
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
High-entropy alloys (HEAs) attract growing interest due to their remarkable properties. However, our knowledge of the mechanism of phase stability of HEAs is still very limited. Herein, CoCrFeNixAl1-x (x = 0.5 and 0.75) were compressed to high pressures to investigate the lattice distortion effect on their phase stability. It was discovered that both bcc CoCrFeNi0.5Al0.5 and fcc CoCrFeNi0.75Al0.25 alloys transform to the hcp structure at high pressures, following a gradual phase transition. The sluggish character of these transitions perhaps originates from the local energy fluctuations caused by the chemical disorder. The phase transitions in both studied compounds commenced when their intrinsic lattice strain had reached the maximum value, indicating that the lattice distortion effect plays a key role in these crystallographic structure-transforming processes. These results provide fundamental information on the phase selection rules of HEAs, which are important for physical metallurgy theories and HEAs design in the future.
I. Introduction
Comparing to conventional alloys containing only one or two principal elements, high-entropy alloys (HEAs) include at least five principal elements whose concentrations range between 5 and 35 at.% [Citation1–3]. This revolutionary metallurgical concept not only dramatically extends the territory of alloys, but also introduces a substantial number of alloys with exceptional properties, such as high fracture toughness [Citation4], excellent strength [Citation5], superconductivity [Citation6], and high resistance to corrosion [Citation7]. The term ‘high-entropy alloys’ stresses the high configuration entropy effect, signifying the underlying reason why HEAs prefer to crystallize in chemical disordered simple structures (body-centered cubic (bcc), face-centered cubic (fcc), or hexagonal close-packed (hcp)) rather than chemically ordered intermetallic compounds [Citation1]. However, a recent investigation reveals that vibrational, electronic, and magnetic entropies, in addition to the configuration entropy, also play important roles in the phase stability of HEAs [Citation8], indicating this category of compounds is more complex than previously expected.
Nowadays, one of the important themes of research on HEAs is understanding the phase selection rules: what are the dominating factors that determine the structures of HEAs? [Citation3]. Knowledge of the phase selection rules of HEAs will not only deepen our understanding of the physical metallurgy principles but also guide the future design of HEAs exhibiting exceptional properties. It has been reported that the atomic size difference (δ) [Citation9], the mixing enthalpy (ΔHmix) [Citation9], the valence electron concentration (VEC) [Citation10], and/or the entropy related parameter (φ) [Citation11], all markedly affect the phase selection rules of HEAs, indicating the complexity of this phenomenon. The values of δ and ΔHmix are defined by formulas [Citation9]:(1)
(1)
(2)
(2) where ci and ri are the atomic fractions and the atomic radius of the ith principal element, respectively, and
is the mixing enthalpy of binary liquid alloy.
HEAs with different combinations of principal elements were selected in order to investigate the δ effect on the phase selection rules. However, the ΔHmix, VEC, and/or φ may change simultaneously, making the conclusions more or less ambiguous. Therefore, for the purpose of the phase selection rules investigation, it is important to disentangle the interplaying effects. As known, pressure represents a fundamental thermodynamic variable, which has the potential to tune the structural, mechanical, and electronic properties of materials pronouncedly [Citation12]. Moreover, it represents a clean tool, which can be selectively applied to a desired effect, hence facilitating the disentanglement of crossover phenomena. Under high pressures, atomic sizes and lattice distortion change pronouncedly, widely influencing the thermodynamic stability and various properties of HEAs [Citation13]. Therefore, investigating the structural evolution of HEAs under compression is expected to provide valuable information about the impact of lattice distortion on their phase selection rules [Citation14–17].
The CoCrFeNixAl1-x system, representing a prototype of HEAs, has been widely studied at ambient pressure [Citation18,Citation19]. Upon an Al (which has a significantly larger atomic size than other composite elements) content increase, the configuration entropy Sconf, the mixing enthalpy ΔHmix, the atomic size difference δ, and the valence electron concentration VEC of CoCrFeNixAl1-x HEAs vary in appreciably large ranges (). Concomitantly, the structure of CoCrFeNixAl1-x transforms from a single fcc phase, through a mixture of bcc and fcc phases, to a single bcc phase [Citation18,Citation19]. Consequently, CoCrFeNixAl1-x HEAs are excellent candidates for studying the lattice distortion effect on phase selection rules in terms of various combinations of Sconf, ΔHmix, δ and VEC values.
Figure 1. The ideal configuration entropy Sconf, the mixing enthalpy ΔHmix [Citation9], the atomic size difference δ [Citation9], and the VEC [Citation10] of CoCrFeNixAl1-x as a function of x values at ambient pressure. The solid dots present the results of CoCrFeNi0.5Al0.5, CoCrFeNi0.75Al0.25, and CoCrFeNi, respectively.
