
Abstract
It is shown that ensemble averages computed in the Gibbs Ensemble with Continuous Fractional Component Monte Carlo (CFCMC GE) are different from those computed in the conventional Gibbs Ensemble (GE). However, it is possible to compute averages corresponding to the conventional GE while performing simulations in the CFCMC GE. In this way, one can benefit from the nice features of CFCMC GE (e.g. more efficient particle exchange) and at the same time compute the ensemble averages that correspond to the conventional GE. As a case study, the equilibrium pressure and densities of the systems of 256 and 512 LJ particles at different reduced temperatures () are computed in the conventional GE and CFCMC GE. The validity of the expressions derived for computation of the thermodynamic pressure and densities corresponding to the conventional GE and computed in the CFCMC GE is examined numerically. The thermodynamic pressure in the conventional GE and CFCMC GE typically differs by at most 4%. It is shown that a very good estimate of the average pressure and densities corresponding to the conventional GE can be obtained by performing simulation in CFCMC GE and ignoring the contributions of the fractional molecule. It is also shown that the fractional molecule does not have an influence on the structure of the liquid, even for very small system sizes (e.g. 40 particles). The approach used here to compute the equilibrium pressure and densities of the conventional GE using the CFCMC GE can be easily extended to other thermodynamic properties and other ensembles.
1. Introduction
Coexistence properties at Vapor–Liquid Equilbria (VLE) are crucial to design many industrial processes.[Citation1–Citation3] Molecular simulations using Monte Carlo algorithms are widely applied to provide information regarding the thermodynamic properties of coexisting phases.[Citation4–Citation6] Since the introduction of Gibbs Ensemble (GE) in 1980s by Panagiotopoulos,[Citation7–Citation9] simulations in this ensemble are frequently used to study VLE of pure components and mixtures.[Citation10–Citation14] Other methods such as histogram reweighting in the grand-canonical ensemble [Citation15,Citation16] can be more efficient to study VLE. However, since the GE is convenient and sufficiently accurate, it is still widely used for simulating phase coexistence of pure components and mixtures.[Citation13,Citation14]
Similar to simulations in the grand-canonical ensemble, GE simulations rely on sufficiently large acceptance probabilities for particle exchanges between the simulation boxes. However, the acceptance probability for particle exchange can be very low when molecules are large or when densities are high (e.g. adsorption close to saturation loading, or liquid phases at low temperatures),[Citation17] even when advanced techniques like Configurational-bias Monte Carlo are used. When the acceptance probability for insertion/deletion is low, it is not straightforward to verify if the two phases have reached equilibrium and that the chemical potentials of a certain component are equal in the simulation boxes. In this case, one should separately check the conditions for chemical equilibrium (equality of pressures, chemical potentials, and temperatures for all components in the two phases). The so-called expanded ensemble methods are among possible solutions to overcome this problem.[Citation18–Citation20] The Continuous Fractional Component Monte Carlo (CFCMC), recently introduced by Shi and Maginn, is one of the most commonly used expanded ensemble approaches.[Citation21–Citation30] Poursaeidesfahani et al. have introduced a more efficient formulation of the GE combined with the CFCMC technique.[Citation31] In this formulation, there is only a single fractional molecule per component which can be in either one of the boxes. The chemical potential can be computed directly without any extra calculations. These authors also showed that the computed chemical potentials are identical to those computed in conventional GE, which was validated for LJ particles and water.[Citation31] For the simple LJ fluid, the acceptance probability for insertion/deletion of particles in CFCMC GE at a reduced temperature is five hundred times larger than in the conventional GE.[Citation31] Although CFCMC improves the acceptance probability of particle exchange, it rises a very important question: How should one relate the properties computed in CFCMC GE simulations to those computed in the conventional GE? As an example, when computing the density of the two phases in CFCMC GE, it is not clear a priori if one should count the fractional molecule or not.[Citation21,Citation22,Citation31] In this paper, we introduce general guidelines on how to relate averages computed in the CFCMC GE to averages in the conventional GE. We consider here thecomputation of pressure and densities in the conventional GE and in the CFCMC GE introduced by Poursaeidesfahani et al. [Citation31]. For both conventional GE and CFCMC GE, we derive equations for thermodynamic pressure of the system. We show that the calculated thermodynamic pressures of the two simulation boxes are exactly equal, and that the thermodynamic pressure of the conventional GE and CFCMC GE are different. We also show that the structure of the liquid is not influenced by the fractional molecule. We show how the expansion of the conventional GE with the fractional molecule affects the average pressure of the two boxes, and how one can compute the pressure corresponding to the conventional GE in the CFCMC GE. The pressure is chosen because of its importance in verification of the equilibrium between the two phases.
