
Abstract
In this work we set out to evaluate the computational performance of several popular Monte Carlo simulation programs, namely Cassandra, DL Monte, Music, Raspa and Towhee, in modelling gas adsorption in crystalline materials. We focus on the reference case of adsorption in IRMOF-1 at 208 K. To critically assess their performance, we first establish some criteria which allow us to make this assessment on a consistent basis. Specifically, the total computational time required for a program to complete a simulation of an adsorption point, consists of the time required for equilibration plus time required to generate a specific number of uncorrelated samples of the property of interest. Our analysis shows that across different programs there is a wide difference in the statistical value of a single MC step, however their computational performance is quite comparable. We further explore the use of energy grids and energy bias techniques, as well as the efficiency of the parallel execution of the simulations. The test cases developed are made openly available as a resource for the community, and can be used for validation and as a template for further studies.
1. Introduction
Recent advances in synthesis of novel porous materials, such as metal–organic frameworks (MOFs), zeolitic imidazolate frameworks (ZIFs) and polymers with intrinsic microporosity (PIMs) have a profound impact on the way we now approach design of technologies and applications based on these materials. Indeed, it is not possible to test the thousands of already discovered MOFs, ZIFs, PIMs and related materials in the context of each potential application, while the best material for a particular purpose may exist among those not yet synthesised, but hypothetically possible structures within these classes. Hence the idea of computational screening of materials, the new starting point of process design and optimisation, which aims to identify the best material or group of materials for a particular application before the actual experimental effort is committed.
Broadly speaking, computational screening can be separated into two phases. The first phase involves building a database of possible structures, both real and hypothetical. The modular nature of these new materials allows one to guess the structure of not yet synthesised materials, using a systematic variation and assembly of the building blocks through what can be best described as molecular Lego approaches. In the second phase, computational methods are used to assess the key characteristics of the materials within the database, based on the performance metrics associated with a particular application. Although in principle this strategy can be employed in the context of any application, the tunable porosity and surface area of MOFs and ZIFs makes them particularly interesting for adsorption applications, such as methane storage and carbon capture, and this is what most of the recent screening studies have been focused on. Prominent examples of this approach include studies from Snurr and co-workers [Citation1], Smit and co-workers [Citation2] and Sholl and co-workers [Citation3]. For comprehensive reviews in the field of molecular simulation of adsorption processes in MOFs see Refs. [Citation4–Citation8].
Application of these virtual screening strategies is associated with several challenges. Firstly, the screening algorithms must be computationally efficient to be able to sieve through potentially millions of structures under a number of conditions of interest; secondly, the accuracy of the molecular simulation methods crucially depends on the availability of accurate forcefields. Although several groups have made substantial contributions to the development of the parameters for several important classes of materials [Citation9–Citation13], a fully comprehensive and transferrable forcefield for MOFs, ZIFs and related materials remains elusive. Finally, the third challenge is associated with the transition from the predictions of the virtual screening to the actual processes and applications [Citation14,Citation15]. At this stage a number of additional factors, such as stability of the materials and cost, become important. This article deals with the first challenge, related to the computational efficiency.
Unless one is interested in adsorption at very low pressures (in other words, in the low loading, Henry’s law regime), calculation of loading in a material at a specific pressure requiresa grand canonical Monte Carlo (GCMC) simulation and its variants, such as the configurational bias GCMC (CB-GCMC) for flexible molecules. A recent special issue of molecular simulation reviewed several of the currently existing Monte Carlo programs, presented by their developers [Citation16]. It is quite clear that different academic groups adopted different philosophies, programming techniques, algorithms, and target problems in the development of their computational tools. The special issue also highlighted an important problem. In the field of molecular dynamics (MD) healthy competition between several programs and the appetite of the biological community for ever longer trajectories and larger systems led to systematic assessment of the computational efficiency of the programs, their propensity to parallelism on different platforms [Citation17] as well as the development of documented case studies that can be used as benchmarks [Citation18].
No such effort has been undertaken in the community using Monte Carlo programs in the context of adsorption problems. Typically, the efficiency of the new programs is tested against the existing in-house programs of a specific group, but the efficiency and the accuracy of the programs from across different groups has not been systematically assessed or explored. We believe this is an important undertaking in order to establish the best starting point and algorithms for the development of the next generation of programs, to share best practices and methodologies, and to establish references cases which can be used by the developers around the world to validate their results.
This defines the remit of the current article. Our original idea was quite simple: to survey the existing, freely available programs for GCMC simulations; explore how accurate they are in reproducing reference data and how fast they are in a sense of the computational resources required to get to the reference data within a certain accuracy. This proved to be a challenging task.
Firstly, it is important to explore how and why different programs can deviate in their predictions and also the possible extent of these deviations. Consider the adsorption of carbon dioxide in a MOF, such as IRMOF-1 [Citation19] which will be our main case study as justified below. Of course for the comparison of two programs we need to set all the parameters for the two runs to identical values. This includes forcefield parameters, mixing rules, distances for the potential cut-off and rules for handling the potential beyond the cut-off distance, number of trial Monte Carlo moves and the distribution of the weights among the available moves, coordinates for the input crystal structure and so on. This nevertheless leaves a substantial amount of technical details outside of what a user of the program can control or may be aware of. This includes conventions on the precision of the irrational numbers and constants, such as Boltzmann constant and ; internal procedures for the control of a trial move acceptance ratio (which may or may not be automatically adjusted to be at the optimum value). Although two programs may use the Ewald summation to calculate the interactions involving partial charges, the way parameters are set for this calculation may vary between the programs (i.e. there is some automatic adjustment depending on the structure or the program uses some fixed, pre-set values based on the experience with similar systems). The actual access and control of these aspects of the Monte Carlo simulation naturally depends on the specific program. From the end user point of view, the understanding of these parameters and methodology space, of what can and cannot be controlled, heavily depends on high quality documentation associated with the program and a decent collection of case studies illustrating the role of different parameters.
Secondly, performance of a program depends on a substantial number of factors that make consistent comparison quite difficult. This include using (or not) pre-calculated potential grids or maps as oppose to the on-the-fly calculation of all interactions; using (or not) some additional biasing techniques; using (or not) cell and neighbour lists; methods for calculation of electrostatic interactions (Ewald and its variants vs. cut-off based methods) and, of course, compilers, algorithms, in other words aspects of the program that we actually want to assess.
Finally, one has to define some meaningful criteria for two simulations performed by two different programs to converge to a result of the same statistical uncertainty. In general, this analysis involves two steps. Firstly, it is important to establish the duration of the equilibration stage of the simulations, within which there is a systematic drift of the running average of the property of interest. Secondly, within the sampling stage, a sufficient number of uncorrelated samples of the property of interest should be accumulated. Here, the main property of interest is the amount of adsorbed.