![Figure 1. The ideal configuration entropy Sconf, the mixing enthalpy ΔHmix [Citation9], the atomic size difference δ [Citation9], and the VEC [Citation10] of CoCrFeNixAl1-x as a function of x values at ambient pressure. The solid dots present the results of CoCrFeNi0.5Al0.5, CoCrFeNi0.75Al0.25, and CoCrFeNi, respectively.](/cms/asset/1af8cfec-2d40-41a1-bf00-5576e6b07e22/ghpr_a_1653865_f0001_oc.jpg)
Recently, Zhang et al. [Citation14] reported an abnormal sluggish phase transition of CoCrFeNi from the fcc to hcp structure, which started at 13.5 GPa and had not been completed at the highest experimental pressure (∼40 GPa). The underlying mechanisms of this sluggish structural transition were not clarified. CoCrFeNi alloy possesses the lowest ideal configuration entropy and the smallest atomic size difference in the CoCrFeNixAl1-x system (). Thus the lattice distortion effect originating from the atomic sizes mismatch is not severe in CoCrFeNi, disregarding it as a proper model to study the lattice distortion effect on the phase selection rules of HEAs.
In the present study, by applying pressure in order to tune the crystal lattice distortion independently, we examined the lattice distortion effect on the phase transition of HEAs, choosing CoCrFeNixAl1-x (x = 0.5, 0.75) as a prototype. In the CoCrFeNixAl1-x system, CoCrFeNi0.5Al0.5 displays the highest configuration entropy. It mainly adopts the bcc structure with a minor presence of the fcc phase. On the other hand, CoCrFeNi0.75Al0.25 exhibits the configuration entropy intermediate between those of CoCrFeNi and CoCrFeNi0.5Al0.5, and crystallizes into a single fcc phase at ambient pressure. We also discovered phase transitions to hcp structures in bcc CoCrFeNi0.5Al0.5 and fcc CoCrFeNi0.75Al0.25 at moderate pressures, both displaying a very sluggish transformation dynamics. The transitions began at the lattice distortions reaching their maximum values, indicating that the distortion plays a key role in the pressure-induced phase transition in the two studied HEAs. These results provide important information on the phase selection rules of HEAs.
II. Materials and methods
The CoCrFeNixAl1-x (x = 0.5, and 0.75) HEAs with nominal molar ratios were prepared by the arc melting method in a high purity argon atmosphere. Diamond anvils with 300 µm culet size were mounted in the symmetric cells used to generate high pressure. The T301 stainless steel gasket with an initial thickness of 250 µm was indented to 30 µm, and a sample chamber with a diameter of 120 µm was drilled in the center of the pre-indented hole. The sample with a typical size of ∼50 µm was scraped from the ingot and loaded in the sample chamber. The sample was sandwiched between two NaCl plates, which served as the pressure transmitting medium and pressure scale [Citation20]. Four runs (two runs for each sample) of in situ angle-dispersive X-ray diffraction experiments were conducted at the High Pressure Station, 4W2 beamline at Beijing Synchrotron Radiation Facility (BSRF). The incident x-ray radiation was monochromatized to 20 keV (wavelength of 0.6199 Å) by using double silicon (111) crystals. The beam was focused to a spot size of 7 µm (vertical) × 13 µm (horizontal) (the full width at half maximum, FWHM) by Kirkpatrick–Baez mirrors. Diffraction patterns were collected by the Pilatus3 2M detector. The distance from the sample to the detector and the geometric parameters of the detector were calibrated by the CeO2 standard from the National Institute of Standards and Technology. All the measurements were conducted at room temperature. The two-dimensional diffraction patterns were integrated by Fit2D [Citation21] to obtain the 2θ–intensity curves for further analysis. Software Fityk [Citation22] was used to fit and decompose the overlapped diffraction peaks applying the Voigt-profile peak function.
III. Results and discussions
A. Structure at ambient pressure
To minimize the potential effect of heterogeneity of the synthesized samples, we performed two independent runs of diffraction experiments for each sample. The results of the two runs are consistent with each other, indicating that the heterogeneity effect is not obvious in the present experiments, at least at the probed length scales. The Debye rings were observed to be smooth, demonstrating the absence of lattice texture in the studied materials.