This paper is organised as follows. In Section 2, the relevant equations for computing the pressures in the conventional GE, the CFCMC GE [Citation31] and the pressure corresponding to the conventional GE calculated in CFCMC GE are derived. Also, guidelines for computing averages corresponding to the conventional GE and computed in the CFCMC GE are presented. The pressures and densities of the two coexisting phases of LJ particles at various temperatures computed in the conventional GE and the CFCMC GE are presented in Section 3. In this section, the influence of the fractional molecule on the structure of the two phases is also investigated. Our findings are summarised in Section 4.
2. Methodology
In the CFCMC GE formulation introduced by Poursaeidesfahani et al. [Citation31], there is only a single fractional molecule per component which is distinguishable from the whole molecules. In the case of LJ pair interactions, the LJ interactions of the fractional molecule are scaled according to [Citation22]:(1)
where is the scaling parameter with
. The partition function of this system is given by [Citation31]:
(2)
where and
is the thermal wavelength. The fractional molecule can be transferred between the boxes and i indicates the box where fractional molecule is in.
and
are the total internal energy of the whole molecules and the internal energy of the fractional molecule in box i, respectively.
is the total volume and
is the volume of box 1.
equals 1 when
and zero otherwise.[Citation31] Except for the trial moves used for the thermalisation of the system and volume changes, three other trial moves are used to facilitate particle exchanges between the simulation boxes:
Changing the scaling parameter
with
Swapping the fractional molecule between the boxes;
Changing the identity of the fractional molecule with a randomly selected whole molecule in the other simulation box, while keeping the value of
Figure 1. (Colour online) Schematic representation of the additional trial moves in CFCMC GE. The red sphere is the fractional molecule and the green spheres are the whole molecules. (a)(b): changing the scaling parameter
with
. (b)
(c): swapping the fractional molecule between the boxes. (c)
(d): changing the identity of the fractional molecule with a randomly selected whole molecule in the other simulation box, while keeping the value of
constant.
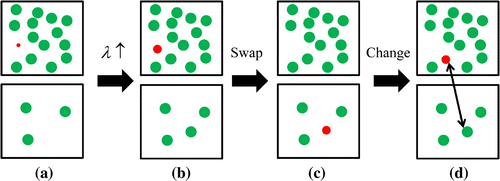
Table 1. Computed pressures and densities in the conventional GE and the CFCMC GE at different reduced temperatures for 256 LJ particles. (Equation (Equation4
(4) )) and
(Equation (Equation5
(5) )) are the pressures in the conventional GE and the CFCMC GE, respectively.
(Equation (Equation6
(6) )) indicates the pressure corresponding to that in the conventional GE and computed in the CFCMC GE.
(Equation (Equation7
(7) )) is the computed pressure in the CFCMC GE, not counting the contributions of the fractional molecule. The exact same definitions apply to the computed densities (Equations (Equation8
(8) )–(Equation11
(11) )). Statistical uncertainties in the last digit are shown in brackets, i.e. 14.21(1) means
. The weight function in the CFCMC GE is calculated iteratively so that the probability distribution
is uniform. The total volume for
and
is
and for
is
.
Table 2. Computed pressures and densities in the conventional GE and the CFCMC GE at different reduced temperatures for 512 LJ particles. (Equation (Equation4
(4) )) and
(Equation (Equation5
(5) )) are the pressures in the conventional GE and the CFCMC GE, respectively.
(Equation (Equation6
(6) )) indicates the pressure corresponding to that in the conventional GE and computed in the CFCMC GE.
(Equation (Equation7
(7) )) is the computed pressure in the CFCMC GE, not counting the contributions of the fractional molecule. The exact same definitions apply to the computed densities (Equations (Equation8
(8) )–(Equation11
(11) )). Statistical uncertainties in the last digit are shown in brackets, i.e. 14.10(1) means
. The weight function in the CFCMC GE is calculated iteratively so that the probability distribution
is uniform. The total volume for
is
and for
,
and for
, the total volume is
.
