserve the topological structure of trajectories in phase space, they are more reliable for long-term integration than non-symplectic integrators. The implemented NVE integrator is the symplectic and time reversible integrator for molecules with an arbitrary level of rigidity, developed by Miller et al. [Citation67] based on a novel quaternion scheme. Thermo- and barostats can be combined with the Miller scheme to control the temperature and pressure.[Citation68,Citation70,Citation71] Figure shows that this type of scheme has excellent energy conservation over many nanoseconds. The CO2 is modelled as rigid and integrated using the quaternion integration scheme of Miller et al. [Citation67]. Separate thermostats are used to thermostat the translation and the rotation of the molecules. The framework–molecule interactions are computed using a grid interpolation scheme.[Citation45,Citation66] The grid spacing was 0.1 Å. A separate grid is used for each van der Waals interaction of the O and the C of the adsorbate molecule, and another grid is used for the real part of the Ewald summation. In the Fourier part of the Ewald summation, the contribution of the rigid framework atoms is pre-computed at the start of the simulation.[Citation10]
![Figure 9 (Colour online) Molecular dynamics of CO2 at 20 molecules per IRMOF-1 unit cell (2 × 2 × 2 system) at room temperature using the quaternion integration scheme of Miller et al. [Citation67]: (a) the individual contributions to the conserved quantity, (b) the instantaneous and average energy drift. The temperature is maintained by using a Nose´–Hoover chain.[Citation68] The molecule–framework interactions are computed using a grid-interpolation scheme [Citation45,Citation66].](/cms/asset/b43009f6-4c00-4c17-b382-43f48eaf7710/gmos_a_1010082_f0009_oc.gif)
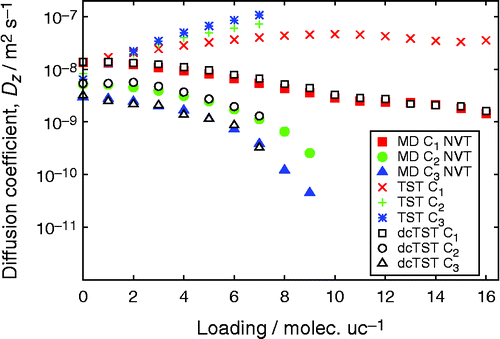
Figure shows the self- and collective diffusivities of small gases (H2, N2, Ar, CH4 and CO2) in IRMOF-1 at 298 K as a function of loading using the force field of Skoulidas and Sholl [Citation69]. The results of RASPA agree quantitatively with previous simulation results of Skoulidas and Sholl. Self-diffusivities can be computed very accurately because it is a single particle property (which can be averaged over all particles). The collective diffusivity is much more difficult to compute because it is a system property. The self-diffusivities are more strongly influenced by correlation effects (kinetic and vacancy correlations) than the collective diffusivities. Correlations between the particles increase with loading.
![Figure 10 (Colour online) Simulated diffusivities of small gases in IRMOF-1 at 298 K. Closed symbols, previous simulation result of Skoulidas and Sholl [Citation69]; open symbols, this work using RASPA.](/cms/asset/3d8562c0-022b-4e78-8103-fe8be85ea35e/gmos_a_1010082_f0010_oc.gif)
7.2 Dynamically corrected transition state theory
For some systems, the molecules move too slowly and the diffusion coefficients cannot be calculated reliably using MD. An alternative approach is to use transition state theory (TST). In the TST approximation, one computes a rate constant for hopping between states A and B by computing the equilibrium particle flux through the dividing surface. The dividing surface should partition the system into two well-defined states along a reaction coordinate, which describes the progress of the diffusion event from state to state
. In many nanoporous materials, the reaction coordinate follows directly from the geometry of the confinement. For example, in Figure the reaction coordinate for methane in LTL (Linde Type L)-type zeolite is shown: the projection of the position on the channel-axis. The location of the dividing surface is denoted by
. In ‘dynamically corrected’ TST, one computes the hopping rate over the barrier in two steps [Citation72,Citation73]:
the relative probability
Figure 11 (Colour online) A typical snapshot of a tagged methane particle (green) in LTL-type zeolite restrained to the barrier 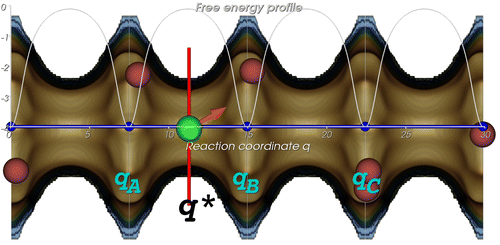
the average velocity at the top of the barrier is computed as
The transmission rate from cage
to cage
is then given by
The dcTST sampling is an example where it is convenient to be able to constrain MC moves. RASPA includes ways to constrain the movement of a component to a line, plane or sub-volume (box, cylinder, etc.). Any attempt to move a particle outside the sub-volume is rejected. Another useful feature is ‘blocking’ volumes that are large enough to contain a molecule, but where that volume is not accessible from the main channel. Yet another common example is a channel system where one would like to always have equal particles for each channel. This can be achieved by creating a different component for each channel, and only allow MC moves to a cylinder that encompasses that channel.