The typical integrated diffraction patterns are shown in . At ambient pressure, the main observed phase of CoCrFeNi0.5Al0.5 was of the bcc structure, while a small diffraction peak from the fcc phase appeared at the left shoulder of the bcc (110) reflection. The CoCrFeNi0.75Al0.25 adopted a single fcc phase at the ambient pressure. Zhang et al. reported that CoCrFeNi also exhibited the single fcc structure at the ambient pressure [Citation14]. It seems that, upon Al concentration increase, the crystal structure of CoCrFeNixAl1-x transforms from the fcc to the bcc symmetry, which is consistent with the previous results [Citation18,Citation19]. Zhang et al. [Citation9] proposed two parameters, the atomic size difference δ, and the mixing enthalpy ΔHmix. It has been predicted that a single-phase solid solution will form when 0 < δ < 0.05, and −15 kJ/mol < ΔHmix < 5 kJ/mol [Citation9]. The values of δ and ΔHmix for CoCrFeNi0.5Al0.5, CoCrFeNi0.75Al0.25, and CoCrFeNi alloys listed in , all these alloys fall in this range and thus should crystallize in a single bcc or fcc phase. However, multiple phases were still found in CoCrFeNi0.5Al0.5, indicating that also other effects, rather than only atomic size difference and mixing enthalpy, should be taken into the account when discussing the phase selection rules of HEAs.
Figure 2. Typical diffraction patterns of CoCrFeNi0.5Al0.5 (a) and CoCrFeNi0.75Al0.25 (b). In the top panels of the figures, we show the deconvolution of the overlapped diffraction peaks using Voigt peak function in the program Fityk [Citation22].
![Figure 2. Typical diffraction patterns of CoCrFeNi0.5Al0.5 (a) and CoCrFeNi0.75Al0.25 (b). In the top panels of the figures, we show the deconvolution of the overlapped diffraction peaks using Voigt peak function in the program Fityk [Citation22].](/cms/asset/3993f60c-9350-4ad3-93c5-079edfd869a9/ghpr_a_1653865_f0002_oc.jpg)
Table 1. The equation of states and thermodynamic variables of samples and their principal elements.
B. Sluggish phase transition
New peaks (hcp(100), and hcp(101)) of the hcp structure appeared ((a)) in the diffraction patterns of CoCrFeNi0.5Al0.5 at 20.12 GPa, indicating the commencement of the bcc-hcp phase transition. For CoCrFeNi0.75Al0.25, the fcc-hcp phase transition was also discovered at 14.64 GPa ((b)). The transition pressure of CoCrFeNi0.75Al0.25 was lower than that of CoCrFeNi0.5Al0.5, but consistent with the reported phase transition pressure (13.5 GPa) of CoCrFeNi in general [Citation14].
Guo et al. proposed that the crystal structure of HEAs can be predicted using the valence electron concentration where (VEC)i is the VEC of the ith element [Citation10]. The HEAs adopted the fcc structure when VEC > 8.0, whereas the bcc structure occurred for VEC > 6.87 [Citation10]. The compounds in the CoCrFeNixAl1-x system follow this rule at ambient pressure – their structures changed from bcc CoCrFeNi0.5Al0.5 (VEC = 7.375), to fcc CoCrFeNi0.75Al0.25 (VEC = 7.813), and to fcc CoCrFeNi (VEC = 8.25). Ye et al. stated that HEAs adopted an hcp structure when VEC = 2.8 ± 0.2 [Citation3]. If this rule is indeed validated at high pressures, the VEC values of CoCrFeNixAl1-x (x = 0.5, 0.75, and 1) should decrease dramatically under compression. However, no prominent electronic change of Co, Cr, Fe, Ni, and Al has been observed in this pressure range. Recently, 4f electrons in addition to the s and d electrons, have also counted as valence electrons [Citation27]. Thus, the VEC values of the hcp HEAs GdTbDyTmLu, HoDyYGdTb, and YGdTbDyLu are 13, 9.8, and 10.6 respectively, being much higher than the value of 3 reported previously [Citation3,Citation28]. Consequently, the electron concentration effect, one of the key factors of Hume-Rothery rules, needs to be assessed carefully when applied to HEAs, especially at high pressures.
It is worth pointing out that all these observed phase transitions were sluggish and not completed at the highest pressures of experiments (41.03 GPa for CoCrFeNi0.5Al0.5, 48.43 GPa for CoCrFeNi0.75Al0.25, and 39 GPa for CoCrFeNi [Citation14]). Zhang et al. attributed the sluggishness of fcc to hcp phase transition to the pressure-assisted inhibition of an atomic motion [Citation14]. Very recently, the pressure-induced sluggish fcc to hcp transition of CrMnFeCoNi HEA has also been reported [Citation15,Citation16]. Tracy et al. considered this sluggish phase transition arose from the increasing energy barrier associated with stacking fault formation with increasing pressure [Citation15], akin to the fcc to hcp transition of xenon [Citation29]. Asides from the sluggish nature of the fcc to hcp phase transition of CoCrFeNi0.75Al0.25, which is similar to those occurring in CoCrFeNi and CrMnFeCoNi, our results for CoCrFeNi0.5Al0.5 reveal the sluggishness also for the bcc to hcp transition and hence suggest that it may represent a universal feature of HEAs.