Figure 2. (Colour online) (a) Radial distribution functions and (b)
for 4 LJ particles at
and
. (c) Radial distribution functions
and (d)
for 40 LJ particles at
and
. To reduce the number of particles and amplify the effect of the fractional molecule, the cutoff radius is reduced to
.
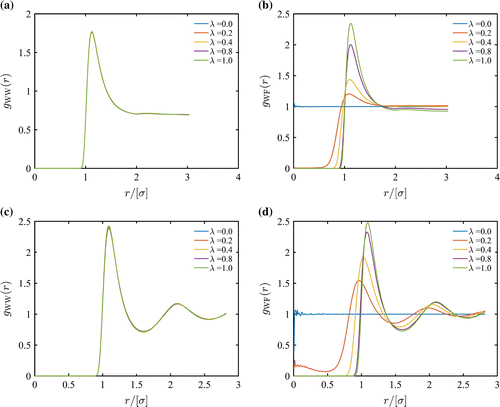
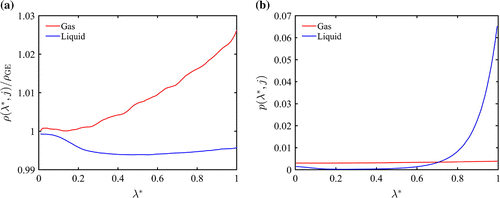
Figure 3. (Colour online) (a) for the two phases as a function
.
is the density of box j averaged over the configurations in which the fractional particle is in box j with
. Note: in calculation of these densities, the fractional molecule was disregarded. (b) Probability distribution of
for the two phases for 256 LJ particles at
.
2.1. Computation of the pressure
In molecular simulations, the thermodynamic pressure is usually computed by averaging over the instantaneous microscopic pressures. In any NVT ensemble, the general expression for the thermodynamic pressure P is [Citation33–Citation35](3)
Considering the fact the GE is a special case of the NVT ensemble, Equation (Equation3(3) ) is applicable to the GE and CFCMC GE. Starting from the partition function of the conventional GE and following the steps presented in the Supporting Information, the pressure in the conventional GE is obtained by the conventional virial equation [Citation35,Citation36]:
(4)
where and
are the distance and the force acting between particles ‘a’ and ‘b’ in box ‘j’ (assuming pair potentials). The first term on the right hand side of Equation (Equation4
(4) ) is the ideal gas contribution and the second term is commonly known as the virial contribution.[Citation35] The labeling of the boxes is arbitrary, therefore, the same equation is obtained for the other box. Since there is only one thermodynamic pressure for the system, the pressures of the two boxes are on average equal. In the same way, as shown in the Supporting Information, the thermodynamic pressure in the CFCMC GE is computed from:
(5)
In this equation, the contribution of the fractional molecule is included in the ideal gas part and in the virial part. Thethermodynamic pressures in the CFCMC GE (Equation (Equation5(5) )) and conventional GE (Equation (Equation4
(4) )) are clearly not identical. As shown in the Supporting Information, it is possible to compute the pressure corresponding to the conventional GE while performing simulations in the CFCMC GE:
(6)
The difficulty associated with computing using Equation (Equation6
(6) ) is that only the states in which the value of
equals zero are contributing to the ensemble average. Therefore, long simulations may be required to obtain reliable pressures especially for the liquid phase. Assuming that there is no correlation between the volume and the number of whole molecules, and also no correlation between the volume and the virial part of the pressure, Equation (Equation6
(6) ) reduces to
(7)
where the notation ‘’ indicates that contribution of fractional molecule in virial part of the pressure should be disregarded. It is important to note that
is an approximation for the pressure corresponding to the GE, and unlike
,
, and
, the quantity
may not be equal for both simulation boxes. In the gas phase, particles are usually far enough from each other that the contribution of the virial part in the total pressure is limited and not correlated with the volume of the box. However, in the liquid phase, stronger correlation between the contribution of the viral part of the pressure and the volume of the box is expected. The validity of the simplification of Equation (Equation7
(7) ) is numerically investigated in the next section. One can use the exact same approach to define different densities:
(8)
(9)
(10)
(11)
where is the average density of box j computed in the conventional GE,
is the average density of box j computed in the CFCMC GE (including the fractional molecule),
is the average density of box j computed in the CFCMC GE only when the value of
equals zero excluding contribution of the fractional molecule, and
is the average density of box j computed in the CFCMC GE excluding the fractional molecule and averaged over all values of
.