The time required for a simulation to equilibrate and the rate at which uncorrelated data are produced is in fact a function of the program, and this will be the basis of our assessment. There is a substantial amount of research on convergence of the Monte Carlo methods [Citation20]. However, the idea to explore statistically independent samples of the system properties, surprisingly, is still not a common practice in the adsorption simulation community.
1.1. Grand canonical Monte Carlo simulations
The problem of interest here is the adsorption of small molecules (, methane, hydrogen) in crystalline porous materials, prompted by the recent surge of interest in computational screening approaches to carbon capture, methane storage and other applications. Within the scope of this study both the adsorbate molecules and the porous material are treated as rigid structures. The volume, V, and temperature, T, of the system are fixed, and the specified value of the chemical potential,
, establishes thermodynamic equilibrium between the system and the bulk reservoir, serving as a source and sink of adsorbate molecules. From the statistical–mechanical point of view, the system corresponds to the grand-canonical ensemble (
), for which Metropolis Monte Carlo method serves as a conventional simulation technique of choice.
Within this method, configurations of the system are generated via a set of standard trial moves; translation, rotation (in case of rigid molecular species), insertion and deletion, with the following acceptance probability applied to ensure the Boltzmann distribution of the generated states:
| (a) | Translation: | ||||
| (b) | Rotation: | ||||
| (c) | Insertion: | ||||
| (d) | Deletion: | ||||
where is the rotational partition function for a single rigid molecule, equal to 1 for a single particle molecule, and
is the thermal de Broglie wavelength:
(6)
where h is Planck’s constant and m is the molecule mass [Citation21].
2. Methodology
2.1. Case study:  adsorption in IRMOF-1
adsorption in IRMOF-1
As has been already discussed in the introduction, computational screening and optimisation of MOFs and ZIFs for carbon capture applications has been a rapidly developing area of research, driven by both the new opportunities emerging in the material science and the societal importance of the problem. For this reason, the adsorption of in IRMOF-1 was selected as the case study.
IRMOF-1 is one of the earliest reported MOFs, with a substantial amount of experimental and simulation data accumulated on its structural and adsorptive properties [Citation22–Citation24].
The study of Walton et al. [Citation24] provides one of the first examples of both experimental and simulation studies of adsorption in a MOF (specifically, IRMOF-1). Six adsorption isotherms were reported at 195, 208, 218, 233, 273 and 298 K. Two isotherms at lower temperatures (195 and 208 K) feature a sharp transition of the adsorbed density associated with the capillary condensation of
within the pores of IRMOF-1. The authors argued that it was the Coulombic term of the fluid–fluid interactions responsible for the shape of the isotherms.
Following this original study, adsorption in IRMOF-1 at 208 K has been used as a tutorial case study in Sarkisov’s group for incoming research students and staff. The location of the transition, as well as the other features of the isotherm, proved to be sensitive to the parameters of the model, cut-off distances, and interaction terms included. For example, in the original study by Walton et al. [Citation24] the Coulombic interactions between
and IRMOF-1 were not considered, and yet, if included, they shift the isotherm toward lower pressure values.
In fact, one of the motivations for this study was the significant amount of effort and attention to detail required for different programs to generate exactly the same result. This highlighted the importance of the consistency between the parameters and methods used, and prompted us to produce and document this comparison for other available programs. Hence, in the case study we will focus specifically on adsorption at 208 K.
Table 1. Summary of the GCMC programs studied.
2.2. Programs under consideration
The special issue of the Molecular Simulation [Citation16], in particular the review by Dubbeldam et al. [Citation25] and our own experience helped us to identify five commonly used, free programs for molecular simulation of adsorption. All programs are distributed under a GNU GPL license, except DL Monte which is distributed under a custom academic license which enables it to be used freely for academic and other non-commercial work. The license for DL Monte does not allow distribution of the source code to third parties. This may lead to obstacles in the future in reproducing scientific data which requires consistency in both the simulation setups as well as in the program used to execute these setups.
The programs studied and their relevent capabilities are summarised in Table , while for the complete description of all capabilities within each program we refer the reader to the respective original publications. It should be emphasised that at no point have we made any alterations to the source code of the programs under study. We have downloaded each program as it is made available to users and treated it strictly as a black box.
All simulations were ran on identical hardware, using single cores of Intel Xeon E5 2360 v3 nodes, running Scientific Linux version 7.2. Each program was compiled using version 16.0 of the Intel compilers with the compilation flags ‘-O3 -xcore-AVX2 -ip -ipo’. This combination of software and hardware is typical of most modern high performance CPU based supercomputers.
2.2.1. Energy grids and energy-bias GCMC
The calculation of pairwise energies between atoms is by far the single most time consuming step in the process of GCMC simulation. For a single fluid atom i the contribution to potential energy can be expressed as:(7)
where U represents the potential energy, the position of an atom, i and j are indices of the atoms, and the two summations are performed over fluid atoms and solid atoms respectively. Since the porous material considered in this study is treated as a rigid structure, it is possible to precalculate and store solid–fluid interactions. As described elsewhere [Citation31], the simulation volume can be divided into a regular grid and for each atom type in the adsorbate the corresponding potential grid is calculated by placing the probe atom onto each grid point and calculating its interaction with the solid framework. In case of Coulombic interactions the probe placed in the grid is a single +1 charge. Although it requires a set of additional ‘upfront’ calculations, this procedure is needed only once for all pressure points and temperatures. The potential energy contribution of a single fluid atom can then be given as:
(8)
where the summation is now only performed over other fluid atoms, while the solid–fluid interaction is approximated by interpolating within the potential grid, alleviating the need for on-the-fly calculations of the solid–fluid interactions. We will refer to this element of the simulation setup as an energy grid, however it is also known as a potential map [Citation28]. Of the programs examined in this work, only Raspa and Music are capable of using this technique.
Using energy grids also opens a possibility for the improvement of sampling efficiency via so-called energy-biasing techniques [Citation32]. Here it is recognised that the solid structure of the framework (zeolite or MOF) may occupy a substantial portion of the simulation cell and choosing a position for the potential molecule insertion at random will likely lead to a large number of rejections (due to the positions overlapping with the structure of the material). Furthermore, certain positions within the available porous space will be preferred for the insertion (at an optimal distance from the atoms of the framework), compared to other locations, such as in the center of a large cavity where the interactions with the framework structure can be quite weak. Hence the idea of the energy-biasing method: bias selection of the trial locations for the molecule insertion towards regions of favourable interaction with the framework structure. For this the location of the insertion is selected from w cubelets according to a weight assigned to each cubelet. Specifically, this weight is based on the energy of interaction of the probe atom placed in the center of cubelet z within the framework:
(9)
where the sum in the denominator is over all grids. This method requires a single energy grid for a probe atom of choice and naturally, if the program uses energy grids in general, it should also invoke energy biasing as described above since it does not require any additional calculation. In particular, this approach is used by Music [Citation28].