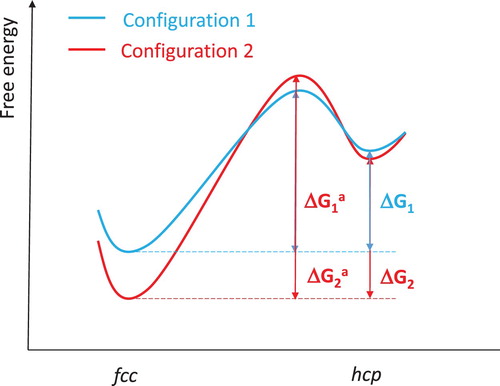
Due to the chemical disorder in HEAs [Citation30–33], the local atom configurations vary between lattice sites, causing variation of local energies. Tsai et al. reported that the difference of mean potential energies of Ni atoms between various lattice sites of fcc CoCrFeMnNi alloy was as high as 60.3 meV [Citation34]. This local energy fluctuation causes the different energy barrier (ΔGa) and/or the different driving force (ΔG) of the phase transition from site to site (). We consider this local energy fluctuation represents the main underlying reason for the observed sluggish phase transitions in CoCrFeNixAl1-x alloys, implying also that this phenomenon would be universal in HEA. To some extent, the larger the amplitude of the local energy fluctuation, the larger the pressure range of coexistence of phases in the transition process.
Figure 3. Local energy fluctuation due to the intrinsic chemical disorder in HEAs. Configurations 1 and 2 correspond to different local atomic arrangements.
Analogous to the HEAs, the local energy fluctuations also exist in metallic glasses, where the variation of the coordination numbers brings an extra contribution to the fluctuations. However, because of the electronic nature of the transition [Citation35–38], several observed phase transitions of metallic glasses completed in a narrow pressure range. Consequently, it is expected that the change in an electronic structure, like 4f electrons delocalization, will accelerate the structural phase transition process in some specific HEAs, for example, in a HEA including element Ce.
Up to date, only about 1% (7 out of 648 reported HEAs) of HEAs crystallize in the hcp structure [Citation13]. Hence, the accumulated knowledge about the hcp HEAs is quite limited. Pressure-tuning turns out to represent an efficient method for the synthesis of hcp HEAs of diverse chemical compositions, allowing to alloy cheap and convenient elements, offering an alternative to expensive lanthanides [Citation14–16]. The results also outline new possibilities to investigate HEAs’ physical and chemical properties. In the future, it is expected that phase selection rules for the hcp HEAs will be progressively emerging with a growing number of studies exploring a sufficiently broad range of compositional variations in this class of materials.
C. Equation of states
The pressure-volume relations of bcc CoCrFeNi0.5Al0.5 and fcc CoCrFeNi0.75Al0.25 are shown in , compared with that of fcc CoCrFeNi. The equilibrium volume (V0) and bulk modulus (K0) at ambient pressure were determined by fitting the experimental data points with the Vinet equation of state (fixing the pressure derivative of bulk modulus at 4):(3)
(3) The results are plotted in and summarized in . The fitting yielded V0 = 11.84(2) Å3/atom, and 11.66(1) Å3/atom for bcc CoCrFeNi0.5Al0.5 and fcc CoCrFeNi0.75Al0.25, respectively. Due to the significantly larger atomic size of aluminum (), the cell parameters increase with a higher Al molar concentration, in agreement with the Vegard’s law [Citation39]. The bulk moduli of bcc CoCrFeNi0.5Al0.5 and fcc CoCrFeNi0.75Al0.25 are 190(4) GPa and 194(2) GPa, respectively, a bit smaller than that of fcc CoCrFeNi, 206.5 GPa [Citation14]. This result is consistent with the conclusion of Zhang et al. that the bulk moduli decrease upon increasing the number of elements in a solid solution [Citation14]. Zhang et al. also suggested that the addition of atoms with a larger size mismatch increased the ‘free volume’ in the solid solution. Consequently, easily squeezing out this volume upon compression lowers the bulk modulus of HEAs [Citation14].