3. Simulation details
To examine the validity of equations provided in the Supporting Information, the VLE of a system with 256 and 512 LJ particles is investigated at three different reduced temperatures (, 0.8, 0.95). The LJ potentials are truncated and shifted at
. Simulations are carried out in the conventional GE and the CFCMC GE. The LJ parameters
and
are used as units of length and energy respectively. Consequently, all calculated properties are in reduced units. A biasing function
is computed iteratively to obtain a flat probability distribution of
and that the fractional molecule is located with equal probability in both boxes. After 2 million equilibration cycles, a long production (500 million cycles) run is carried out to reduce the uncertainties in the values computed for pressures introduced in Equations (Equation4
(4) )–(Equation7
(7) ). The number of Monte Carlo steps per cycle equals the total number of molecules in the system, with a minimum of 20. For more simulation details the reader is referred to Ref. [Citation31].
4. Results
To compute the pressures and densities, simulations are performed in the conventional GE and the CFCMC GE. In Tables and , the average pressures derived in Equations (Equation4(4) )–(Equation7
(7) ) and corresponding densities for the gas and liquid phases are shown for three different reduced temperatures (
, 0.8, 0.95) and for two system sizes (256 and 512 particles).
An important point in Tables and is the fact that the thermodynamic pressures of the two phases computed in the conventional GE () are equal. The thermodynamic pressures of the two phases computed in CFCMC GE (
) are also equal. However, the thermodynamic pressures of the two ensembles, CFCMC GE and the conventional GE (
, and
) are clearly not equal. As discussed in the previous section, the presence of the fractional molecule in the CFCMC GE simulations results in an increase in the thermodynamic pressure. However, the computed values for
and
are nearly identical. In the same way, densities computed in CFCMC GE including the fractional molecule (
) are not equal to those computed in the conventional GE (
). However, densities corresponding to the conventional GE but computed in CFCMC GE (
) are equal to densities computed in the conventional GE (
). This numerically confirms the validity of the derivations provided for computing properties corresponding to the conventional GE in the CFCMC GE. Only the states in which the value of
is zero are contributing to the
. As a result, the uncertainties associated with
values are much larger than the other ensemble averages.
The values of computed for the gas phase are very close to the values computed for
and
(deviation less than 0.2%). This is not the case for
computed for the liquid phase (deviation up to 4%). The gas phase density of the conventional GE can be accurately estimated using
(see Tables and ). Since the contribution of the virial part in the pressure of the gas phase is negligible and the ideal gas part is defined by the density,
for the gas phase can be used as an estimate of
and
.
In the liquid phase, the presence of a fractional molecule (with scaling parameter larger than zero) may influence the density and structure of the liquid phase. Radial Distribution Functions (RDFs) can be used to investigate the effect of the fractional molecule on the structure of the phases. The CFCMC GE system can be considered as a binary system, therefore, there are three different RDFs (Whole–Whole),
(Whole-Fractional) and
(Fractional–Fractional). Since there is only one fractional molecule,
is always zero. In Figure ,
and
are plotted for different densities and values of
. To reduce the number of particles and amplify the effect of the fractional molecule, the cutoff radius is reduced to
and minimum box size and number of particles are used for these simulations. Simulations are performed in the NVT ensemble with only a single fractional molecule with a fixed value of
. As shown in Figure ,
are almost identical for all values of
. To test the extreme case, the interactions of the fractional molecule with the whole molecules were changed in such a way that the fractional molecule is acting as an attraction site without any repulsive potential (
). For
and
, hardly any changes were observed in the RDFs, when the density was close to typical liquid densities (not shown here). This indicates that the structure of the liquid is not affected by the fractional molecule.
In Figure (a), the dependency of the density (excluding the fractional molecule) to the value of is investigated. An interesting point is that the densities corresponding to the conventional GE are only recovered when the value of
is close to zero. It can be observed that the density of the gas phase increases and the density of the liquid phase decreases as
changes from 0 to 1. In Figure (b), the unbiased probability distribution of
in the two phases is shown. The fractional molecule is most of the times in the liquid phase. As a result,
for the gas phase is close to
for the gas phase. It can be seen that the fractional molecule is most of the times in the liquid phase with
close to one. In this case, the density of the liquid phase in underestimated. Therefore, one would expect the
to be slightly lower than the values of
for the liquid phase. This is confirmed by data presented in Tables and . Underestimation of the density of the liquid phase can influence both the ideal part and virial contribution of
. This explains why the values reported for
are slightly off.
