
Abstract
The ELBA coarse-grained model describes a water molecule as a single-site Lennard-Jones particle embedded with a point dipole. ELBA was previously reported to capture several properties of real water with relatively high accuracy, while being up to two orders of magnitude more computationally efficient than atomistic models. Here, we ‘stress test’ the ELBA model by investigating the temperature and pressure dependences of two most important water properties, the liquid density and the self-diffusion coefficient. In particular, molecular dynamics simulations are performed spanning temperatures from 268 K up to 378 K and pressures from 1 atm up to 4000 atm. Comparisons are made with literature data from experiments and from simulations of traditional three-site atomistic models. Remarkably, the ELBA results show an overall similar (and sometimes higher) accuracy with respect to the atomistic data. We also calculate a number of additional thermodynamic properties at ambient conditions, namely isothermal compressibility, shear viscosity, isobaric heat capacity, thermal expansion coefficient and melting point. The accuracy of ELBA is relatively good compared to atomistic and other coarse-grained models.
1. Introduction
The modelling and simulation of water is an area of great interest for both academic research and industrial applications. Numerous water models, with various characteristics and capabilities, have been developed over several decades now.Citation1Citation2Citation3Citation4 A popular approach involves atomistic models where each atom in the water molecule is represented by a simulated site,Citation5Citation6Citation7 although optimised models also exist which include one or two extra sites.Citation8Citation9Citation10 While the simulations of atomistic models can provide accurate and realistic results, they are also highly demanding of computational resources. To reduce the computational cost, simplified models can be developed by representing one or several water molecules with lower resolution single sites; in this approach, known as ‘coarse-graining’, some atomic-level details are sacrificed to increase simulation efficiency.Citation11Citation12Citation13Citation14Citation15
Recently, the ELBA coarse-grained model has been developed to attempt striking a new balance between physical accuracy and simulation efficiency.Citation16Citation17Citation18 In the ELBA model, the three atoms of a water molecule are reduced to a single particle, with an electrical point dipole affixed to its centre to capture the well-known dipolar nature of water. Previous molecular dynamics simulations have shown relatively good accuracy of the ELBA model under ambient conditions of temperature and pressure. In particular, ELBA reproduced several fundamental water properties as accurately as the best atomistic models, including density, potential energy, self-diffusion coefficient, heat of vaporisation, critical point, surface tension and the liquid–vapour interface structure.[Citation18] A significant issue was however observed for the radial distribution function, which showed qualitative and quantitative differences compared with experimental and atomistic data.[Citation18] Notably, ELBA can also be used to hydrate atomistic solutes, including small organic molecules and proteins; in particular, uniquely for a coarse-grained model, ELBA was shown to be directly compatible with common atomistic force fields, meaning that no additional or ad hoc scaling factors, intermediate regions or extra sites were required.[Citation19] In terms of computational cost, ELBA proves to be between one to two orders of magnitude more efficient to simulate than traditional multi-site atomistic models.[Citation18]
While ambient conditions are obviously of primary importance, there is also substantial interest in applying molecular simulations to investigate phenomena which take place under a variety of temperatures and pressures, such as protein folding processes [Citation20,Citation21] and water filtration.[Citation22] Hence, it is important to assess how well a water model can predict real water behaviour at non-ambient conditions. In this work, the performance of the ELBA model (with no modification to the original parameters) is examined over a wide range of temperatures (268–378 K) and pressures (1–4000 atm). We investigate the temperature and pressure dependences of two fundamental water properties, the liquid phase density and self-diffusion, which ELBA reproduces accurately under ambient conditions.[Citation18] Comparisons are made with corresponding experimental data and simulation results for the atomistic models SPC,[Citation5] SPC/E [Citation7] and TIP3P,[Citation6] which represent the most widely used water models in molecular simulation. As extra findings, we also compute the pressure dependence of liquid density and self-diffusion coefficient at room temperature for, respectively, TIP3P and SPC. To our knowledge, these properties have not been previously published.
In addition, five thermodynamic properties at ambient conditions are reported here for ELBA for the first time, namely, isobaric heat capacity, thermal expansion coefficient, isothermal compressibility, melting point and shear viscosity. For these properties, literature data exist also from simulation of other coarse-grained models, which are thus included in the comparison.