2.2.2. Parallelism
Two of the programs investigated have the ability to accelerate simulations through using multiple CPU cores. Cassandra uses OpenMP to distribute the calculation of contributions to the total energy, both Lennard-Jones and electrostatics. For the Lennard-Jones contribution within the summation of Equation (Equation7(7) ), it is possible to assign cores to different j indices and calculate each contribution simultaneously. For electrostatics an Ewald summation is used, and it is possible to calculate different k-vectors independently across different cores. OpenMP is a shared memory protocol, which limits the extent to which the problem can be split to the number of cores on a single node, typically around 12–24. DL Monte uses a similar strategy, however it parallelises calculation of the electrostatic interactions only and uses MPI, rather than OpenMP. Towhee also uses MPI, but instead of parallel execution of a single simulation, it runs several parallel simulations for each point on the adsorption isotherm in a so-called jobfarm or task-based parallelism fashion. MPI is a distributed memory paradigm, and so there is no upper limit on the number of cores which can be included, although there will be inevitably diminishing returns in terms of computational efficiency.
2.3. Forcefield and simulation setup details
For the selected system of in IRMOF-1 at 208 K we consider three different forcefield setups. In the first setup only Lennard-Jones interactions between
molecules and between
and IRMOF-1 are considered. In the second setup, we further include Coulombic interactions between the molecules of
. The final setup has all interaction terms considered, including the Coulombic
–IRMOF-1 contribution. This three-level approach allows us to revisit the issue of the role of the different terms in the adsorption behavior of
in IRMOF-1 and individually benchmark the computational cost associated with the different terms of the interaction energy.
Pressures of 5, 10, 20, 30, 40, 50, 60, and 70 kPa are modelled, with the Peng–Robinson equation of state [Citation33] used to calculate the fugacity and chemical potential of the fluid phase across all programs. The solid framework consists of replicas of the crystal unit cell for IRMOF-1, resulting in 3392 atoms of the framework, and up to 1600
molecules at the highest loadings. Here we use DREIDING [Citation34] parameters for the atoms of IRMOF-1, charges for IRMOF-1 from Yazaydın et al. [Citation35] and TraPPE [Citation36] parameters for
.
While the exact implementation of the MC moves is beyond our control, we have attempted to make the simulation setups as consistent as possible across all programs. All analysis in this work is based on the number of MC steps. Raspa instead defines simulation length as a number of cycles, where a cycle is attempted MC moves, where N is the number of molecules in the system. Throughout this work we have translated cycles to steps through knowing the number of adsorbed molecules. Each MC step has an equal probability of performing an insertion, deletion, rotation or translation move. All the parameters and the details of the simulation setups are provided as case studies in Supplementary Material.
3. Defining computational performance
For any meaningful comparison of the performance of different simulation programs, we first need to precisely define how it is measured. In benchmarks of MD simulations it is typical to express performance as a measure of the number of time steps that a program can complete in a given time [Citation37]. Two different programs using the time step of the same size and simulate the same number of time steps should in principle explore the same volume of the phase space and arrive at the same statistical averages of the properties of interest. From this point of view, the number of MD steps performed per unit time can be seen as a direct measurement of data generated per unit time.
The situation is different in Monte Carlo simulations, where each step represents an attempted change in the system which may or may not be accepted. Equations (Equation1(1) )–(Equation4
(4) ) provide the foundation for the most basic Metropolis algorithm, however different programs may have more advanced acceptance criteria which aim to increase the sampling efficiency through various type of biasing. Although, more complex biasing moves may come with an additional computational cost, the resulting simulation scheme may be much more efficient in sampling the phase space.
Therefore depending on the exact implementation of MC moves within a program a fixed number of steps may traverse a differing volume of the phase space. This means that a direct comparison of the rate with which an MC program performs a fixed number of steps, similarly to MD simulations, is an incomplete metric of performance. Instead, we must measure both the rate with which steps are performed as well as the rate with which these steps traverse the phase space to arrive to the expected statistical averages. As we will further argue later in this section, it is a product of these two rates which quantifies the performance of the program.
Each GCMC simulation consists of two stages. In the first, so-called equilibration stage, the properties of the system drift from the initial conditions until they stabilise around some average values. Once the system reached this point, we can commence the second, sampling stage, where statistical averages of the properties of interest are accumulated. To assess computational performance of the program we need to measure how long it takes for the program to reach the sampling stage and then apply our definition of the performance of a Monte Carlo simulation in terms of data generated per MC step in the second stage of the simulation.
In the following three sections we will describe our methodology for measuring computational performance, from defining the equilibration and sampling stages of a simulation, defining the rate at which sampling occurs and the method by which we timed these processes. We have endeavoured to make this analysis as automated as possible, both to make the results as impartial as possible and to ensure that this analysis can be reproduced independently by other researchers. All preparation of inputs and implementation of the analysis was achieved using a combination of the Python packages datreant [Citation38], matplotlib [Citation39], MDAnalysis [Citation40,Citation41], numpy [Citation42] and pandas [Citation43]. Examples of the analysis performed are also made available in Supplementary Material.
3.1. Equilibration stage
In this section we explain how we determine the duration of the equilibration stage in a GCMC simulation. Figure shows a typical evolution of the number of adsorbed molecules in the system as a function of the number of MC steps. Initially the system is empty, and as the simulation progresses the number of molecules rapidly increases to about 135 molecules. For the remaining part of the run, the instantaneous values of the number of molecules in the system fluctuate around some average value. The visual inspection of Figure gives us a fairly good understanding of the boundary between the equilibration and sampling stages. In fact, in the MC community such a visual inspection is still commonly used to identify the number of steps required for equilibration. However, we are interested in having an automated procedure to identify this boundary.
Figure 1. (Colour online) Illustration of the procedure for the location of the equilibration stage in an MC simulation. Data taken from a simulation at 70 kPa, forcefield setup 1, performed using Music. The blue line represents the rolling mean of the number of adsorbed molecules (over 20,000 steps) as a function of the number of MC steps. Instantaneous values of the number of adsorbed molecules are not shown for clarity. The vertical dotted line indicates the final half of the data-set, the horizontal black dashed line represents the value of the mean from this region while the red dashed line underneath corresponds to two standard deviations below the mean. The red dot delineates the equilibration and sampling stages, according to the procedure described in the text.