5. Conclusions
In this study, we showed that there are differences between the averages computed in the CFCMC GE and those computed in the conventional GE. Although these differences may be limited for many properties, it is important to know that they exist. For example, the thermodynamic pressures in the conventional GE and CFCMC GE are different and typically differ by at most 3% for a system of 256 LJ particles. We also introduced guidelines for computing the averages corresponding to the conventional GE and computed in the CFCMC GE. We showed analytically and numerically that these values are identical to values computed in the conventional GE. As an example, we computed the pressure and density in the conventional GE and CFCMC GE introduced by Poursaeidesfahani et al. [Citation31]. The pressure and densities corresponding to the conventional GE and computed in the CFCMC GE are equal to the pressure and densities computed in the conventional GE. However, due to the limited sampling (only when ) of these averages in CFCMC GE, long simulations are required to obtain reliable results. For the gas phase, the pressure is predominately defined by the ideal gas part. Therefore, using the estimation provided by Equation (Equation7
(7) ) (i.e. ignoring the fractional molecule in the ideal gas part and the virial part), one can compute the pressure corresponding to the conventional GE from the gas phase of a CFCMC GE simulation and still sampling for all values of
. We also showed that the structures of the two phases are not influenced by the fractional molecule.
Supplemental data
Download PDF (269.3 KB)Additional information
Funding
Notes
No potential conflict of interest was reported by the authors.
Related Research Data
References
- Walas SM. Phase equilibria in chemical engineering. Boston: Butterworth; 1985.
- Nebig S, Gmehling J. Prediction of phase equilibria and excess properties for systems with ionic liquids using modified UNIFAC: typical results and present status of the modified UNIFAC matrix for ionic liquids. Fluid Phase Equilib. 2011;302:220–225.
- Neroorkar K, Schmidt D. Modeling of vapor-liquid equilibrium of gasoline-ethanol blended fuels for flash boiling simulations. Fuel. 2011;90:665–673.
- Potoff JJ, Siepmann JI. Vapor-liquid equilibria of mixtures containing alkanes, carbon dioxide, and nitrogen. AlChE J. 2001;47:1676–1682.
- Errington JR, Boulougouris GC, Economou IG, et al. Molecular simulation of phase equilibria for water-methane and water-ethane mixtures. J Phys Chem B. 1998;102:8865–8873.
- Vorholz J, Harismiadis V, Rumpf B, et al. Vapor-liquid equilibrium of water, carbon dioxide, and the binary system, water+carbon dioxide, from molecular simulation. Fluid Phase Equilib. 2000;170:203–234.
- Panagiotopoulos AZ. Direct determination of phase coexistence properties of fluids by Monte Carlo simulation in a new ensemble. Mol Phys. 1987;61:813–826.
- Baus M, Rull L, Ryckaert J. Observation, prediction and simulation of phase transitions in complex fluids. Vol. 460, Edinburgh: Science \ & Business Media; 2012.
- Panagiotopoulos AZ, Quirke N, Stapleton M, et al. Phase equilibria by simulation in the Gibbs ensemble: alternative derivation, generalization and application to mixture and membrane equilibria. Mol Phys. 1988;63:527–545.
- Neubauer B, Boutin A, Tavitian B, et al. Gibbs ensemble simulations of vapour-liquid phase equilibria of cyclic alkanes. Mol Phys. 1999;97:769–776.
- De Pablo JJ, Prausnitz JM. Phase equilibria for fluid mixtures from Monte-Carlo simulation. Fluid Phase Equilib. 1989;53:177–189.
- Green DG, Jackson G, de Miguel E, et al. Vapor-liquid and liquid-liquid phase equilibria of mixtures containing square-well molecules by Gibbs ensemble Monte Carlo simulation. J Chem Phys. 1994;101:3190–3204.
- Fetisov EO, Siepmann JI. Prediction of vapor-liquid coexistence properties and critical points of polychlorinated biphenyls from Monte Carlo simulations with the TraPPE force field. J Chem Eng Data. 2015;60:3039–3045.
- Dinpajooh M, Bai P, Allan DA, et al. Accurate and precise determination of critical properties from Gibbs ensemble Monte Carlo simulations. J Chem Phys. 2015;143:114113.
- Ferrenberg AM, Swendsen RH. Optimized Monte Carlo data analysis. Phys Rev Lett. 1989;63:1195–1198.