Figure 4. The measured average atomic volumes of bcc CoCrFeNi0.5Al0.5 and fcc CoCrFeNi0.75Al0.25 as a function of pressure, comparing with the results of fcc CoCrFeNi [Citation14]. The lines represent the fitted equation of states with fixed at 4, using Vinet equation of state.
![Figure 4. The measured average atomic volumes of bcc CoCrFeNi0.5Al0.5 and fcc CoCrFeNi0.75Al0.25 as a function of pressure, comparing with the results of fcc CoCrFeNi [Citation14]. The lines represent the fitted equation of states with K0′ fixed at 4, using Vinet equation of state.](/cms/asset/18cc7ea1-d779-431d-acf6-470085999d66/ghpr_a_1653865_f0004_oc.jpg)
D. Lattice distortion effect
Lattice distortion originates from the deviation of equilibrium atom positions away from the ideal lattice sites because of the large atomic size mismatch. Though it influences the structure and properties of HEA significantly, there is no widely accepted quantitative definition of the lattice distortion to date. The great challenge remains to evaluate the lattice distortion of HEAs experimentally [Citation40,Citation41], even more so at high pressures. Because HEA has several principal elements, the atomic configurations in the unit cells vary from one unit cell to another, so does the lattice distortions. This variation leads to a distribution of local distortion, similar to the interatomic distance distribution [Citation42]. To investigate the lattice distortion effect on the bulk material, the lattice distortion of a single unit cell is meaningless, and a statistical method is needed to describe the lattice distortion of the whole bulk material. Unfortunately, it is almost impossible to measure this distribution at the present stage.
The direct outcome of the lattice distortion is that the unit cell parameters are not equal to each other anymore (for cubic unit cells). Therefore, if we can measure the standard deviation of the lattice parameters, this deviation can also represent the magnitude of lattice distortion of HEA. Furthermore, lattice constants represent experimentally measurable structural parameters of HEA, thus it is reasonable to use them for evaluating the magnitude of lattice distortion:(4)
(4) where ai are the exact real edge length of the distorted unit cell, and ai0 are the corresponding lattice constants of the undistorted ideal unit cell. The measurement of ai is still almost impossible, and the x-ray diffraction technique determines the average value of all ai, i.e. a. Consequently, for cubic HEAs, Equation (4) can be rewritten as:
(5)
(5) The undistorted ideal unit cell is imaginary. Therefore Zhou et al. used the lattice constant of original elemental crystals aOEC to replace a0, and got [Citation43,Citation44]
(6)
(6)
The physical implication of this model is to evaluate the magnitude of the deviation of the HEA from the undistorted cubic lattice. Zhou et al. proposed the following four criteria for selecting the original elemental crystals: (1) being the principal element of the HEA; (2) crystallizing in the same structure with the HEA; (3) being homogeneously distributed in the HEA, and (4) possessing the moderate atomic radius among all the principal elements [Citation44]. Comparing with Equation (6), Equation (1) ignores the atomic size alteration during the alloying process, as well as it is difficult to measure the atomic size at high pressures. Equation (6) not only includes the alteration of the atomic size and crystal structure effects, but is also expedient to perform calculations at high pressures. Therefore, in order to study the effect of severe lattice distortion effect on the phase transition, we use the intrinsic lattice strain (Equation (6)) for evaluating the magnitude of lattice distortion of HEAs at high pressures.
According to the criteria, the equation of states of bcc Fe and fcc Ni, which were used to calculate the εintr of CoCrFeNixAl1-x, were taken from Ref. [Citation24] and are listed in . The εintr of bcc CoCrFeNi0.5Al0.5, fcc CoCrFeNi0.75Al0.25, and fcc CoCrFeNi as a function of pressure are shown in (a)–(c). The εintr of bcc CoCrFeNi0.5Al0.5 and fcc CoCrFeNi0.75Al0.25 reach their maximum values at 18.85 and 11.93 GPa, respectively. These pressures agree well with the experimentally observed onset pressures of the phase transitions of bcc CoCrFeNi0.5Al0.5 and fcc CoCrFeNi0.75Al0.25 alloys ((d)), respectively. The agreement suggests that the lattice distortion effect plays a key role in these phase transformations. The intrinsic lattice strains of bcc CoCrFeNi0.5Al0.5 and fcc CoCrFeNi0.75Al0.25 reached values 0.0043 and 0.0220, respectively, when the phase transitions commenced. The evaluated strains are much smaller than previous lattice instability criterions (10% as proposed by Lindemann [Citation45], 5% as proposed by Ye et al. [Citation46], or 6% according to Egami et al. [Citation47]). The reason is that different methods were applied for evaluating the lattice strain.