8. Material properties
8.1 Surface area, void fraction and pore-size distribution
Surface area is the most basic property of porous materials. Along with pore volume, surface area has become the main benchmark characterisation method for any porous material. The surface area is usually determined for experimental samples by measuring a nitrogen isotherm at 77 K and then applying the Brunauer–Emmett–Teller (BET) model. Walton and Snurr [Citation75] examined the consistency of the surface areas obtained from the BET model with those calculated geometrically from the crystal structure for several prototypical MOFs with varying pore sizes. Geometric surface areas can be calculated by using a simple MC integration technique in which a nitrogen probe (3.681 Å) molecule is rolled along the surface of the framework.[Citation26,Citation76,Citation77] Walton and Snurr provided compelling evidence for the importance of calculating the BET surface area from the proper region of the adsorption isotherm.[Citation75] Commercial ‘BET’ instruments are typically set to automatically choose a fixed range for BET fitting. The operator must ensure that the range results in consistent BET model parameters.
The pore-size distribution (PSD) can be calculated geometrically in RASPA using the method of Gelb and Gubbins [Citation77,Citation78]. For every point in the void volume, the largest sphere is found that encloses the point but does not overlap with any framework atoms. This yields the cumulative pore volume curve. Let be the volume of the void space ‘coverable’ by spheres of radius r or smaller; a point x is in
only if we can construct a sphere of radius
that overlaps
and does not overlap any substrate atoms. This volume is equivalent to that enclosed by the pore's ‘Connolly surface’.
is a monotonically decreasing function of
and is easily compared with the ‘cumulative pore volume’ curves often calculated in isotherm-based PSD methods. The derivative
is the fraction of volume coverable by spheres of radius
but not by spheres of radius
and is a direct definition of the pore size distribution. The
function can be calculated by a MC volume integration in RASPA.
Knowledge of the density of porous materials is critical for full characterisation of the adsorbents and for fixed-bed adsorber design studies. Talu and Myers [Citation25] proposed a simulation methodology that mimics the experimental procedure. For consistency with experiment, the helium void fraction is determined by probing the framework with a helium molecule using the Widom particle insertion method:
Widom insertion uses a probe particle that is inserted at random positions to measure the energy required for or obtained by insertion of the particle in the system. Usually a reference temperature of 25°C (298 K) is chosen for the determination of the helium void volume. Computationally, one can also use the limit of the pore size distribution to evaluate the void fraction. The helium void fraction is needed to convert absolute loadings to excess values (or vice versa).
8.2 Thermal and mechanical properties
Structural flexibility is a well-known property of MOFs.[Citation79] For example, MIL-53 exhibits breathing [Citation6] and IRMOFs exhibit negative thermal expansion.[Citation34] RASPA allows a wide variety of flexible models for the framework. Flexible models are needed to obtain properties such as thermal expansion of the framework itself. Thermal and mechanical transport properties are calculated either from equilibrium Green–Kubo relations or by setting up a small non-equilibrium flux across the system. Thermal expansion can be calculated from MD simulations using a flexible framework.
Figure plots the unit cell size of IRMOF-1 as a function of temperature.[Citation34] The structure become smaller with increasing temperature, in contrast to most materials which expand when you heat them. The classical models, quantum mechanical results and experimental data are qualitatively consistent. The quantum results at zero Kelvin depend very much on the used level of theory and basis sets. The negative thermal expansion of IRMOFs is a direct result of the inherent structure of MOFs: linker molecules connected via metal corners. The wiggling of the linkers becomes larger with increasing temperature leading to a reduced ‘projected’ length. Viewed oppositely, when the temperature is lowered the linkers ‘stretch out’. Therefore, negative thermal expansion is very likely a generic property of many MOFs.
![Figure 13 (Colour online) Unit cell size of IRMOF-1 as a function of temperature; experimental data, quantum simulations and three classical models. For further details see text and Ref. [Citation34].](/cms/asset/195cf08b-e33e-4aa5-a69e-1b46cd8cf646/gmos_a_1010082_f0013_oc.gif)
9. Zero Kelvin modelling
9.1 Unit cells
To study the shape and size of unit cells, one needs a model for the framework itself. Core-shell models are very suitable for minerals, metal-oxides and zeolites. The core-shell model introduces charged, massless shells around each core. For minimisation of core-shell models, the generalised Hessian matrix contains both the cores and the shells because the shells need to be minimised with respect to the cores, too. In the shell model, the short-range repulsion and van der Waals interactions are taken to act between the shell particles.
In Table we compare our results to those obtained with GULP [Citation80,Citation81] for the minimisation of various zeolites and minerals using Baker's mode-following technique.[Citation83] Using mode-following minimisation, the gradients on the cell and particles can be lowered arbitrarily close to zero and a true minimum energy is obtained (i.e. all positive eigenvalues of the Hessian matrix). The results of the RASPA and GULP codes are identical. The structures are minimised in space group P1 (no symmetry) with a cut-off of 12 Å using the mode-following technique with full unit cell fluctuation, i.e. all cell lengths and angles are allowed to change. The GULP simulations are computed using GULP 3.1 and experimental data are taken from Schröder and Sauer [Citation82]. The RASPA simulations were fully converged to forces smaller than 10 − 8 K/Å and 10 − 8 K/strain (1 degree Kelvin is eV).