2. Methods
2.1 ELBA coarse-grained model
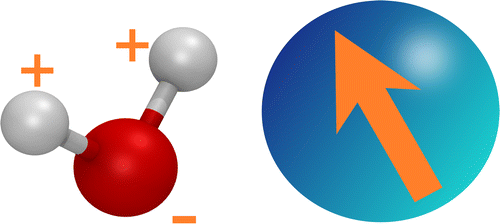
The ELBA model describes a water molecule as a single Lennard-Jones particle embedded with a point dipole (Figure ).[Citation16,Citation18] The potential energy for a pair of water particles i and j is the sum of a Lennard-Jones interaction term
and a dipole interaction term
:
Both terms are in the ‘shifted-force’ form.[Citation23,Citation24] whereby the potential energy and its derivative (the force) go to zero smoothly at the cut-off point. The shifted-force scheme removes cut-off-related artefacts and simulation stability problems,[Citation23,Citation24] which are especially severe for orientation-dependent potentials such as the point dipole potential.[Citation25] For the Lennard-Jones part, the following expression proposed by Stoddard and Ford [Citation26] is adopted:
2.2 General simulation details
Molecular dynamics simulations were performed using the software LAMMPS (version 11 Nov 2013).[Citation28,Citation29] Input scripts are available on our group website.[Citation30] The simulation systems contained 8000 water sites in a cubic region. Conventional periodic boundary conditions were adopted. Conditions of constant temperature and pressure were applied in most of the simulations; in these cases, the edge length of the starting simulation box was set to 6.2 nm, yielding an initial water density of approximately 1 g cm − 3 (consistent with the density of real water at standard ambient conditions). However, in some cases (detailed below), we fixed the volume, and hence also the system density, to a constant value. In all simulations, the temperature was controlled using the Langevin thermostat,[Citation31] with a collision frequency of 1 ps − 1. The pressure was maintained (when needed) using the barostat by Berendsen et al., [Citation32] with a damping time of 1 ps and a bulk modulus of 2.174 × 104 atm. The integration time step for the ELBA simulations was 10 fs. At every time step, the net momentum of the mass centre of the entire system was removed to prevent drifting during the simulation. The interaction cut-off radius was 12 Å and no long range interactions (beyond the cut-off distance) were included for the ELBA model. All these settings are consistent with previous work. [Citation18] Additional atomistic simulations were also run to study the pressure dependence of the TIP3P density and the SPC diffusion coefficient, as no corresponding results were found in the literature. For these simulations the time step was 2 fs. Bonds and angles were constrained using the SHAKE algorithm [Citation33] with a relative tolerance of 10 − 4. The non-bonded cut-off distance was 10 Å for SPC [Citation18] and 13 Å for TIP3P.[Citation18,Citation34] Long range electrostatic interactions were included with the particle-particle particle-mesh (PPPM) method [Citation35] with a relative tolerance of 10 − 5.
Specific details of individual calculations are given in Section 2.3.
2.3 Details of individual calculations
To investigate density, self-diffusion, isothermal heat capacity, thermal expansion coefficient and isothermal compressibility, we ran series of 7 ns long simulations. The initial 2 ns was regarded as equilibration, while the subsequent 5 ns was considered as production; during production, the relevant properties were sampled every 0.1 ps. For each simulation, three independent repeats were run by assigning the initial velocities with different random seeds.
Calculations of density and self-diffusion coefficient followed standard procedures. [Citation2,Citation24] Regarding isothermal heat capacity, thermal expansion coefficient and isothermal compressibility, details are given in the following Sections 2.3.1, 2.3.2 and 2.3.3, respectively. The calculations of shear viscosity and melting point were carried out according to the protocols described below in Sections 2.3.4 and 2.3.5, respectively.
2.3.1 Isobaric heat capacity
The isobaric heat capacity Cp at 298 K was estimated as [Citation36]:
2.3.2 Thermal expansion coefficient
The thermal expansion coefficient α at 298 K was estimated using the finite-difference expression method [Citation37,Citation39]:
2.3.3 Isothermal compressibility
The isothermal compressibility was calculated from two sets of constant-volume (and hence also constant-density) simulations, by applying the finite-difference equation as follows [Citation36,Citation37]:
2.3.4 Shear viscosity
The shear viscosity was calculated according to the Green–Kubo formula,[Citation40,Citation41] in which the autocorrelation function of the stresses is integrated as
where V is the system volume,
is the Boltzmann constant, T is the system temperature and
are the off-diagonal components of the stress tensor
. The calculation was carried out over 1 ns production stages following 1 ns equilibration stages.[Citation42] A total of six independent simulations were run by setting different initial velocities.
2.3.5 Melting point
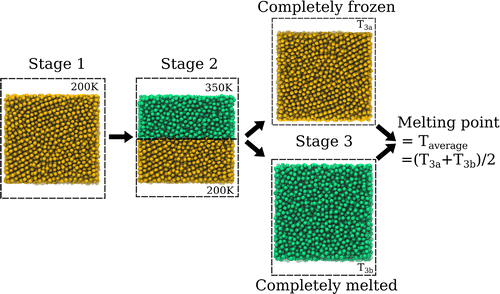
The liquid–solid coexistence method [Citation43,Citation44] was followed in order to determine the melting point. The simulation system consisted of a cubic region containing 8000 water sites in total (as described above, in Section 2.2). The cube was divided into two equal regions, designated liquid and ice region, respectively. Then three separate simulation stages were performed. In the first stage, lasting 1 ns, the whole system was cooled down to 200 K to achieve complete freezing of both regions. In the second stage, lasting 1 ns, particles assigned to the ice region were immobilised (thus retaining their frozen state), while the temperature for the liquid region was raised to 350 K to ensure complete melting. Finally, in the third stage, lasting 10 ns, the whole system was equilibrated to a series of target temperatures, with all particles freely moving. During freezing, the thermostat subtracts energy from the system, therefore the total energy decreases. Conversely, during melting, the thermostat adds energy to the system, thus the total energy increases.[Citation44] By examining the total energy evolution over time and the final state of these trial simulations, the melting point was gradually approached. In the simulations where the total energy neither increased nor decreased and both ice and liquid phases coexisted at a specific temperature, this temperature was considered to be the melting point. Alternatively, if no coexistence appeared, but the temperature difference between a completely melted and a completely frozen system was equal to 1 K, the melting point was estimated as the average between them (see Figure , and Section 1 in the Supplementary Material). In total, the three-stage procedure described was repeated five times with different initial conditions.