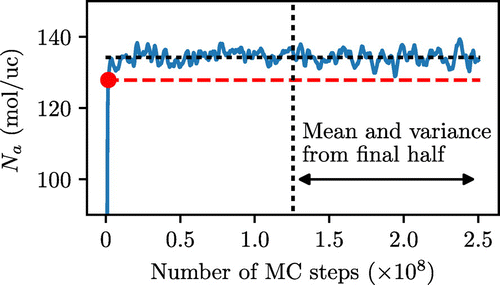
This procedure works as follows. We first run a very long simulation, typically around 250 MC steps. The results of the second half of the long simulation are used to estimate the mean and standard deviation of the number of molecules at this pressure. At this stage it is important to emphasise that calculating the standard deviation in this fashion significantly underestimates the true value, as there will be correlation between data points, as we will discuss later in the article. However, for the purposes of the algorithm that simply intends to locate the equilibration stage this crude approach suffices. As an additional test to ensure that the sampling stage is reached, a straight line of best fit is constructed through the data in the second half of the simulation and the difference between the starting point of this line and final point of this line must be less than 5% of the mean value.
We then consider a rolling mean average of the number of molecules in the system using a window width of 20,000 MC steps. Again, this number may seem rather arbitrary and the statistical quality of this mean for different programs will be different, but extensive testing of the approach on a number of systems shows that it works well in practice. We then move this rolling average backwards from the halfway point until it falls below two standard deviations from the estimated mean (Figure ). According to our protocol, this point delineates equilibrium and sampling stages. All MC steps before this point belong to the equilibration stage, while all subsequent points are considered to be within the sampling stage.
This process gives us, for each simulation condition in each program, a measure of the number of MC steps required to reach the sampling stage. We acknowledge that the criteria for defining the two stages are fairly arbitrary and alternative algorithms could have been used [Citation44]. However it provided consistent and sensible results across the data we examined. Moreover it could be used in a fully automised fashion, removing any human interaction in interpreting the results.
3.2. Sampling stage
As has been discussed, comparison of the computational performance of GCMC programs cannot be based on a fixed number of MC steps, since it will produce averages of different statistical quality and uncertainty. Instead we base this analysis on the number of steps required for a program to generate a certain number of independent, uncorrelated configurations.
For this we consider the normalised autocorrelation function (ACF) of the number of molecules adsorbed, C(n), shown in Equation (Equation10(10) ).
(10)
where angular brackets denote an average over the ensemble, n is the number of the MC steps and denotes the starting step.
Figure 2. (Colour online) Illustration of the fitting of the ACF to an exponential decay, using the same data shown in Figure . The blue section indicates the portion used for the fitting, while the red section indicates the discarded portion of the function. The truncation point is shown as black dashed lines. Black dotted line indicates the function estimated by the fitting procedure.
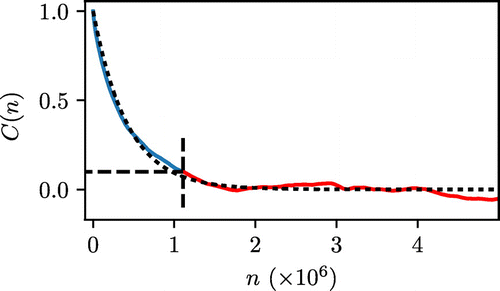
The ACF can then be fitted using a least squares regression to an exponential decay with a constant , Equation (Equation11
(11) ). As the ACF is inherently noisy for low values of C(n), we identify the first point at which it falls below a value of 0.1 and only the initial portion of the function is used for fitting. An example of this procedure is illustrated in Figure .
(11)
This then allows us to define the so-called statistical inefficiency, g [Citation45,Citation46]:(12)
The value g is a measure of the number of MC steps required to move to a statistically novel point in the simulation, with any measurement inbetween being correlated and therefore yielding no new information. If each step in the data were completely decorrelated from the previous step, would be zero yielding
, ie that each step is statistically novel. With the increasing correlation between subsequent data points,
will also increase, meaning that one must look further ahead in the data for a novel, uncorrelated sample.
Another approach to estimating autocorrelation in data are the block standard error (BSE) method [Citation47,Citation48]. In this approach the simulation trajectory of n MC steps is split into M blocks of m steps, and the standard deviation of the block averages is calculated (). This is repeated many times to measure BSE (B) as a function of m.
(13)
For small values of m the standard error will be underestimated due to significant correlation between the blocks. As m is increased to larger values than the memory in the data series (), the estimate of B will converge to the true value of the standard error. An example of this function is shown in Figure .
Figure 3. (Colour online) Example of the BSE approach applied to the same data shown in Figures and .
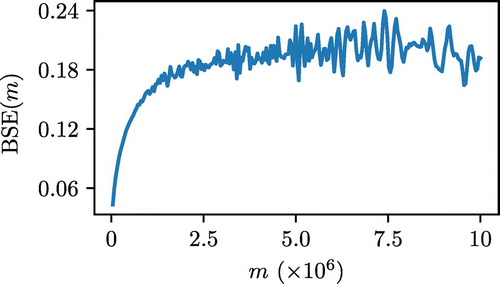
Through comparing Figures and , which operate on the same data, we can see that both approaches yield approximately the same value for g of around . However, we chose not to use the BSE approach in our analysis for the following reasons. In principle, for the BSE as a function of m, gradual increase in B is to be followed by a plateau region. At higher values of m a significant scattering in B is observed due to the small number of blocks available. This makes it difficult, especially in a fully automated procedure, to accurately locate the start of the plateau region. We also observe that in the systems under consideration much longer simulations are required to arrive at the same estimate of
compared to the direct calculation of the ACF. Finally, one of the original arguments advocating the use of BSE was the high computational cost associated with directly calculating the ACF [Citation47]. However, through the Wiener–Khinchin theorem [Citation49] it is possible to calculate this function in a much faster way using the Fourier transformations and on a modern desktop workstation it is now possible to do this in a negligible amount of time. Overall, we find the approach based on the ACF analysis easy to implement in a fully automatic fashion, and this convenience provides the final decisive argument in its favour.
3.3. Standard simulation length
Building on the ideas presented in the previous sections, we can now define standard simulation length of a program:(14)
where is the number of MC steps required for equilibration. Both
and g depend on the GCMC program and conditions of the system. We arbitrarily adopt 20 as a factor for how many statistically independent samples an MC program should generate [Citation48]. Other values could be used to achieve a different level of certainty in the result, however this would affect the timing of all programs equally. This definition of
makes it possible to measure how long it takes for an MC program to reach a result with a consistent statistical quality between the programs, which in turn allows us to compare MC programs on the same basis.
For the reasons advocated throughout the article, we deliberately avoid any discussion or presenting any results on the acceptance ratio of the Monte Carlo moves. This property is specific for each program implementation and bears no information on the actual sampling efficiency of the simulation [Citation50].