Figure 5. The intrinsic lattice strain and its pressure derivative for bcc CoCrFeNi0.5Al0.5 (a), fcc CoCrFeNi0.75Al0.25 (b), and fcc CoCrFeNi (c) as a function of pressure. The cyan lines are the fitting lines using the 3rd order polynomial function. The orange lines correspond to the pressure derivative of the lattice strain. For fcc CoCrFeNi, the 4th order polynomial function was used when fitting the intrinsic lattice strain, because of insufficient quality when using the 3rd order function (blue dash line). Results of both 3rd and 4th order function are presented in (c) and indicate the intrinsic lattice strain reaches the maximum value at 29.53 and 32.33 GPa, respectively. The possible pressure ranges of phase transitions (open squares are the phase transition pressures determined in experiments, the horizontal bars are the pressure points before phase transition in experiments, the asterisks represent pressures where intrinsic lattice strains reach the maximum) and the pressures where the intrinsic lattice strain reaches maximum are summarized in (d). The vertical green line in (d) shows the x value (0.8) where the atomic concentration of Al reaches 5%, signifying the limiting boundary of the HEA concept.
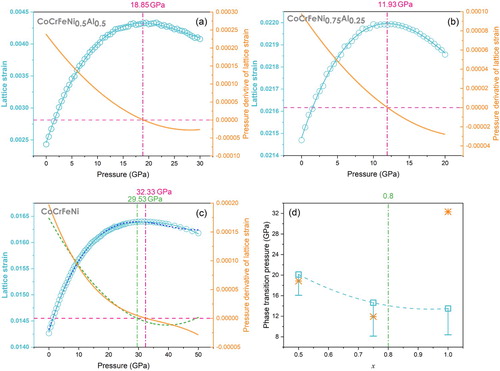
However, for the fcc CoCrFeNi, the onset pressure of phase transition is 13.5 GPa [Citation14], while its intrinsic lattice strain reaches the maximum value of 0.0163 at 32.33 GPa, indicating that effects other than the lattice distortion effect play the main role in its fcc to hcp phase transition. In the CoCrFeNixAl1-x system, as is evident from values listed in , Co, Cr, Fe, and Ni atoms have almost the same atomic sizes (∼1.24–1.25 Å), while Al atom is of a much larger atomic radius (∼1.43 Å). Accordingly, most part of the lattice distortion is ascribed to the addition of Al atoms. From , one can see that the atomic size difference δ of the aluminum-free alloy CoCrFeNi is much smaller (0.0030) in comparison with the values of CoCrFeNi0.5Al0.5 (0.0482), and CoCrFeNi0.75Al0.25 (0.0357). This indicates that the lattice distortion is not the dominant effect in CoCrFeNi. Zhang et al. considered that the driving force of the fcc to hcp phase transition of CoCrFeNi is the vibrational entropy difference between the two crystal structures [Citation14]. Apparently, several different effects compete in the phase selection rules of HEAs, which makes the application of these rules less straightforward and calls for a further investigation.
Taking into the account that the intrinsic lattice strain is proportional to the total elastic energy stored in the whole lattice [Citation46], the obtained results signify that the phase transitions took place when a maximally possible amount of accumulated elastic energy has been reached in the distorted lattice. For the bcc CoCrFeNi0.5Al0.5 and fcc CoCrFeNi0.75Al0.25 alloys, because of the strain relaxation, the elastic energy decreased gradually above the onset pressure of transition. However, the elastic energy of the fcc CoCrFeNi continued to increase until 32.33 GPa, a value much higher than the transition pressure.