3. Results and discussion
3.1 Temperature dependence
The density and the self-diffusion coefficient of the ELBA water have been calculated at temperatures ranging from 268 and 378 K, under a constant pressure of 1 atm. Results for both properties are compared with previously published results from experiment and from simulations of the widely used three-site atomistic water models SPC, SPC/E and TIP3P.Citation6Citation45Citation46Citation47Citation48
Figure shows the density data as a function of temperature. Compared with the experimental data, it is clear that the ELBA results exhibit a larger rate of change with temperature in comparison to real water. Specifically, the ELBA results underestimate the density above room temperature while overestimate it below room temperature. Note, however, that the discrepancies observed are not particularly severe; the largest error, at around 373 K, is ∼0.03 g cm − 3, which corresponds to a relative error of only ∼3% with respect to the experimental measurement. When compared to the data from the atomistic models, the ELBA results are remarkably accurate (where accurate is intended to mean close to the experimental value). In particular, ELBA proves more accurate than SPC for temperatures above ∼285 K. ELBA is also more accurate than TIP3P for temperatures above ∼290 K. However, ELBA is less accurate than SPC/E for all temperatures.
![Figure 3 (Colour online) Density as a function of temperature at 1 atm. The standard deviation of the ELBA data is less than 0.00005 g cm− 3 for all values; corresponding error bars are smaller than the size of the symbols. The experimental data are from the NIST database.[Citation73] Atomistic data are from Paschek. [Citation74]](/cms/asset/192fbbb3-f068-4b5f-bb55-e4a01d9e9e66/gmos_a_1047367_f0003_oc.gif)
The self-diffusion coefficient data as a function of temperature are displayed in Figure . It can be seen that the ELBA results exhibit a lower rate of change with temperature in comparison to real water. In terms of absolute values, the accuracy of ELBA is rather good in a fairly large region centred around room temperature (approximately from 270 to 320 K). In this region, ELBA is substantially more accurate than SPC and TIP3P, and largely as accurate as SPC/E. For temperatures above ∼320 K, the ELBA values increasingly diverge from the experimental curve, indicating that the mobility of the ELBA water is less sensitive than real water to the temperature increase.
![Figure 4 (Colour online) Self-diffusion coefficient as a function of temperature at 1 atm. The standard deviation for the ELBA results is at most 0.05 × 10− 9 m2 s− 1; corresponding error bars are of the order of the size of the symbols. The experimental data,[Citation75] as well as results for SPC,[Citation48] SPC/E [Citation48] and TIP3P [Citation47] are shown for comparison.](/cms/asset/635593ee-d97d-41aa-af76-54b20fef0639/gmos_a_1047367_f0004_oc.gif)
3.2 Pressure dependence
The pressure dependence of ELBA was investigated by applying external isotropic pressures from 1 to 4000 atm while maintaining a standard ambient temperature of 298 K. From Figure , it can be seen that the ELBA results are close to experiment only in the initial region around ambient condition, while they increasingly underestimate the experimental values with increase in pressure. This implies that ELBA is less compressible by high pressure than real water. Compared with the atomistic models, ELBA is markedly less accurate than both SPC/E and TIP3P, but it proves more accurate than SPC for pressures below 1000 atm.
![Figure 5 (Colour online) Density as a function of pressure at 298 K of ELBA, together with experimental data [Citation73] and simulation results for SPC,[Citation76] SPC/E [Citation76] and TIP3P (this work). The standard deviations for the ELBA and TIP3P results are at most 0.00005 and 0.00007 g cm− 3, respectively; corresponding error bars are smaller than the size of the symbols.](/cms/asset/b2f6def9-80a4-4a22-aa52-74aea0ad39ee/gmos_a_1047367_f0005_oc.gif)
Regarding the self-diffusion coefficient, Figure shows that the accuracy of ELBA is rather high at ambient pressure, but it decreases with increase in pressure, as the experimental values are more and more underestimated. Nevertheless, ELBA proves markedly more accurate than both SPC and (especially) TIP3P, while the SPC/E data almost match the experimental values.