Table 1 Comparison of RASPA vs GULP for two core-shell models: the model of Catlow and Gale as provided in GULP [Citation80,Citation81] and Schröder and Sauer [Citation82].
9.2 Elastic constants
Elastic constants express the degree to which a material possesses elasticity and mechanical stability (Born criteria). The elasticity tensor is the second derivative of the energy with respect to strain
, and can be described in terms of fluctuations in the stress tensor
[Citation84]
At zero Kelvin, the elastic constants reduce to the Born term minus a ‘relaxation term’ [Citation85]
Note that the derivative needs to be evaluated at constant zero gradient , which is an algebraic relation between the coordinates at zero temperature. That is, the state before and after a strain is applied must be in a state of zero net force. When more than one particle is present in the system, this requires a ‘relaxation’ of the atoms relative to one another when the system is strained.[Citation85] The zero temperature limit of the stress fluctuation term in Equation (8) is the relaxation term (and the ideal gas term vanishes in this limit). All expressions in Equation (10) are contained in the generalised Hessian matrix, which is the central quantity used in Baker's minimisation scheme. The elastic constants at 0 K can therefore be computed with very high accuracy. In Table we show the elastic constants at 0 K computed from RASPA and GULP for several zeolites and other silicates. The results are identical. Note that RASPA uses the upper triangular matrix for the simulation cell with the
direction of the lattice always aligned with the
-axis. This means that during the minimisation, the cell does not change orientation. This is convenient when computing elastic constants (which are directional) because the elastic constants are computed along the Cartesian axes (so the crystal should be aligned with these axes).
Table 2 Comparison of elastic constants at zero Kelvin in units of GPa as computed by GULP and RASPA.
9.3 Approach angles
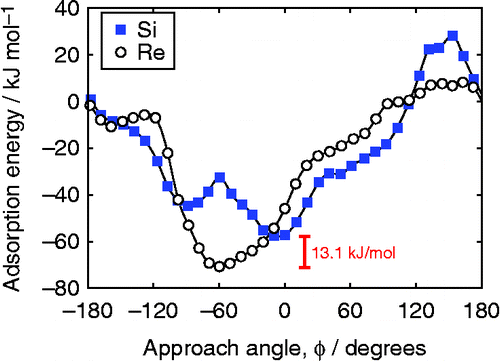
In catalysis, insight into chirality transfer from catalyst to reactant can be gained by performing constrained minimisations of the reactant–catalyst complex. Because the mechanism of asymmetric induction for epoxidation of olefins by (salen)Mn catalysts is thought to involve steric interactions between the olefin and the catalyst, the direction of olefin approach has been studied by Oxford et al. [Citation86] using hybrid MC simulations combined with classical optimisations. Four main directions of approach to the Mn-oxo moiety have been proposed in the literature (Figure (a)); see Ref. [Citation86] and references therein. To examine the approach of 2,2-dimethyl-2H-chromene to the active site of (salen)Mn = O, the potential energy surface for rotation around the dihedral angle of approach was mapped using constrained classical optimisations. In these optimisations using RASPA, the distance between the oxo ligand and C1 atom was constrained to 2.0 Å, and the angle defined by the manganese atom, oxoligand and C1 atom was constrained to 122 (see Figure (b)) because this geometry is similar to that expected in the transition state.[Citation87] Hard constraints were employed, using the r 2-SHAKE, cos2-SHAKE and the
-SHAKE algorithms [Citation88] for the bond, bend and torsion angle constraints, respectively. The minimisation method used guaranteed that the minimum found was a true minimum (all eigenvalues of the Hessian matrix are positive).
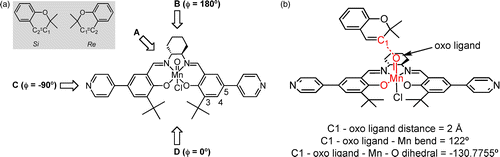
Figure shows adsorption energies of 2,2-dimethyl-2H-chromene on the homogeneous catalyst as a function of the approach angle. The approach angle was the dihedral angle defined by the midpoint of the oxygen atoms in the salen catalyst, the manganese atom, the oxo ligand and the C
atom of the reactant that would be forming a bond with the oxo ligand in the first transition state to the radical intermediate. The C
atom of chromene was chosen assuming that the phenyl group would stabilise the radical on the C
atom. The Si and Re enantiofaces of chromene are defined by the chirality of the C
atom in the reactant–catalyst complex. The reactant–catalyst complex was optimised at 10
intervals in
. The simulations predict the Re enantioface to be the preferred enantioface, in agreement with experiments. The Re enantioface favours the approach from
(approximately equivalent to the approach from direction C in Figure ), while the Si enantioface prefers to approach from direction D, with a minimum in energy at
.
10. Visualisation
It is difficult to determine the connectivity, shape and size of a channel/cage system just by examining the atomic positions of the framework. Early simulation work therefore used visualisations of energy contour plots and 3D density distributions, e.g. for benzene in silicalite,[Citation90] to obtain siting information. The visualization toolkit (VTK) is an open-source, freely available software system for 3D computer graphics, image processing and visualisation.[Citation91] RASPA has a stand-alone utility, written in C++, which visualises output files written by RASPA using VTK. RASPA produces 3D VTK files for visualising channel structures and 3D VTK files of the histograms of molecule positions during adsorption. For mixtures, a 3D histogram is produced for each component. This allows one to check and study ‘segregation’ of molecules.[Citation92] Figures and have been made using RASPA and VTK.