Materials stabilize into a structure whose Gibbs free energy reaches a (local) minimum at a specific pressure and temperature. For HEAs exhibiting a severe lattice distortion, the distortion-induced elastic energy ΔE is not trivial and the Gibbs free energy at constant temperature can be written as [Citation44](7)
(7) where Gp is the Gibbs free energy of a perfect lattice and can be expressed by the formula:
(8)
(8)
Here, E0 is the zero-point energy. Fel, Fmag, and Fvib are electronic, magnetic, and vibrational contributions to the free energy, respectively, and Sconf represents the ideal configuration entropy. The elastic energy can be expressed as [Citation48,Citation49](9)
(9) where ε(P) is the elastic strain arising from atomic size mismatch at pressure P, and D is the atomic packing efficiency (D is 0.68, 0.74, and 0.74 for bcc, fcc and hcp structures, respectively). Ye et al. proposed a sophisticated geometric model to calculate ε at ambient pressure [Citation46]. However, it is not a convenient model to apply, especially at high pressures. It is reasonable to assume that the elastic strain ε is proportional to the atomic size mismatch δ. The atomic size difference δ of fcc CoCrFeNi is less than 10% of that of bcc CoCrFeNi0.5Al0.5 and fcc CoCrFeNi0.75Al0.25 alloys (). Hence the distortion-induced elastic energy ΔE of fcc CoCrFeNi is ∼1% of that of bcc CoCrFeNi0.5Al0.5 and fcc CoCrFeNi0.75Al0.25 alloys. For the fcc CoCrFeNi, the ΔE contribution to the Gibbs free energy is negligible, while the magnitudes of contributions Fel, Fmag, and/or Fvib are decisive for its phase transition at high pressures. Ab initio calculations revealed that a vibrational contribution dominated the Gibbs free energy increase, thus controlling the phase transition of CoCrFeNi at high pressures [Citation14]. For bcc CoCrFeNi0.5Al0.5 and fcc CoCrFeNi0.75Al0.25 alloys, their lattice distortion and elastic strain ε(P) are pronounced, as indicated by the large atomic size difference δ. Consequently, upon compression, the increasing δ(P) which is proportional to εintr(P), contributes to a major part of the Gibbs free energy increment, eventually destabilizing structures and triggering phase transitions. Thus, we suggest that the observed pressure-induced phase transitions in bcc CoCrFeNi0.5Al0.5 and fcc CoCrFeNi0.75Al0.25 alloys are controlled by the lattice distortion.
At last, it should be pointed out that while δ is a good indicator of the elastic strain ε of the distorted lattice, εintr is not. The absolute value of δ can be used to evaluate the absolute value of the elastic strain ε: the materials with a larger δ also have a larger ε. However, the absolute values of εintr of materials with different structures should not be compared with each other. For example, εintr of bcc CoCrFeNi0.5Al0.5 is much smaller than that of fcc CoCrFeNi. However, δ and ε of bcc CoCrFeNi0.5Al0.5 are obviously larger than those of fcc CoCrFeNi. The reason is that different original elemental crystals are used to calculate εintr of bcc CoCrFeNi0.5Al0.5 and fcc CoCrFeNi. Moreover, because the average lattice parameters of HEAs are used to calculate εintr, a major part of the fluctuation of atomic size is smeared out. For instance, εintr of fcc CoCrFeNi is ∼ 70% of that of fcc CoCrFeNi0.75Al0.25, but δ or ε of fcc CoCrFeNi are less than 10% of those of fcc CoCrFeNi0.75Al0.25. Fortunately, for a specific HEA, the value of εintr increases when δ or ε increase. This is the reason why one can use εintr for evaluating the variation of ε or lattice distortion under pressure. Anyway, for future investigations, a handy model for a reliable assessment of ε needs to be worked out, especially for high pressure analyses.
IV. Conclusions
In the present study, CoCrFeNi0.5Al0.5 and CoCrFeNi0.75Al0.25 HEAs were compressed to pressures reaching 40–50 GPa (volume compression ∼15%) in order to investigate the effect of lattice distortion on the phase selection rules of HEAs. Both bcc CoCrFeNi0.5Al0.5 and fcc CoCrFeNi0.75Al0.25 transformed into an hcp structure at high pressure, despite the different starting crystal structures at ambient pressure. The phase transitions of CoCrFeNi0.5Al0.5 and CoCrFeNi0.75Al0.25 took place when the intrinsic lattice strain had reached a maximum value, implying that the lattice distortion played a key role in triggering the transformation processes. By spanning the pressure range of a few tens of GPa, the observed transitions turned out to be very sluggish and had not been completed even at the highest experimental pressures. The local energy fluctuations arising from the chemical disorder are suggested to represent the underlying reason for the sluggishness, making it an inherent and universal feature of the pressure-induced phase transitions in HEAs.
Acknowledgments
We thank Yi Zhang, Qiyue Hou, and Shourui Li for their assistance with the experiments. This work is supported by the National Natural Science Foundation of China (Grant No. 11504354). The experiments were performed at the 4W2 high pressure station of BSRF, which is supported by the Chinese Academy of Sciences (Grant Nos. 11474281, KJCX2-SWN03 and KJCX2-SW-N20).