![Figure 6 (Colour online) Self-diffusion coefficient as a function of pressure at 298 K. Results from experiment [Citation77] and for SPC (this work), SPC/E [Citation78] and TIP3P [Citation79] are shown for comparison. The standard deviation for the ELBA and SPC values are at most 0.018 × 10− 9 and 0.23 × 10− 9 m2 s− 1, respectively; corresponding error bars are of the order of the size of the symbols.](/cms/asset/6105664e-ea67-476d-be47-68847789ee66/gmos_a_1047367_f0006_oc.gif)
3.3 Thermodynamic properties at ambient conditions
Results for five thermodynamic properties of the ELBA model at 298 K and 1 atm are presented in Table , together with literature data from experiments, three-site atomistic models and also other coarse-grained models. The atomistic models are SPC,[Citation5] SPC/E [Citation7] and TIP3P,[Citation6] already used as standards for comparison in the temperature and pressure dependences reported in the previous section. Regarding the other coarse-grained models, comparable results have been reported for single-site models (like ELBA) and for ‘coarser’ descriptions, whereby several water molecules are reduced to a smaller number of coarse-grained particles. Considering single-site models, relevant data have been reported in the literature for the soft sticky dipole (SSD), the model of water (mW) and multiscale coarse-grained (MS-CG) models. SSD is a model comprising a Lennard-Jones sphere, a point dipole and an octupolar term used to capture hydrogen bonding. SSD has been used to study water in the liquid and ice phases,Citation49Citation50Citation51Citation52 and as a solvent for different systems.Citation52Citation53Citation54Citation55Citation56 The mW model is characterised by a three-body term that captures tetrahedral coordination.[Citation57] MS-CG is a model consisting of an effective potential obtained by the force-matching method.[Citation58] The big multipole water (BMW) model [Citation59] is a coarser model that reduces four water molecules to three charged sites, with the Born–Mayer–Huggins potential replacing the conventional Lennard-Jones potential. BMW has been applied to study the solvation and aggregation of peptides.[Citation60] MARTINI is a popular model based on the Lennard-Jones potential, without explicit electrostatics, where each site represents four water molecules.[Citation61] P-MARTINI [Citation62] refers to a polarisable variant of MARTINI which comprises three sites, two of which are oppositely charged. The GROMOS model [Citation36] maps five water molecules to two oppositely charged sites. This model has also been used as a solvent for proteins.[Citation63] WT4 is a model that represents clusters of 11 water molecules by tetrahedral particles comprising four charged sites.[Citation64] WT4 has been used as a solvent for ions and nucleic acids.[Citation64]
Table 1 Thermodynamic properties at ambient conditions.a
Regarding the isobaric heat capacity Cp, the ELBA result is lower than the experimental value. This could be expected since there is no explicit hydrogen-bonding network in the ELBA model. In fact, the high heat capacity of real liquid water is believed to be partly due to the presence of hydrogen bonds, because when water is heated, the corresponding energy is partly used to bend or break the hydrogen bonds, rather than being totally transferred to the kinetic energy of the water molecules.[Citation65,Citation66] Quantitatively, the difference between ELBA and the experimental value is ∼10 , corresponding to a relative error of 14.5%. Compared with the atomistic models, ELBA proves slightly more accurate than SPC/E, for which the relative error is 15%; however, SPC and TIP3P are in turn markedly more accurate than ELBA, with relative errors of 0.3% and 4%, respectively. As for the other coarse-grained models, it can be seen that mW is characterised by a Cp value that is less than half the experimental value. The WT4 model also underestimates
, being 43% lower than the experimental measurement. Both the SSD and GROMOS models instead overestimate
, although with rather different relative errors of, respectively, 60% and 7%.
Regarding the thermal expansion coefficient α, it can be seen that the ELBA value is not very accurate, as it is almost four times larger than the experimental value. However, in comparison with the atomistic models, the accuracy of ELBA is relatively good, being within the range reported for SPC and TIP3P (which have α values about three to five times larger than the experimental value). The SPC/E result is the most accurate, even though it is still over twice as large as the experimental result. It should be pointed out that these results are consistent with the temperature-dependent density data reported in Figure , where all densities from simulations show a larger rate of change with temperature with respect to the experimental data. Specifically, as the density of the models decreases faster than that experimentally observed with increasing temperature, the thermal expansion coefficient is expected to be overestimated. Compared with the other coarse-grained models, ELBA proves slightly more accurate than WT4 and substantially more accurate than GROMOS and MS-CG.
As for the isothermal compressibility , the ELBA model underestimates the experimental value by ∼36%, while the atomistic models all prove more accurate, with relative errors in the range from 4% to 28%. Compared with the other coarse-grained models, ELBA is instead more accurate than all but one model (BMW).