Figure (a) shows the MFI-type zeolite. The orthorhombic unit cell has edge lengths Å,
Å and
Å. The visualisation shows two straight channels, two ‘zig–zag’ channels and four intersections per unit cell. The depicted surface is how a methane molecule would feel the adsorption surface. To visualise molecules inside the structure, the pore walls can be rendered transparent. RASPA generates 3D energy landscapes using the free energy obtained from the Widom insertion method. The simulation cell is divided up into, e.g. 150 × 150 × 150 voxels. The adsorbate is randomly inserted millions of times and the voxels corresponding to the atom positions of the adsorbate are updated with the current Boltzmann weight. The resulting data-set has regions with value
, which correspond to overlap with the structure. The ratio of the non-zero values to the total number of voxels is the void fraction. Multiplying by the volume of the unit cell, we can compute the pore volume.
![Figure 16 (Colour online) Energy landscape of MFI. The MFI unit cell has edge lengths a = 20.022 Å, b = 19.899 Å and c = 13.383 Å, with cell angles . The MFI pore system (a) consists of straight channels running in the c-direction, which are connected via ‘zig–zag’ channels. About 29% of the structure is void. Colour code: oxygen (red), silicon (yellow). The snapshot (b) and density plot (c) are at 433 K and 100 kPa. Pictures adapted from Ref. [Citation89].](/cms/asset/338fa336-7337-4aec-a51e-0fde266a310f/gmos_a_1010082_f0016_oc.gif)
Figure (b) shows a snapshot of 2,3-dimethylbutane in MFI. To see the molecules themselves, the framework has to be either cut open or rendered transparent. The combination of the snapshot and the transparent framework allows for an analysis of adsorption sites, molecular positions and orientations, and molecule–molecule correlations. Snapshots are useful to detect differences in adsorption sites of the various species. For example, in this system the linear alkanes predominantly adsorb in the channels while the dibranched molecules adsorb first in the intersections.
Snapshots are very useful, but sometimes one needs to examine a large number of them to start to see a pattern. By keeping track of the atomic positions using 3D histograms the ‘density’ can be visualised. Figure (c) is the average of many snapshots. Therefore, the density is very convenient to obtain the siting information at the unit cell level. The picture is made using atomic positions (you could also use the centre of mass position) and, therefore, gives information on the average configuration (position and orientation). Using this type of approach we previously showed that average positions and occupations of the adsorption sites of argon and nitrogen in IRMOF-1 match well with experiments.[Citation93]
11. Conclusions
We have provided an overview of the algorithms that RASPA implements and showed examples of its application in computing coexistence properties, adsorption isotherms for single and multiple components, self- and collective diffusivities and reaction systems. RASPA is provided as source code under the GPL. The login information for the ‘git’-server can be obtained by emailing one of the authors of this manuscript. RASPA is provided without any kind of support or warranty. It should be viewed as an educational ‘research-code’ that could be useful for researchers working in the field.
Acknowledgements
The partition function values for the RxMC propene metathesis were computed by Sayee Prasaad Balaji; Diego A. Gómez-Gualdrón provided Figure ; Ariana Torres Knoop provided Figure ; R. Krishna provided the IAST computation in Figure . We would like to thank the following people for their help and input to improve the program and for the very helpful discussions about the algorithms: Sayee Prasaad Balaji, Youn-Sang Bae, Xiaoying Bao, Rocío Bueno Pérez, Nicholas C. Burtch, Tom Caremans, Ana Martín Calvo, Yamil Colon, Juan Manuel Castillo Sanchez, Allison Dickey, Tina Düren, Titus van Erp, Denise Ford, Houston Frost, Rachel Getman, Pritha Ghosh, Elena García Pérez, Gloria Oxford, Sudeep Punnathanam, Almudena Garcia Sanchez, Juan Jose Gutierrez Sevillano, John J. Low, Patrick Merkling, Patrick Ryan, Lev Sarkisov, Ben Sikora, Ariana Torres Knoop, Krista S. Walton, Chris Wilmer, Ozgur Yazaydin and Decai Yu. Very special thanks to Thijs Vlugt.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Notes
References
- Gupta A, Chempath S, Sanborn MJ, Clark LA, Snurr RQ. Object-oriented programming paradigms for molecular modeling. Mol. Simulat.. 2003;29:29–46. doi:10.1080/0892702031000065719.
- Chempath S, Düren T, Sarkisov L, Snurr RQ. Experiences with the publicly available multipurpose simulation code, Music. Mol. Simulat. 2013;39:1223–1232. doi:10.1080/08927022.2013.819103.
- Snurr RQ, Hupp JT, Nguyen ST. Prospects for nanoporous metal-organic materials in advanced separations processes. AIChE J. 2004;50:1090–1095. doi:10.1002/aic.10101.
- Long JR, Yaghi OM. The pervasive chemistry of metal-organic frameworks. Chem. Soc. Rev. 2009;38:1213–1214. doi:10.1039/b903811f.