3.4. Benchmarking of the programs
With the properties introduced in the previous sections, we can now describe the rules and formulae for the benchmarking tests. In general we split benchmarking into measuring the time required for the program to complete the equilibration stage and the time required to generate a certain number (20 as a convention here) of uncorrelated samples of the properties of interest. The time required for equilibration is the CPU time, , needed by a program to perform
steps, as defined in the previous sections. Furthermore, within the sampling stage we can define the average CPU time per MC step:
(15)
In this case, the time required to generate a single uncorrelated sample of the property of interest is:(16)
And therefore the total CPU time associated to perform a standard length simulation for a given point on the adsorption isotherm is:(17)
All these benchmarks depend on the conditions within the system, and particularly on the adsorbed density. For these reasons the benchmarking tests are performed individually for each point on the adsorption isotherm for each program for each setup of the forcefields, and repeated three times to exclude any possibility of transient abnormal performance variation of the CPU. It is also important to note that while for the estimation of g and other aspects of the protocol, the data were saved very frequently, in the benchmarking tests the data were saved at much lower frequencies (but consistent among the programs) to avoid heavy computational overheads of the I/O operations.
Figure 4. (Colour online) Adsorption isotherms for in IRMOF-1 at 208 K, simulation data taken from the Raspa results. Solid black: experimental isotherm [Citation24], Dash dotted red: Forcefield setup 1, Dashed blue: Forcefield setup 2 and Dotted green: Forcefield setup 3.
![Figure 4. (Colour online) Adsorption isotherms for in IRMOF-1 at 208 K, simulation data taken from the Raspa results. Solid black: experimental isotherm [Citation24], Dash dotted red: Forcefield setup 1, Dashed blue: Forcefield setup 2 and Dotted green: Forcefield setup 3.](/cms/asset/630a9878-4995-4c18-8d69-82a928bd625a/gmos_a_1375492_f0004_oc.gif)
Two programs also have the option of using energy grids. Additional benchmarking tests were performed for these two programs. In these tests we do not account for the time required to generate the grids, as in a long term where they are reused many times for many simulations, this additional penalty is not important anymore.
4. Results
4.1. Adsorption isotherms
Table provides a complete summary of adsorption data for all programs for each pressure point and forcefield setup considered. Given a small number of samples, all five programs show a high degree of consistency with each other across all conditions. This a very reassuring result as it provides an independent validation for the existing programs and builds confidence in their application. This also provides a valuable set of reference data for further development and validation of new programs.
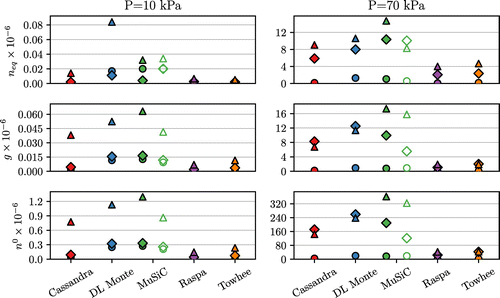
Figure 5. (Colour online) Comparison of the number of steps to reach equilibrium (), statistical inefficiency (g) and standard simulation length (
) for all programs. Lower values indicate fewer steps required to equilibrate and sample. Circles: Setup 1; Diamonds: Setup 2; Triangles: Setup 3. Unfilled markers indicate the use of energy biasing through energy grids. Results for two pressures are shown, with the full results given in Supplementary Material.
Adsorption isotherms for different forcefield setups are plotted in Figure along with the original experimental result from Walton et al. [Citation24] for completeness. Here we use the results from Raspa, as on the scale of the graph the isotherms for all five programs would be essentially indistinguishable. The most complete forcefield (setup 3) includes both fluid–fluid and solid–fluid electrostatic interaction terms, in addition to the ubiquitous default Lennard-Jones interaction between all species. This isotherm features a sharp step in the adsorbed amount, occurring between 10 and 20 kPa, which corresponds to the capillary condensation of in the pores. In the absence of the solid–fluid electrostatic term (setup 2) the isotherm maintains its shape but is shifted by 10 kPa to the right of the graph. The experimental isotherm also features a sharp transition, but compared to the result from the forcefield setup 2 it is further shifted to higher pressures. The fluid–fluid electrostatic interactions are indeed crucial for the capillary condensation step and in the absence of this term (setup 1), the resulting adsorption isotherm exhibits gradual increase in the adsorbed amount without any transitions. From the confined phase behaviour perspective, this is likely because the quasi-
fluid (
molecules without electrostatic interactions) is either very close or above the confined critical point at 208 K.
Figure 6. (Colour online) Top panel: time required to reach equilibrium, . Middle panel: time required to perform 1 M sampling steps. Bottom panel: time required to produce g sampling steps. Legend as in Figure , unfilled markers indicate the use of energy grids. Results for two pressures are shown, with the full results given in Supplementary Material.
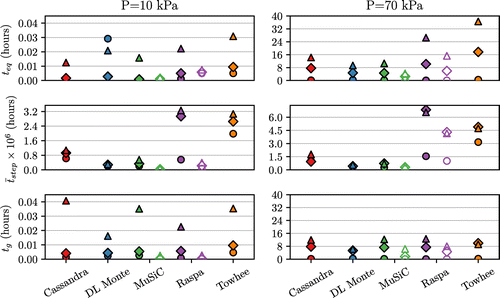
Table 2. Adsorption isotherm data for each program, reported as molecules per unit cell together with the standard error (, where
is the standard deviation and n the sample size). All results were generated using data from a standard length simulation (
).
4.2. Analysis of and g
The top two panels of Figure show the behaviour of and g for different programs as a function of pressure across all forcefield setups. For denser systems at higher pressures it takes a larger number of steps to equilibrate and also to accumulate sufficient number of uncorrelated samples. This is not surprising, as in this regime acceptance ratios for all types of MC moves tend to drop and the system remains in the same configuration for longer periods of sampling. It is also clear from Figure that the addition of electrostatics in general have a significant impact on the sampling metrics of the programs.
It is interesting to note that for Cassandra and DL Monte at high pressure the order in the trend for g values of setups 2 and 3 is reversed, however the values are very close to each other (the difference is comparable to the size of the symbol). Given some uncertainty of the ACF fitting procedure (Equation (Equation11(11) )), we believe this is most likely the source of this effect.
The standard simulation length, , is summarised for all programs in the bottom panel of Figure . From this figure it becomes apparent that Raspa and Towhee both require relatively fewer MC steps to produce the required amount of data. Both from Figure and the analysis of statistical inefficiency g, it is clear that different programs require different number of steps to produce the comparable amount of useful data. This highlights the importance of performing a proper statistical analysis of the data, based for example on ACFs as is done here, to define the characteristic correlations within a simulation. From this perspective, reporting simulation length in terms of the total number of MC steps is ineffective and misleading, as this is not a transferable metric. Instead, a much better metric is the multiple of the statistical inefficiency (g) of the property of interest, for example 20 g as in this study.