Regarding the melting temperature, we obtained for ELBA a value of 301.7 ± 4.4 K, which is 28 K higher than the experimental value. By considering only the magnitude of the error with respect to the experimental temperature, ELBA would seem to be more accurate than the atomistic models, which all underestimate the true melting temperature with larger absolute errors. However, it must be stressed that underestimating the melting temperature is a much less serious problem than overestimating it. In most applications, simulations are carried out at (or around) room temperature, where water must be in the liquid phase; in this context, models overestimating the melting temperature may instead freeze. In particular, considering those coarse-grained models overestimating the melting temperature (ELBA, MARTINI, P-MARTINI and SSD), the solid phase is predicted to be the most thermodynamically stable phase for temperatures close to room temperature (or even substantially higher in the case of SSD); as noted, this is in principle very problematic. In practice however, since freezing is an activated process,[Citation67] unless a model is already in the solid phase, simulated liquid water can remain in the (supercooled) liquid phase for temperatures well below the predicted melting temperature. Regarding ELBA, we have run microsecond long simulations at decreasing temperatures, and we have observed spontaneous freezing only for temperatures below 250 K (see Section 2 in the Supplementary Material). Moreover, for any other system we have previously simulated, including the water–vacuum interface,[Citation18] hydrated lipid bilayers in the lamellar and inverse hexagonal phases,[Citation16,Citation17] and hydrated small molecules and proteins,[Citation19] the ELBA water has consistently remained in the liquid phase. To lower the melting temperature, ELBA could be reparametrised. In particular, decreasing the dipole magnitude should have the effect of decreasing the melting temperature, consistently with what has been reported for generic ‘Lennard-Jones plus point dipole’ models in reduced units.[Citation68,Citation69] However, changing the dipole will of course have repercussions not only on the melting point, but also on all other physical properties; a reparametrisation of the whole force field could therefore be needed, with no guarantee of eventually obtaining an overall better model.
Finally, regarding the shear viscosity , ELBA yielded a result only 7% larger than the experimental value; the corresponding accuracy is higher than that of the atomistic models (which have relative errors between 18% and 64%) as well as the other coarse-grained models BMW (relative error from 13% to 81%) and GROMOS (relative error of 316%). Such a high accuracy of ELBA in predicting the shear viscosity was not unexpected, as the shear viscosity is related to the self-diffusion coefficient (through the Stokes–Einstein expression), and ELBA was shown previously to reproduce closely the experimental self-diffusion coefficient.[Citation18]
4. Conclusion
We have studied the temperature and pressure dependences of density and self-diffusion for the coarse-grained ELBA water model. Notably, the accuracy of ELBA was found to be overall comparable to that of the standard three-site atomistic models, in that the relative errors with respect to experiment obtained with ELBA were mostly within the range of errors characterising the atomistic models. In fact, in some cases ELBA was shown to be even more accurate than some of the atomistic models.
Moreover, we computed five properties at standard ambient conditions, namely isothermal compressibility, shear viscosity, isobaric heat capacity, thermal expansion coefficient and melting point, for which comparisons could also include other coarse-grained models. For the thermal expansion coefficient and the isothermal compressibility, ELBA proved less accurate than the atomistic models, but more accurate than the other coarse-grained models. Regarding the heat capacity, ELBA was shown to be as accurate as SPC/E and more accurate than most available coarse-grained models. For the shear viscosity, the ELBA prediction was even closer to experiment than the results reported for the atomistic models.
Together with the recent demonstration of the direct compatibility of ELBA with atomistic force fields for organic molecules and proteins,[Citation19] the work presented here opens up a range of opportunities for mixed atomistic/coarse-grained simulations under non-ambient conditions of temperature and pressure.
Supplementary data
Supplemental data for this article can be accessed doi:10.1080/08927022.2015.104736710.1080/08927022.2015.1047367.
Supplemental
Download PDF (452.6 KB)Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Notes
1. Email: [email protected]
2. Email: [email protected]
References
- Vega C, Abascal L. Simulating water with rigid non-polarizable models: a general perspective. Phys Chem Chem Phys. 2011;13(44):19663–19688. doi:10.1039/c1cp22168j.
- Guillot B. A reappraisal of what we have learnt during three decades of computer simulations on water. J Mol Liq. 2002;101(1–3):219–260. doi:10.1016/S0167-7322(02)00094-6.
- He X, Shinoda W, DeVane R, Klein ML. Exploring the utility of coarse-grained water models for computational studies of interfacial systems. Mol Phys. 2010;108(15):2007–2020. doi:10.1080/00268976.2010.503197.
- Hadley KR, McCabe C. Coarse-grained molecular models of water: a review. Mol Simul. 2012;38(8–9):671–681. doi:10.1080/08927022.2012.671942.
- Berendsen HJ, Postma J, Van Gunsteren W, Hermans J. Intermolecular forces. Dordrecht: Springer; 1981. p. 331–342.
- Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. J Chem Phys. 1983;79: 926–935.
- Berendsen H, Grigera J, Straatsma T. The missing term in effective pair potentials. J Phys Chem. 1987;91(24):6269–6271. doi:10.1021/j100308a038.