- Li JR, Ma YG, McCarthy MC, Sculley J, Yu JM, Jeong HK, Balbuena PB, Zhou C. Carbon dioxide capture-related gas adsorption and separation in metal-organic frameworks. Coordin. Chem. Rev. 2011;255:1791–1823.
- Serre C, Millange F, Thouvenot C, Noguès M, Marsolier G, Louër D, Férey G. Very large breathing effect in the first nanoporous chromium(iii)-based solids: Mil-53 or Cr-III(OH) {O2C-C6H4-CO2} {HO2C-C6H4-CO2H}x H2Oy. J. Am. Chem. Soc. 2002;124:13519–13526. doi:10.1021/ja0276974.
- Dubbeldam D, Torres-Knoop A, Walton KS. On the inner workings of Monte Carlo codes. Mol. Simulat. 2013;39:1253–1292.
- Dubbeldam D, Snurr RQ. Recent developments in the molecular modeling of diffusion in nanoporous materials. Mol. Simulat. 2007;33:305–325. doi:10.1080/08927020601156418.
- Todorov IT, Smith W, Trachenko K, Dove MT. Dl_poly_3: new dimensions in molecular dynamics simulations via massive parallelism. J. Mater. Chem. 2006;16:1911–1918. doi:10.1039/b517931a.
- Vlugt TJH, Garcia-Perez E, Dubbeldam D, Ban S, Calero S. Computing the heat of adsorption using molecular simulations: The effect of strong coulombic interactions. J. Chem. Theory. Comput. 2008;4:1107–1118. doi:10.1021/ct700342k.
- Linstrom PJ, Mallard WG, editors. NIST Chemistry WebBook, NIST Standard Reference Database Number 69. Gaithersburg MD: National Institute of Standards and Technology; 2014.
- Martin MG, Siepmann JI. Transferable potentials for phase equilibria. 1. United-atom description of n -alkanes. J. Phys. Chem. B. 1998;102:2569–2577. doi:10.1021/jp972543+.
- Martin MG, Siepmann JI. Novel configurational-bias Monte Carlo method for branched molecules. transferable potentials for phase equilibria. 2. united-atom description of branched alkanes. J. Phys. Chem. B. 1999;103:4508–4517. doi:10.1021/jp984742e.
- Martin MG. MCCCS Towhee: a tool for Monte Carlo molecular simulation. Mol. Simulat. 2013;39:1212–1222. doi:10.1080/08927022.2013.828208.
- Martin MG, Biddy MJ. Monte Carlo molecular simulation predictions for the heat of vaporization of acetone and butyramide. Fluid Phase Equilibria. 2005;236:53–57. doi:10.1016/j.fluid.2005.06.003.
- Panagiotopoulos AZ. Adsorption and capillary condensation of fluids in cylindrical pores by Monte Carlo simulation in the Gibbs ensemble. Mol. Phys. 1987;62:701–719. doi:10.1080/00268978700102501.
- Panagiotopoulos AZ, Quirke N, Stapleton NM, Tildesley DJ. Phase equilibria by simulation in the Gibbs ensemble - alternative derivation, generalization and application to mixture and membrane equilibria. Mol. Phys. 1988;63:527–545. doi:10.1080/00268978800100361.
- Garcia-Sanchez A, Ania CO, Parra JB, Dubbeldam D, Vlugt TJH, Krishna R, Calero S. Transferable force field for carbon dioxide adsorption in zeolites. J. Phys. Chem. C. 2009;113:8814–8820.
- Gutiérrez-Sevillano JJ, Martín-Calvo A, Dubbeldam D, Calero S, Hamad S. Adsorption of hydrogen sulphide on metal-organic frameworks. RSC Adv. 2013;3:14737–14749. doi:10.1039/c3ra41682h.
- Finsy V, Verelst H, Alaerts L, de Vos DE, Jacobs PA, Baron GV, Denayer JEM. Pore-filling-dependent selectivity effects in the vapor-phase separation of xylene isomers on the metal–organic framework MIL-47. J. Am. Chem. Soc. 2008;130:7110–7118. doi:10.1021/ja800686c.
- Dąbrowski A. Adsorption - from theory to practice. Adv. Colloid Interface Sci. 2001;93:135–224. doi:10.1016/S0001-8686(00)00082-8.
- McGrother SC, Gubbins E. Constant pressure Gibbs ensemble Monte Carlo simulations of adsorption into narrow pores. Mol. Phys. 1999;97:955–965. doi:10.1080/00268979909482897.
- Torres-Knoop A, Krishna R, Dubbeldam D. Separating xylene isomers by commensurate stacking of p-xylene within channels of MAF-X8. Angew. Chem. Int. Ed. 2014;53:7774–7778. doi:10.1002/anie.201402894.
- Castillo JM, Vlugt TJH, Calero S. Molecular simulation study on the separation of xylene isomers in MIL-47 metal–organic frameworks. J. Phys. Chem. C. 2009;113:20869–20874. doi:10.1021/jp908247w.
- Talu O, Myers AL. Molecular simulation of adsorption: Gibbs dividing surface and comparison with experiment. AIChE. J. 2001;47:1160–1168. doi:10.1002/aic.690470521.