We also note a clear relationship across all programs between and g especially at higher pressures. For example in Figure the programs with equilibration length of around 10 M MC steps also have a similar g length, while the programs with shorter equilibration lengths also perform a statistical decorrelation faster. This prompts us to speculate that the sampling rate in MC steps of a program can be roughly assessed from
alone.
4.3. Program timing
Now that we have defined the required number of MC steps to perform equivalent simulations, we can proceed to measuring the time this will take to calculate, this is shown in Figure . Immediately apparent is that simulations at higher loadings take longer to complete. This is not surprising as at higher loadings larger number of intermolecular interactions must be calculated with each MC move.
The time to produce sampling MC steps is shown in Figure and from this we see again a large difference between programs to perform a seemingly similar task. This time however the order of programs is reversed compared to Figure , which clearly indicates that whilst programs such as Raspa and Towhee are able to produce more data with a fixed number of steps, it also takes longer to calculate such steps. Conversely we can see that the two programs with the longest decorrelation times, Music and DL Monte, are also the two fastest programs to complete a fixed number of sampling steps. Clearly there is a difference between the various programs in the definition of what constitutes a single MC step, as without any biasing should be identical between programs as it relies on random insertions. This again underlines the importance of the steps to decorrelation (g) as previously measured, by naively benchmarking the time to perform a fixed number of MC steps we might arrive at the wrong conclusion.
5. Final benchmarks
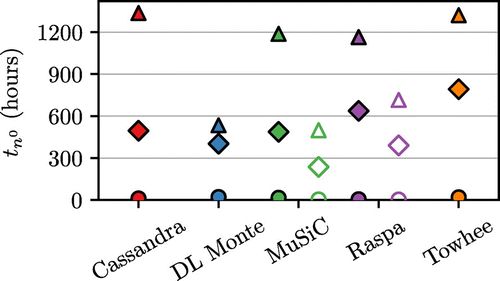
The total time required to obtain a complete adsorption isotherm is presented in Figure . We note that simulations take much longer than what would typically be expected from our experience, particularly for setup 3 that includes a complete set of electrostatic interactions. Overall the combination of sampling rate and CPU time per step has brought the programs remarkably close together compared to the differences seen in Figures and . For example the relatively high computational cost per step () in Raspa has been balanced by the lower value of g which gives each MC step performed a higher statistical value. Between the programs, DL Monte, Music and Raspa (when using an energy grid) all have comparable performance, with Cassandra and Towhee performing slower.
5.1. Effect of using energy grids on the computational performance
Based on the results shown in Figure it is clear that the use of energy grids provides a significant performance boost in all cases. Calculating the required energy grids, two for Lennard-Jones interactions and one for the electrostatic interactions, takes 3.6 and 1.6 h in Raspa and Music respectively, which is an insignificant amount of time when compared to the total time of even a single simulation.
Overall Raspa simulations run approximately 1.6 times faster when using energy grids across all forcefield setups. Music on the other hand is about 4.0, 2.0 and 2.4 times faster when using energy grids in forcefield setups 1, 2 and 3 respectively.
Figure 7. (Colour online) The total time required to run an entire isotherm of eight pressure points using standard simulation length. Legend as in Figure , with unfilled markers showing the results for simulations using energy grids.
6. Computational performance from parallel execution
As has previously been described, two of the programs investigated can use multiple cores for parallel execution. Whilst using additional cores will almost always decrease the time needed to perform a simulation, it is important to consider how efficiently additional computational resources are being used. The strong scaling efficiency, , of a program running in parallel is defined as [Citation51]:
(18)
where c is the number of cores and the runtime of a program as a function of the number of computer cores used.
As an alternative to running a single simulation in parallel, we can consider running different portions of the total number of MC steps of a long run using different instances of a program. In the context of adsorption simulation using GCMC methods, we also need to be aware that in the system which is always initiated from an empty unit cell, a certain portion of a single run will be spent on the equilibration stage. For example a simulation of equilibration steps and
sampling steps could be split into two simulations, consisting of
equilibration steps and
sampling steps each. As long as the smaller parallel runs and one long run produce the same number of uncorrelated samples, these two modes of execution are equivalent and this approach is a common practice in the MD community [Citation52]. In the previous example of splitting a long run into two runs, this doubles the number of steps and computational effort spent on equilibration.
This is implemented in Towhee, however this can also be done outside of the program by simply setting up and running multiple simulations as independent tasks. Mathematically, assuming is simply proportional to the number of MC steps, the efficiency of these parallel tasks can be estimated as
(19)
where refers to the number of equilibration steps, and
the number of sampling steps. The efficiency at a given number of cores can be seen to rely on the ratio of sampling steps to equilibration steps, with relatively longer equilibration periods leading to less efficient usage of parallel cores.
Splitting single simulations into separate tasks has previously been dismissed due to the long equilibration times [Citation27]. Our previous results however show that typically and g are of the same order of magnitude, therefore long equilibration stages simply indicate that long sampling stages are also required. In these circumstances, task based parallelism is a valid routeto accelerate simulations. Based on our previously discussed observations and our defined standard simulation length (
), we have used a
ratio of 20 to estimate the efficiency of task based parallelism.
Figure 8. (Colour online) Comparison of the efficiency of parallel computation in different programs, measured for forcefield Setup 3 at 70 kPa. Task based efficiency (as defined in Equation (Equation19(19) )) is shown in solid black for a
ratio of 20. Cassandra: red dashed line; DL Monte: blue dotted line; Towhee: orange dash-dotted line.
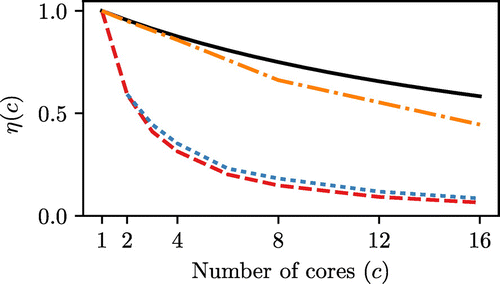
From Figure it can be seen that both Cassandra and DL Monte do not efficiently use parallel cores, and these results are in agreement with the previously published results for DL Monte [Citation27]. Under these circumstances, for the simulation of a single pressure point, computational resources would be much better used by running many serial tasks and collating the results at the end of the simulations. This is shown in the results for Towhee, where the efficiency is much greater than the other two programs.