- Jorgensen WL, Madura JD. Temperature and size dependence for Monte Carlo simulations of TIP4P water. Mol Phys. 1985;56(6):1381–1392. doi:10.1080/00268978500103111.
- Mahoney MW, Jorgensen WL. A five-site model for liquid water and the reproduction of the density anomaly by rigid, nonpolarizable potential functions. J Chem Phys. 2000;112(20): 8910–8922.10.1063/1.481505.
- Abascal JL, Vega C. A general purpose model for the condensed phases of water: TIP4P/2005. J Chem Phys. 2005;123(23):234505. doi:10.1063/1.2121687.
- Nielsen SO, Lopez CF, Srinivas G, Klein ML. Coarse grain models and the computer simulation of soft materials. J Phys Condens Matter. 2004;16(15):R481–R512. doi:10.1088/0953-8984/16/15/R03.
- Orsi M, Haubertin DY, Sanderson WE, Essex JW. A quantitative coarse-grain model for lipid bilayers. J Phys Chem B. 2008;112(3):802–815. doi:10.1021/jp076139e.
- Wang H, Junghans C, Kremer K. Comparative atomistic and coarse-grained study of water: What do we lose by coarse-graining? Eur Phys J. 2009;E 28:221–229.
- Orsi M, Michel J, Essex JW. Coarse-grain modelling of DMPC and DOPC lipid bilayers. J Phys Condens Matter. 2010;22(15):155106. doi:10.1088/0953-8984/22/15/155106.
- Riniker S, Allison JR, Van Gunsteren F. On developing coarse-grained models for biomolecular simulation: a review. Phys Chem Chem Phys. 2012;14(36):12423–12430. doi:10.1039/c2cp40934h.
- Orsi M, Essex JW. The ELBA force field for coarse-grain modeling of lipid membranes. PLoS One. 2011;6(12):e28637. doi:10.1371/journal.pone.0028637.
- Orsi M, Essex JW. Physical properties of mixed bilayers containing lamellar and nonlamellar lipids: insights from coarse-grain molecular dynamics simulations. Faraday Discuss. 2013;161:249–272. doi:10.1039/C2FD20110K.
- Orsi M. Comparative assessment of the ELBA coarse-grained model for water. Mol Phys. 2014;11:1.
- Orsi M, Ding W, Palaiokostas M. Direct mixing of atomistic solutes and coarse-grained water. J Chem Theory Comput. 2014; 10(10):4684–4693.10.1021/ct500065k.
- Levitt M. Protein folding by restrained energy minimization and molecular dynamics. J Mol Biol. 1983;170(3):723–764. doi:10.1016/S0022-2836(83)80129-6.
- Karplus M, McCammon JA. Molecular dynamics simulations of biomolecules. Nat Struct Mol Biol. 2002;9(9):646–652. doi:10.1038/nsb0902-646.
- Elimelech M, Phillip WA. The future of seawater desalination: energy, technology, and the environment. Science. 2011;333(6043):712–717. doi:10.1126/science.1200488.
- Allen MP, Tildesley DJ. Computer simulation of liquids. 1st ed. Oxford: Oxford Science Publications; 1987.
- Rapaport DC. The art of molecular dynamics simulation. Cambridge: Cambridge University Press; 2004.
- Adams DJ, Adams EM, Hills GJ. The computer simulation of polar liquids. Mol Phys. 1979;38(2):387–400. doi:10.1080/00268977900101751.
- Stoddard SD, Ford J. Numerical experiments on the stochastic behavior of a Lennard-Jones gas system. Phys Rev A. 1973;8(3):1504–1512. doi:10.1103/PhysRevA.8.1504.
- Price S, Stone A, Alderton M. Explicit formulae for the electrostatic energy, forces and torques between a pair of molecules of arbitrary symmetry. Mol Phys. 1984;52(4):987–1001. doi:10.1080/00268978400101721.
- Plimpton S. Fast parallel algorithms for short-range molecular dynamics. J Comput Phys. 1995;117(1):1–19. doi:10.1006/jcph.1995.1039.
- LAMMPS molecular dynamics simulator; [accessed 2014 Oct 10]. Available from: http://lammps.sandia.gov 2014.
- Orsi group website. ; 2014 [accessed 2014 Oct 10]. Available from: http://www.orsi.sems.qmul.ac.uk.
- Schneider T, Stoll E. Molecular-dynamics study of a three-dimensional one-component model for distortive phase transitions. Phys Rev B. 1978;17(3):1302–1322. doi:10.1103/PhysRevB.17.1302.
- Berendsen HJ, Postma JPM, Van Gunsteren WF, DiNola A, Haak J. Molecular dynamics with coupling to an external bath. J Chem Phys. 1984;81(8):3684–3690. doi:10.1063/1.448118.
- Ryckaert JP, Ciccotti G, Berendsen HJ. Numerical integration of the Cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J Comput Phys. 1977;23(3):327–341. doi:10.1016/0021-9991(77)90098-5.