- Düren T, Sarkisov L, Yaghi OM, Snurr RQ. Design of new materials for methane storage. Langmuir. 2004;20:2683–2689. doi:10.1021/la0355500.
- Hansen N. Multiscale modeling of reaction and diffusion in Zeolites. [PhD thesis], Technische Universität Hamburg, Hamburg, Germany. 2010.
- Vlugt TJH, Smit B. The BIGMAC: A configurational Bias Monte Carlo Program. Amsterdam: University of Amsterdam; 1998.
- Bai P, Tsapatsis M, Siepmann JI. Multicomponent adsorption of alcohols onto silicalite-1 from aqueous solution: isotherms, structural analysis, and assessment of ideal adsorbed solution theory. Langmuir. 2012;28:15566–15576. doi:10.1021/la303247c.
- Myers AL, Prausnitz JM. Thermodynamics of mixed-gas adsorption. AIChE J. 1965;11:121–127. doi:10.1002/aic.690110125.
- Krishna R, Long JR. Screening metal-organic frameworks by analysis of transient breakthrough of gas mixtures in a fixed bed adsorber. J. Phys. Chem. C. 2011;115:12941–12950. doi:10.1021/jp202203c.
- Walton KS, Millward AR, Dubbeldam D, Frost H, Low JJ, Yaghi OM, Snurr RQ. Understanding inflections and steps in carbon dioxide adsorption isotherms in metal–organic frameworks. J. Am. Chem. Soc. 2008;130:406–407. doi:10.1021/ja076595g.
- Millward AR. Adsorption of environmentally significant gases (H2, CO2, H2S, CH4) in metal-organic frameworks. [PhD thesis]. The University of Michigan, the United States of America. 2006.
- Dubbeldam D, Walton KS, Ellis DE, Snurr RQ. Exceptional negative thermal expansion in isoreticular metal-organic frameworks. Angew. Chem. Int. Ed. 2007;46:4496–4499. doi:10.1002/anie.200700218.
- Wolf RJ, Lee MW, Davis RC, Fay PJ, Ray JR. Pressure-composition isotherms for palladium hydride. Phys. Rev. B. 1993;48:12415–12418. doi:10.1103/PhysRevB.48.12415.
- Spyriouni T, Economou IG, Theodorou DN. Phase equilibria of mixtures containing chain molecules predicted through a novel simulation scheme. Phys. Rev. Lett. 1998;80:4466–4469. doi:10.1103/PhysRevLett.80.4466.
- Duane S, Kennedy AD, Pendleton BJ, Roweth D. Hybrid Monte Carlo. Phys. Lett. B. 1987;195:216–222. doi:10.1016/0370-2693(87)91197-X.
- Chempath S, Clark LA, Snurr RQ. Two general methods for grand canonical ensemble simulation of molecules with internal flexibility. J. Chem. Phys. 2003;118:7635–7643. doi:10.1063/1.1562607.
- Rosenbluth MN, Rosenbluth AW. Monte carlo calculation of the average extension of molecular chains. J. Chem. Phys. 1955;23:356–359. doi:10.1063/1.1741967.
- Siepmann JI. A method for the direct calculation of chemical potentials for dense chain systems. Mol. Phys. 1990;70:1145–1158. doi:10.1080/00268979000101591.
- Laso M, de Pablo JJ, Suter UW. Simulation of phase-equilibria for chain molecules. J. Phys. Condens. Matter. 1992;97:2817–2819.
- Shi W, Maginn EJ. Continuous fractional component Monte Carlo: an adaptive biasing method for open system atomistic simulations. J. Chem. Theory Comput. 2007;3:1451–1463. doi:10.1021/ct7000039.
- Shi W, Maginn EJ. Improvement in molecule exchange efficiency in Gibbs ensemble Monte Carlo: Development and implementation of the continuous fractional component move. J. Comput. Chem. 2008;29:2520–2530. doi:10.1002/jcc.20977.
- Rosch TW, Maginn EJ. Reaction ensemble Monte Carlo simulation of complex molecular systems. J. Chem. Theory Comput. 2011;7:269–279. doi:10.1021/ct100615j.
- Torres-Knoop A, Prasaad Balaji S, Vlugt T, Dubbeldam D. A comparison of advanced Monte Carlo methods for open systems: CFCMC vs. CBMC. J. Chem. Theor. Comp. 2014;10:942–952.
- van Erp TS, Caremans TP, Dubbeldam D, Martin-Calvo A, Calero S, Martens JA. Enantioselective adsorption in achiral zeolites. Angew. Chem. Int. Edit. 2010;49:3010–3013. doi:10.1002/anie.200906083.
- Qiao Z, Torres-Knoop A, Dubbeldam D, Fairen-Jimenez D, Zhou J, Snurr RQ. Advanced Monte Carlo simulations of the adsorption of chiral alcohols in a homochiral metal-organic framework. AIChE J. 2014;60:2324–2334. doi:10.1002/aic.14415.
- Gómez-Gualdrón DA, Wilmer CE, Farha OK, Hupp JT, Snurr RQ. Exploring the limits of methane storage and delivery in nanoporous materials. J. Phys. Chem. C. 2014;118:6941–6951. doi:10.1021/jp502359q.