In the case of DL Monte, where only the electrostatic interactions are parallelised, the poor efficiency can be explained by considering Amdahl’s Law [Citation53] which states that the limit of speedup (s) for a program is given as: (20)
where p represents the fraction of the program’s runtime which can be parallelised. If for instance the electrostatics took up 75% of the programs run time [Citation27], this would limit the potential speed up to only 4, in the limit of infinite cores.
In the case of Cassandra, which parallelises all force calculations with OpenMP, the same argument cannot be made. Previous attempts at a similar parallelisation scheme in MD simulations have shown similar lacklustre results for systems of a similar size, where it was observed that efficiency depended heavily on system size [Citation54].
7. Conclusion
The fact that all the programs tested produced the same results for all conditions with a high level of consistency may seem trivial, yet we believe it is very important, as such a comprehensive comparison has not been attempted earlier and it is not done routinely by the MC program users. Furthermore, this study provides a set of well documented program setups and case studies, that can be used for further development and validation. Finally, it creates certain confidence in the programs currently employed by the MC community.
The benchmarking process did reveal some differences in the overall performance of different programs, however this variation was relatively small when considering the broader picture of making a choice of which program to use. Whilst in this work each program performed an identical simulation, they also each have a multitude of different other capabilities and compatibility with other programs within the wider simulation ecosystem. Also not assessed here was the entirely subjective topic of how easy a given program is to correctly set up and use. Equally as research projects get more mature, it is important to consider how difficult a program will be to modify to allow it to meet future, more specific needs. When selecting a MC program to use all these various factors must also be taken into consideration alongside considering the computational performance.
Whilst it is common to report the lengths of MC simulations as a number of steps, we have shown that this metric is not transferable between different programs, and therefore between research groups who have built an intuition for a given program. Instead, we argue it would be much more useful and important for reproducibility of the results to report simulation length as a multiple of g, along with the value of g for each specific simulation, which reflects the quantity of sampling that has been performed. When analysing a MC simulation, calculating the length of the simulation in terms of g allows us to define if enough data have been gathered and for the level of confidence in the result to be quantified. We have found that for systems of this type that g can be estimated as approximately the same order of magnitude as , and this can be used to estimate the required length of additional simulation required once a simulation has reached equilibrium.
The varying relative value in data of an MC step simply reflects the fact that there are different strategies to implement an MC move. From a program development point of view it would be very misleading to concentrate on the required walltime for a single move in order to optimise the performance of a program. Instead, the larger picture of the entire sampling process must be considered. The choice of which MC moves to use and the proportion of each move to use remains open for investigation, however using the metrics developed in this work it should now be possible to accurately quantify the effects these moves make. This will become especially true when considering more complicated MC moves, such as configuration bias moves for flexible molecules.
Our further recommendations can be summarised as follows. Energy grids must definitely be used when possible as they clearly provide a quick and efficient way to substantially increase computational performance of the MC programs in the context of adsorption problems. The scope for increasing performance through parallelism within a program seems limited, due mostly to the inherently small system sizes, and measured performances of existing implementations show poor efficiency. In fact, we argue that in the context of adsorption problems and computational screening of materials, parallel execution of multiple instances of the process offers much better efficiency and overall speed up for a fixed amount of computational resources.
Supplementary_file.pdf
Download PDF (2.1 MB)Acknowledgements
The authors would like to thank Dr. David Dubbeldam, Dr. Martin Sweatman, Dr. Daniel Siderius and Prof. Randy Snurr for useful discussions and advice. The work has made use of resources provided by the Edinburgh Compute and Data Facility (ECDF; www.ecdf.ed.ac.uk)
Additional information
Funding
Notes
No potential conflict of interest was reported by the authors.
Supplemental data for this article can be accessed here. [https://doi.org/10.1080/08927022.2017.1375492]
Related Research Data
References
- Wilmer CE, Leaf M, Lee CY, et al. Large-scale screening of hypothetical metal--organic frameworks. Nat Chem. 2012 Feb;4:83–89.
- Lin LC, Berger AH, Martin RL, et al. In silico screening of carbon-capture materials. Nat Mater. 2012;11:633–641.
- Keskin S, Sholl DS. Efficient methods for screening of metal organic framework membranes for gas separations using atomically detailed models. Langmuir. 2009;25:11786–11795.
- Jiang J, Babarao R, Hu Z. Molecular simulations for energy, environmental and pharmaceutical applications of nanoporous materials: from zeolites, metal--organic frameworks to protein crystals. Chem Soc Rev. 2011;40:3599–3612.
- Yang Q, Liu D, Zhong C, et al. Development of computational methodologies for metal--organic frameworks and their application in gas separations. Chem Rev. 2013;113:8261–8323.
- Jiang J. Molecular simulations in metal--organic frameworks for diverse potential applications. Mol Simul. 2014;40:516–536.
- Fischer M, Gomes JR, Jorge M. Computational approaches to study adsorption in MOFs with unsaturated metal sites. Mol Simul. 2014;40:537–556.
- Erucar I, Manz TA, Keskin S. Effects of electrostatic interactions on gas adsorption and permeability of MOF membranes. Mol Simul. 2014;40:557–570.
- Chen L, Morrison CA, Düren T. Improving predictions of gas adsorption in metal--organic frameworks with coordinatively unsaturated metal sites: model potentials, ab initio parameterization, and GCMC simulations. J Phys Chem C. 2012;116:18899–18909.
- Dzubak AL, Lin LC, Kim J, et al. Ab initio carbon capture in open-site metal-organic frameworks. Nat Chem. 2012;4:810–816.
- Kim J, Lin LC, Lee K, et al. Efficient determination of accurate force fields for porous materials using ab initio total energy calculations. J Phys Chem C. 2014;118:2693–2701.
- Haldoupis E, Borycz J, Shi H, et al. Ab initio derived force fields for predicting CO2 adsorption and accessibility of metal sites in the metal-organic frameworks M-MOF-74 (M = Mn Co, Ni, Cu). J Phys Chem C. 2015;119:16058–16071.
- Becker TM, Heinen J, Dubbeldam D, et al. Polarizable force fields for CO2 and CH4 adsorption in M-MOF-74. J Phys Chem C. 2017;121:4659–4673.
- Banu AM, Friedrich D, Brandani S, et al. A multiscale study of mofs as adsorbents in h2 psa purification. Ind Eng Chem Res. 2013;52:9946–9957.
- Hasan MF, First EL, Floudas CA. Discovery of novel zeolites and multi-zeolite processes for p-xylene separation using simulated moving bed (smb) chromatography. Chem Eng Sci. 2017;159:3–17.
- Sweatman MB. Preface to the special issue on ‘Monte Carlo codes, tools and algorithms’. Mol Simul. 2013;39:1123–1124.
- Abraham MJ, Murtola T, Schulz R, et al. GROMACS: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX. 2015;1–2:19–25.