- Price DJ, Brooks CL III. A modified TIP3P water potential for simulation with Ewald summation. J Chem Phys. 2004;121(20):10096–10103. doi:10.1063/1.1808117.
- Hockney RW, Eastwood JW. Computer simulation using particles. New York: Adam Hilger; 1989.
- Riniker S, Van Gunsteren WF. A simple, efficient polarizable coarse-grained water model for molecular dynamics simulations. J Chem Phys. 2011;134(8):084110. doi:10.1063/1.3553378.
- Glättli A, Daura X, Van Gunsteren WF. Derivation of an improved simple point charge model for liquid water: SPC/A and SPC/L. J Chem Phys. 2002;116:9811–9828.
- Horn HW, Swope WC, Pitera JW, Madura JD, Dick TJ, Hura GL, Head-Gordon T. Development of an improved four-site water model for biomolecular simulations: TIP4P-Ew. J Chem Phys. 2004;120(20):9665–9678. doi:10.1063/1.1683075.
- Tironi IG, Van Gunsteren WF. A molecular dynamics simulation study of chloroform. Mol Phys. 1994;83(2):381–403. doi:10.1080/00268979400101331.
- Green MS. Markoff random processes and the statistical mechanics of time-dependent phenomena. II. Irreversible processes in fluids. J Chem Phys. 1954;22(3):398–413. doi:10.1063/1.1740082.
- Kubo R. Statistical-mechanical theory of irreversible processes. I. General theory and simple applications to magnetic and conduction problems. J Phys Soc Jpn. 1957;12(6):570–586. doi:10.1143/JPSJ.12.570.
- Nevins D, Spera F. Accurate computation of shear viscosity from equilibrium molecular dynamics simulations. Mol Simul. 2007;33(15):1261–1266. doi:10.1080/08927020701675622.
- Wang J, Yoo S, Bai J, Morris JR, Zeng XC. Melting temperature of ice Ih calculated from coexisting solid–liquid phases. J Chem Phys. 2005;123(3):036101. doi:10.1063/1.1950647.
- Fernández RG, Abascal JL, Vega C. The melting point of ice Ih for common water models calculated from direct coexistence of the solid–liquid interface. J Chem Phys. 2006;124:144506.
- Jorgensen WL, Jenson C. Temperature dependence of TIP3P, SPC, and TIP4P water from NPT Monte Carlo simulations: seeking temperatures of maximum density. J Comput Chem. 1998;19(10):1179–1186. 10.1002/(SICI)1096-987X(19980730)19:10 < 1179:AID-JCC6>3.0.CO;2-J.
- B´ez LA, Clancy P. Existence of a density maximum in extended simple point charge water. J Chem Phys. 1994;101(11):9837–9840. doi:10.1063/1.467949.
- Vega C, Abascal JLF, Conde MM, Aragones JL. What ice can teach us about water interactions: a critical comparison of the performance of different water models. Faraday Discuss. 2009;141:251–276. doi:10.1039/B805531A.
- Guevara-Carrion G, Vrabec J, Hasse H. Prediction of self-diffusion coefficient and shear viscosity of water and its binary mixtures with methanol and ethanol by molecular simulation. J Chem Phys. 2011;134(7):074508. doi:10.1063/1.3515262.
- Chandra A, Ichiye T. Dynamical properties of the soft sticky dipole model of water: molecular dynamics simulations. J Chem Phys. 1999;111(6):2701–2709. doi:10.1063/1.479546.
- Fennell CJ, Gezelter JD. On the structural and transport properties of the soft sticky dipole and related single-point water models. J Chem Phys. 2004;120(19):9175–9184. doi:10.1063/1.1697381.
- Fennell CJ, Gezelter JD. Computational free energy studies of a new ice polymorph which exhibits greater stability than ice Ih. J Chem Theory Comput. 2005;1(4):662–667. doi:10.1021/ct050005s.
- Liu Y, Ichiye T. Soft sticky dipole potential for liquid water: a new model. J Phys Chem. 1996;100(7):2723–2730. doi:10.1021/jp952324t.
- Michel J, Orsi M, Essex JW. Prediction of partition coefficients by multiscale hybrid atomic-level/coarse-grain simulations. J Phys Chem B. 2008;112(3):657–660. doi:10.1021/jp076142y.
- Orsi M, Sanderson WE, Essex JW. Permeability of small molecules through a lipid bilayer: a multiscale simulation study. J Phys Chem B. 2009;113:12019–12029.
- Orsi M, Essex JW. Permeability of drugs and hormones through a lipid bilayer: insights from dual-resolution molecular dynamics. Soft Matter. 2010;6(16):3797–3808. doi:10.1039/c0sm00136h.
- Orsi M, Noro MG, Essex JW. Dual-resolution molecular dynamics simulation of antimicrobials in biomembranes. J R Soc Interface. 2011;8(59):826–841. doi:10.1098/rsif.2010.0541.
- Molinero V, Moore EB. Water modeled as an intermediate element between carbon and silicon. J Phys Chem B. 2008;113:4008–4016.