- Wilmer CE, Leaf M, Lee CY, Farha OK, Hauser BG, Hupp JT, Snurr RQ. Large-scale screening of hypothetical metal-organic frameworks. Nat. Chem. 2012;4:83–89. doi:10.1038/nchem.1192.
- Wilmer CE, Farha OK, Bae YS, Hupp JT, Snurr RQ. Structure-property relationships of porous materials for carbon dioxide separation and capture. Energy Env. Sci. 2012;5:9849–9856. doi:10.1039/c2ee23201d.
- Colón YJ, Snurr RQ. High-throughput computational screening of metal-organic frameworks. Chem. Soc. Rev. 2014;43:5735–5749. doi:10.1039/C4CS00070F.
- Sikora BJ, Winnegar R, Proserpio DM, Snurr RQ. Textural properties of a large collection of computationally constructed MOFs and zeolites. Microporous Mesoporous Mater. 2014;186:207–213. doi:10.1016/j.micromeso.2013.11.041.
- Dubbeldam D, Krishna R, Calero S, Yazaydın O. Computer-assisted screening of ordered crystalline nanoporous adsorbents for separation of alkane isomers. Angew. Chem. Int. Ed. 2012;51:11867–11871. doi:10.1002/anie.201205040.
- Johnson JK, Panagiotopoulos AZ, Gubbins KE. Reactive canonical Monte Carlo: a new simulation technique for reacting or associating fluids. Mol. Phys. 1994;81:717–733. doi:10.1080/00268979400100481.
- Smith WR, Triska B. The reaction ensemble method for the computer simulation of chemical and phase equilibria. i. theory and basic examples. J. Chem. Phys. 1994;100:3019. doi:10.1063/1.466443.
- Hansen N, Jakobtorweihen S, Keil FJ. Reactive Monte Carlo and grand-canonical Monte Carlo simulations of the propene metathesis reaction system. J. Chem. Phys. 2005;122:164705. doi:10.1063/1.1884108.
- Wittcoff HA, Reuben BG, Plotkin JS. Industrial Organic Chemicals. Hoboken NJ: Wiley; 2004.
- Jakobtorweihen S, Hansen N, Keil FJ. Combining reactive and configurational-bias Monte Carlo: Confinement influence on the propene metathesis reaction system in various zeolites. J. Chem. Phys. 2006;125:224709. doi:10.1063/1.2404658.
- Dubbeldam D, Ford DC, Ellis DE, Snurr RQ. A new perspective on the order-n algorithm for computing correlation functions. Mol. Simulat. 2009;35:1084–1097.
- Reed DA, Ehrlich G. Surface diffusivity and the time correlation of concentration fluctuations. Surf. Sci. 1981;105:603–628. doi:10.1016/0039-6028(81)90021-2.
- Krishna R, van Baten JM. Describing binary mixture diffusion in carbon nanotubes with the Maxwell–Stefan equations. An investigation using molecular dynamics simulations. Ind. Eng. Chem. Res. 2006;45:2084–2093. doi:10.1021/ie051126d.
- Krishna R, van Baten JM. Diffusion of alkane mixtures in zeolites: validating the Maxwell–Stefan formulation using md simulations. J. Phys. Chem. B. 2005;109:6386–6396. doi:10.1021/jp044257l.
- Theodorou DN, Snurr RQ, Bell AT. Molecular dynamics and diffusion in microporous materials. In: Alberti G, Bein T, editors. Comprehensive Supramolecular Chemistry. volume 7, chapter 18 Oxford: Pergamon Oxford; 1996. p. 507–548.
- June RL, Bell AT, Theodorou DN. Molecular dynamics study of methane and xenon in silicalite. J. Phys. Chem. 1990;94:8232–8240. doi:10.1021/j100384a047.
- Snurr RQ, June RL, Bell AT, Theodorou DN. Molecular simulations of methane adsorption in silicalite. Mol. Simulat. 1991;8:73–92. doi:10.1080/08927029108022468.
- Lekien F, Marsden J. Tricubic interpolation in three dimensions. Int. J. Numer. Methods Eng. 2005;63:455–471. doi:10.1002/nme.1296.
- Miller TF, Eleftheriou M, Pattnaik P, Ndirango A, Newns D, Martyna GJ. Symplectic quaternion scheme for biophysical molecular dynamics. J. Chem. Phys. 2002;116:8649–8659. doi:10.1063/1.1473654.
- Tuckerman ME, Alejandre J, López-rendón R, Jochim AL, Martyna GJ. A Liouville-operator derived measure-preserving integrator for molecular dynamics simulations in the isothermal–isobaric ensemble. J. Phys. A. 2006;39:5629–5651. doi:10.1088/0305-4470/39/19/S18.
- Skoulidas AI, Sholl DS. Self-diffusion and transport diffusion of light gases in metal-organic framework materials assessed using molecular dynamics simulations. J. Phys. Chem. B. 2005;109:15760–15768. doi:10.1021/jp051771y.
- Martyna GJ, Tuckerman M, Tobias DJ, Klein ML. Explicit reversible integrators for extended systems dynamics. Mol. Phys. 1996;87:1117–1157. doi:10.1080/00268979600100761.