- Eastman P, Friedrichs MS, Chodera JD, et al. OpenMM 4: a reusable, extensible, hardware independent library for high performance molecular simulation. J Chem Theory Comput. 2013;9:461–469.
- Li H, Eddaoudi M, O’Keeffe M, et al. Design and synthesis of an exceptionally stable and highly porous metal--organic framework. Nature. 1999 Nov;402:276–279.
- Frenkel D, Smit B. Understanding molecular simulation. 2nd ed. Orlando (FL): Academic Press; 2001.
- Allen MP, Tildesley DJ. Computer simulation of liquids. New York (NY): Clarendon Press; 1989.
- Babarao R, Hu Z, Jiang J, et al. Storage and separation of CO2 and CH4 in silicalite, C168 schwarzite, and IRMOF-1: a comparative study from Monte Carlo simulation. Langmuir. 2007;23:659–666.
- Liu B, Smit B. Comparative molecular simulation study of CO2/N2 and CH4/N2 separation in zeolites and metal-organic frameworks. Langmuir. 2009;25:5918–5926.
- Walton KS, Millward AR, Dubbeldam D, et al. Understanding inflections and steps in carbon dioxide adsorption isotherms in metal-organic frameworks. J Am Chem Soc. 2008;130:406–407.
- Dubbeldam D, Torres-Knoop A, Walton KS. On the inner workings of Monte Carlo codes. Mol Simul. 2013;39:1253–1292.
- Shah JK, Maginn EJ. A general and efficient Monte Carlo method for sampling intramolecular degrees of freedom of branched and cyclic molecules. J Chem Phys. 2011;135:134121.
- Purton J, Crabtree J, Parker S. DL\_Monte: a general purpose program for parallel Monte Carlo simulation. Mol Simul. 2013 Dec;39:1240–1252.
- Gupta A, Chempath S, Sanborn MJ, et al. Object-oriented programming paradigms for molecular modeling. Mol Simul. 2003 Jan;29:29–46.
- Dubbeldam D, Calero S, Ellis DE, et al. RASPA: molecular simulation software for adsorption and diffusion in flexible nanoporous materials. Mol Simul. 2016 Jan;42:81–101.
- Martin MG. MCCCS Towhee: a tool for Monte Carlo molecular simulation. Mol Simul. 2013 Dec;39:1212–1222.
- Kim J, Rodgers JM, Athènes M, et al. Molecular monte carlo simulations using graphics processing units: to waste recycle or not? J Chem Theory Comput. 2011;7:3208–3222.
- Snurr RQ, Bell AT, Theodorou DN. Prediction of adsorption of aromatic hydrocarbons in silicalite from grand canonical Monte Carlo simulations with biased insertions. J Phys Chem. 1993;97:13742–13752.
- Peng DY, Robinson DB. A new two-constant equation of state. Ind Eng Chem Fundam. 1976;15:59–64.
- Mayo SL, Olafson BD, Goddard WA. DREIDING: a generic force field for molecular simulations. J Phys Chem. 1990;94:8897–8909.
- Yazaydın AÖ, Snurr RQ, Park TH, et al. Screening of metal--organic frameworks for carbon dioxide capture from flue gas using a combined experimental and modeling approach. J Am Chem Soc. 2009;131:18198–18199.
- Potoff JJ, Siepmann JI. Vapor--liquid equilibria of mixtures containing alkanes, carbon dioxide, and nitrogen. AIChE J. 2001;47:1676–1682.
- Plimpton SJ, Thompson AP. Computational aspects of many-body potentials. MRS Bulletin. 2012 May;37:513–521.
- Dotson David L, Seyler Sean L, Linke Max, et al. datreant: persistent, Pythonic trees for heterogeneous data. In: Benthall S, Rostrup S, editors. Proceedings of the 15th Python in Science Conference; Austin, TX; 2016. p. 51–56.
- Hunter JD. Matplotlib: a 2d graphics environment. Comput Sci Eng. 2007;9:90–95.
- Michaud-Agrawal N, Denning EJ, Woolf TB, et al. MDAnalysis: a toolkit for the analysis of molecular dynamics simulations. J Comput Chem. 2011;32:2319–2327.
- Gowers Richard J, Linke Max, Barnoud Jonathan, et al. MDAnalysis: a Python package for the rapid analysis of molecular dynamics simulations. In: Benthall S, Rostrup S, editors. Proceedings of the 15th Python in science conference; Austin, TX; 2016. p. 98–105.
- van der Walt S, Colbert SC, Varoquaux G. The NumPy array: a structure for efficient numerical computation. Comput Sci Eng. 2011 Mar;13:22–30.
- McKinney W. Data structures for statistical computing in python. In: van der Walt S, Millman J, editors. Proceedings of the 9th Python in Science Conference; Austin, TX; 2010. p. 51–56.
- Chodera JD. A simple method for automated equilibration detection in molecular simulations. J Chem Theory Comput. 2016;12:1799–1805.
- Janke W. Statistical analysis of simulations: data correlations and error estimation. In: Grotendorst J, Marx D, Muramatsu A, editors. Quantum simulations of complex many-body systems: from theory to algorithms. Vol. 10, Kerkrade, The Netherlands: Springer; 2002. p. 423–445.
- Shirts MR, Chodera JD. Statistically optimal analysis of samples from multiple equilibrium states. J Chem Phys. 2008;129:124105.
- Flyvbjerg H, Petersen HG. Error estimates on averages of correlated data. J Chem Phys. 1989;91:461–466.
- Grossfield A, Zuckerman DM. Quantifying uncertainty and sampling quality in biomolecular simulations. Annu Rep Comput Chem. 2009 Jan;5:23–48.
- Chatfield C. The analysis of time series: an introduction. 6th ed. London: CRC Press; 2004.
- Frenkel D. Simulations: the dark side. Eur Phys J Plus. 2013;128:10.
- Kalé L, Skeel R, Bhandarkar M, et al. NAMD2: greater scalability for parallel molecular dynamics. J Comput Phys. 1999;151:283–312.
- Balasubramanian V, Treikalis A, Weidner O, et al. EnsembleMD Toolkit: scalable and flexible execution of ensembles of molecular simulations. CoRR. 2016. abs/1602.00678.
- Amdahl GM. Validity of the single processor approach to achieving large scale computing capabilities. In: Proceedings of the April 18–-20, 1967, Spring Joint Computer Conference; Atlantic City, New Jersey; AFIPS ’67 (Spring). New York (NY): ACM; 1967.p. 483–485.
- Tarmyshov KB, Müller-Plathe F. Parallelizing a molecular dynamics algorithm on a multiprocessor workstation using OpenMP. J Chem Inf Model. 2005;45:1943–1952.