- Izvekov S, Voth GA. Multiscale coarse graining of liquid-state systems. J Chem Phys. 2005;123(13):134105. doi:10.1063/1.2038787.
- Wu Z, Cui Q, Yethiraj A. A new coarse-grained model for water: the importance of electrostatic interactions. J Phys Chem B. 2010;114(32):10524–10529. doi:10.1021/jp1019763.
- Wu Z, Cui Q, Yethiraj A. Driving force for the association of hydrophobic peptides: the importance of electrostatic interactions in coarse-grained water models. J Phys Chem Lett. 2011;2(14):1794–1798. doi:10.1021/jz2006622.
- Marrink SJ, Risselada HJ, Yefimov S, Tieleman DP, De Vries AH. The MARTINI force field: coarse grained model for biomolecular simulations. J Phys Chem B. 2007;111(27):7812–7824. doi:10.1021/jp071097f.
- Yesylevskyy SO, Schäfer LV, Sengupta D, Marrink SJ. Polarizable water model for the coarse-grained MARTINI force field. PLoS Comput Biol. 2010;6(6):e1000810. doi:10.1371/journal.pcbi.1000810.
- Riniker S, Eichenberger AP, Van Gunsteren WF. Solvating atomic level fine-grained proteins in supra-molecular level coarse-grained water for molecular dynamics simulations. Eur Biophys J. 2012;41(8):647–661. doi:10.1007/s00249-012-0837-1.
- Darré L, Machado MR, Dans PD, Herrera FE, Pantano S. Another coarse grain model for aqueous solvation: WAT FOUR? J Chem Theory Comput. 2010;6:3793–3807.
- Dougherty RC, Howard LN. Equilibrium structural model of liquid water: evidence from heat capacity, spectra, density, and other properties. J Chem Phys. 1998;109(17):7379–7393. doi:10.1063/1.477344.
- Lishchuk SV, Malomuzh NP, Makhlaichuk PV. Contribution of H-bond vibrations to heat capacity of water. Phys Lett A. 2011;375(27):2656–2660. doi:10.1016/j.physleta.2011.05.049.
- Radhakrishnan R, Trout BL. Nucleation of hexagonal ice (Ih) in liquid water. J Am Chem Soc. 2003;125(25):7743–7747. doi:10.1021/ja0211252.
- Groh B, Dietrich S. Density-functional theory for the freezing of Stockmayer fluids. Phys Rev E. 1996;54:1687.
- Wang J, Apte PA, Morris JR, Zeng XC. Freezing point and solid–liquid interfacial free energy of Stockmayer dipolar fluids: A molecular dynamics simulation study. J Chem Phys. 2013;139(11):114705. doi:10.1063/1.4821455.
- Braun D, Boresch S, Steinhauser O. Transport and dielectric properties of water and the influence of coarse-graining: Comparing BMW, SPC/E, and TIP3P models. J Chem Phys. 2014;140(6):064107. doi:10.1063/1.4864117.
- Marrink SJ, De Vries AH, Mark AE. Coarse grained model for semiquantitative lipid simulations. J Phys Chem B. 2004;108(2):750–760. doi:10.1021/jp036508g.
- Darré L, Tek A, Baaden M, Pantano S. Mixing atomistic and coarse grain solvation models for MD simulations: let WT4 handle the bulk. J Chem Theory Comput. 2012;8:3880–3894.
- Lemmon E, McLinden M, Friend D. Thermophysical properties of fluid systems. In: Mallard WG, Linstrom P, editors. NIST chemistry webbook, NIST standard reference database. Gaithersburg, MD; 2011. http://webbook.nist.gov.
- Paschek D. Temperature dependence of the hydrophobic hydration and interaction of simple solutes: An examination of five popular water models. J Chem Phys. 2004;120(14):6674–6690. doi:10.1063/1.1652015.
- Holz M, Heil SR, Sacco A. Temperature-dependent self-diffusion coefficients of water and six selected molecular liquids for calibration in accurate 1H NMR PFG measurements. Phys Chem Chem Phys. 2000;2(20):4740–4742. doi:10.1039/b005319h.
- Raabe G, Sadus RJ. Molecular dynamics simulation of the dielectric constant of water: the effect of bond flexibility. J Chem Phys. 2011;134(23):234501. doi:10.1063/1.3600337.
- Harris KR, Newitt PJ. Self-diffusion of water at low temperatures and high pressure. J Chem Eng Data. 1997;42(2):346–348. doi:10.1021/je9602935.
- Raabe G, Sadus RJ. Molecular dynamics simulation of the effect of bond flexibility on the transport properties of water. J Chem Phys. 2012;137(10):104512. doi:10.1063/1.4749382.
- Ghosh T, García AE, Garde S. Molecular dynamics simulations of pressure effects on hydrophobic interactions. J Am Chem Soc. 2001;123(44):10997–11003. doi:10.1021/ja010446v.