- Ferrenberg AM, Swendsen RH. New Monte Carlo technique for studying phase transitions. Phys Rev Lett. 1988;61:2635–2638.
- Poursaeidesfahani A, Torres-Knoop A, Rigutto M, et al. Computation of the heat and entropy of adsorption in proximity of inflection points. J Phys Chem. C. 2016;120:1727–1738.
- Rane KS, Murali S, Errington JR. Monte Carlo simulation methods for computing liquid-vapor saturation properties of model systems. J Chem Theory Comput. 2013;9:2552–2566.
- Escobedo FA, de Pablo JJ. Monte Carlo simulation of the chemical potential of polymers in an expanded ensemble. J Chem Phys. 1995;103:2703–2710.
- Lyubartsev AP, Martsinovski AA, Shevkunov SV, et al. New approach to Monte Carlo calculation of the free energy: method of expanded ensembles. J Chem Phys. 1992;96:1776–1783.
- Shi W, Maginn EJ. Continuous fractional component Monte Carlo: an adaptive biasing method for open system atomistic simulations. J Chem Theory Comput. 2007;3:1451–1463.
- Shi W, Maginn EJ. Improvement in molecule exchange efficiency in Gibbs ensemble Monte Carlo: development and implementation of the continuous fractional component move. J Comput Chem. 2008;29:2520–2530.
- Shi W, Maginn EJ. Atomistic simulation of the absorption of carbon dioxide and water in the ionic liquid 1-n-Hexyl-3-methylimidazolium bis(trifluoromethylsulfonyl)imide ([hmim][Tf2N]. J Phys Chem B. 2008;112:2045–2055.
- Maginn EJ. Atomistic simulation of the thermodynamic and transport properties of ionic liquids. Acc Chem Res. 2007;40:1200–1207.
- Kelkar MS, Shi W, Maginn EJ. Determining the accuracy of classical force fields for ionic liquids: atomistic simulation of the thermodynamic and transport properties of 1-Ethyl-3-methylimidazolium ethylsulfate ([emim][EtSO4]) and its mixtures with water. Ind Eng Chem Res. 2008;47:9115–9126.
- Zhang X, Huo F, Liu Z, et al. Absorption of CO2 in the ionic liquid 1-n-hexyl-3-thylimidazolium tris(pentafluoroethyl)trifluorophosphate ([hmim][FEP]): a molecular view by computer simulations. J Phys Chem B. 2009;113:7591–7598.
- Chen Q, Balaji SP, Ramdin M, et al. Validation of the CO2/N2O analogy using molecular simulation. Ind Eng Chem Res. 2014;53:18081–18090.
- Ramdin M, Balaji SP, Vicent-Luna JM, et al. Solubility of the precombustion gases CO2, CH4, CO, H2, N2, and H2S in the ionic liquid [bmim][Tf2N] from Monte Carlo simulations. J Phys Chem C. 2014;118:23599–23604.
- Ramdin M, Balaji SP, Torres-Knoop A, et al. Solubility of natural gas species in Ionic liquids and commercial solvents: experiments and Monte Carlo simulations. J Chem Eng Data. 2015;60:3039–3045.
- Shi W, Siefert NS, Morreale BD. Molecular simulations of CO2, H2, H2O, and H2S gas absorption into hydrophobic poly(dimethylsiloxane) (PDMS) solvent: solubility and surface tension. J Phys Chem C. 2015;119:19253–19265.
- Poursaeidesfahani A, Torres-Knoop A, Dubbeldam D, et al. Direct free energy calculation in the continuous fractional component Gibbs ensemble. J Chem Theo Comput. 2016;12:1481–1490.
- Poursaeidesfahani A, Torres-Knoop A, Dubbeldam D, et al. 4TU Datacenter. doi:10.4121/uuid:14e501bc-2408-41bc-90d5-5f4588604849.
- de Miguel E, Jackson G. The nature of the calculation of the pressure in molecular simulations of continuous models from volume perturbations. J Chem Phys. 2006;125:164109.
- Gray CG, Gubbins KE, Joslin CG. Theory of molecular fluids 2: applications. International series of monographs on chemistry. New York (NY): Oxford University Press; 2011.
- Allen MP, Tildesley DJ. Computer simulation of liquids. New York (NY): Oxford University Press; 1989.
- Frenkel D, Smit B. Understanding molecular simulation: from algorithms to applications. San Diego (CA): Academic Press; 2002.