- Yu T-Q, Alejandre J, López-rendón R, Martyna GJ, Tuckerman ME. Measure-preserving integrators for molecular dynamics in the isothermal-isobaric ensemble derived from the Liouville operator. Chem. Phys. 2010;370:294–305. doi:10.1016/j.chemphys.2010.02.014.
- Frenkel D, Smit B. Understanding molecular simulation 2nd ed. London: Academic Press; 2002.
- Beerdsen E, Smit B, Dubbeldam D. Molecular simulation of loading dependent slow diffusion in confined systems. Phys. Rev. Lett. 2004;93: 248301.10.1103/PhysRevLett.93.248301.
- Dubbeldam D, Beerdsen E, Vlugt TJH, Smit B. Molecular simulation of loading-dependent diffusion in nanoporous materials using extended dynamically corrected transition state theory. J Chem Phys. 2005;122: 224712.10.1063/1.1924548.
- Walton KS, Snurr RQ. Applicability of the BET method for determining surface areas of microporous metal–organic frameworks. J. Am. Chem. Soc. 2007;129:8552–8556. doi:10.1021/ja071174k.
- Düren T, Millange F, Férey G, Walton KS, Snurr RQ. Calculating geometric surface areas as a characterization tool for metal–organic frameworks. J. Phys. Chem. C. 2007;111:15350–15356. doi:10.1021/jp074723h.
- Sarkisov L, Harrison A. Computational structure characterisation tools in application to ordered and disordered porous materials. Mol. Phys. 2011;37:1248–1257.
- Gelb LD, Gubbins KE. Pore size distributions in porous glasses: a computer simulation study. Langmuir. 1999;15:305–308. doi:10.1021/la9808418.
- Fletcher AJ, Thomas KM, Rosseinsky MJ. Flexibility in metal-organic framework materials: Impact on sorption properties. J. Solid State Chem. 2005;178:2491–2510. doi:10.1016/j.jssc.2005.05.019.
- Gale JD. Gulp: A computer program for the symmetry-adapted simulation of solids. J. Chem. Soc. Faraday Trans. 1997;93:629–637. doi:10.1039/a606455h.
- Gale JD, Rohl AL. The general utility lattice program (gulp). Mol. Sim. 2003;29:291–341. doi:10.1080/0892702031000104887.
- Schröder K-P, Sauer J. Potential functions for silica and zeolite catalysts based on ab initio calculations. 3. a shell model ion pair potential for silica and aluminosilicates. J. Phys. Chem. 1996;110:11043–11049.
- Dubbeldam D, Krishna R, Snurr RQ. Method for analyzing structural changes of flexible metal − organic frameworks induced by adsorbates. J. Phys. Chem. C. 2009;113:19317–19327. doi:10.1021/jp906635f.
- van Workum K, Gao G, Schall JD, Harrison JA. Expressions for the stress and elasticity tensors for angle-dependent potentials. J. Chem. Phys. 2006;125:144506. doi:10.1063/1.2338522.
- Lutsko JF. Generalized expressions for the calculation of elastic constants by computer simulation. J. Appl. Phys. 1989;65:2991–2997. doi:10.1063/1.342716.
- Oxford GAE, Dubbeldam D, Broadbelt LJ, Snurr RQ. Elucidating steric effects on enantioselective epoxidation catalyzed by (salen)Mn in metal-organic frameworks. J. Mol. Catal. A. 2011;334:89–97. doi:10.1016/j.molcata.2010.11.001.
- Jacobsen H, Cavallo L. A possible mechanism for enantioselectivity in the chiral epoxidation of olefins with [Mn(salen)] catalysts. Chem. Eur. J. 2001;7:800–807. 10.1002/1521-3765(20010216)7:4 < 800:AID-CHEM800>3.0.CO;2-1.
- Dubbeldam D, Oxford GAE, Krishna R, Broadbelt LJ, Snurr RQ. Distance and angular holonomic constraints in molecular simulations. J. Chem. Phys. 2010;133: 034114.10.1063/1.3429610.
- Dubbeldam D, Walton KS. On the application of classical molecular simulations of adsorption in metal-organic frameworks. In: Jianwen J, editor. Metal-organic frameworks: materials modeling towards engineering applications. Pan Stanford Publishing Pte Ltd; 2014.
- Snurr RQ, Bell AT, Theodorou DN. Prediction of adsorption of aromatic hydrocarbons in silicalite from grand canonical Monte Carlo simulations with biased insertions. J. Phys. Chem. 1993;97:13742–13752. doi:10.1021/j100153a051.
- Schroeder W, Martin K, Lorensen B. The Visualization Toolkit: an object-oriented approach to 3D graphics. Upper Saddle River, New Jersey: Prentice-Hall,Inc; 1996. p. 07458.
- Dubbeldam D, Galvin CJ, Walton KS, Ellis DE, Snurr RQ. Separation and molecular-level segregation of complex alkane mixtures in metal − organic frameworks. J. Am. Chem. Soc. 2008;130:10884–10885. doi:10.1021/ja804039c.
- Dubbeldam D, Frost H, Walton KS, Snurr RQ. Molecular simulation of adsorption sites of light gases in the metal-organic framework IRMOF-1. Fluid Phase Equilibria. 2007;261:152–161. doi:10.1016/j.fluid.2007.07.042.