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Yeh JW, Chen SK, Lin SJ, et al. Nanostructured high-entropy alloys with multiple principal elements: novel alloy design concepts and outcomes. Adv Eng Mater. 2004;6:299–303. doi: 10.1002/adem.200300567
- Zhang Y, Zuo TT, Tang Z, et al. Microstructures and properties of high-entropy alloys. Prog Mater Sci 2014;61:1–93. doi: 10.1016/j.pmatsci.2013.10.001
- Ye YF, Wang Q, Liu J, et al. High-entropy alloy: challenges and prospects. Mater Today. 2016;19:349–362. doi: 10.1016/j.mattod.2015.11.026
- Gludovatz B, Hohenwarter A, Catoor D, et al. A fracture-resistant high-entropy alloy for cryogenic applications. Science. 2014;345:1153–1158. doi: 10.1126/science.1254581
- Youssef KM, Zaddach AJ, Niu CN, et al. A novel low-density, high-hardness, high-entropy alloy with close-packed single-phase nanocrystalline structures. Mater Res Lett. 2015;3:95–99. doi: 10.1080/21663831.2014.985855
- Kozelj P, Vrtnik S, Jelen A, et al. Discovery of a superconducting high-entropy alloy. Phys Rev Lett. 2014;113:107001. doi: 10.1103/PhysRevLett.113.107001
- Lee CP, Chen YY, Hsu CY, et al. The effect of Boron on the corrosion resistance of the high entropy alloys Al[sub 0.5]CoCrCuFeNiB[sub x]. J Electrochem Soc. 2007;154:C424. doi: 10.1149/1.2744133
- Ma D, Grabowski B, Körmann F, et al. Ab initio thermodynamics of the CoCrFeMnNi high entropy alloy: importance of entropy contributions beyond the configurational one. Acta Mater. 2015;100:90–97. doi: 10.1016/j.actamat.2015.08.050
- Zhang Y, Zhou YJ, Lin JP, et al. Solid-Solution phase formation rules for Multi-component alloys. Adv Eng Mater. 2008;10:534–538. doi: 10.1002/adem.200700240
- Guo S, Ng C, Lu J, et al. Effect of valence electron concentration on stability of fcc or bcc phase in high entropy alloys. J Appl Phys 2011;109:103505. doi: 10.1063/1.3587228
- Ye YF, Wang Q, Lu J, et al. Design of high entropy alloys: a single-parameter thermodynamic rule. Scr Mater. 2015;104:53–55. doi: 10.1016/j.scriptamat.2015.03.023
- Shen G, Mao HK. High-pressure studies with x-rays using diamond anvil cells. Rep Prog Phys 2017;80:016101. doi: 10.1088/1361-6633/80/1/016101
- Miracle DB, Senkov ON. A critical review of high entropy alloys and related concepts. Acta Mater. 2017;122:448–511. doi: 10.1016/j.actamat.2016.08.081
- Zhang FX, Zhao SJ, Jin K, et al. Pressure-induced fcc to hcp phase transition in Ni-based high entropy solid solution alloys. Appl Phys Lett. 2017;110:011902. doi: 10.1063/1.4973627
- Tracy CL, Park S, Rittman DR, et al. High pressure synthesis of a hexagonal close-packed phase of the high-entropy alloy CrMnFeCoNi. Nature Comm. 2017;8:15634. doi: 10.1038/ncomms15634
- Zhang F, Wu Y, Lou H, et al. Polymorphism in a high-entropy alloy. Nature Comm. 2017;8:15687. doi: 10.1038/ncomms15687
- Yu PF, Zhang LJ, Ning JL, et al. Pressure-induced phase transitions in HoDyYGdTb high-entropy alloy. Mater Lett 2017;196:137–140. doi: 10.1016/j.matlet.2017.02.136
- Wang WR, Wang WL, Yeh JW. Phases, microstructure and mechanical properties of AlxCoCrFeNi high-entropy alloys at elevated temperatures. J Alloys Comp. 2014;589:143–152. doi: 10.1016/j.jallcom.2013.11.084
- Jasiewicz K, Cieslak J, Kaprzyk S, et al. Relative crystal stability of AlxFeNiCrCo high entropy alloys from XRD analysis and formation energy calculation. J Alloys Comp. 2015;648:307–312. doi: 10.1016/j.jallcom.2015.06.260
- Dorogokupets PI, Dewaele A. Equations of state of MgO, Au, Pt, NaCl-B1, and NaCl-B2: internally consistent high-temperature pressure scales. High Press Res 2007;27:431–446. doi: 10.1080/08957950701659700
- Hammersley AP, Svensson SO, Hanfland M, et al. Two-dimensional detector software: from real detector to idealised image or two-theta scan. High Press Res 1996;14:235–248. doi: 10.1080/08957959608201408
- Wojdyr M. Fityk: a general-purpose peak fitting program. J Appl Cryst. 2010;43:1126–1128. doi: 10.1107/S0021889810030499
- Senkov ON, Miracle DB. Effect of the atomic size distribution on glass forming ability of amorphous metallic alloys. Mater Res Bull. 2001;36:2183–2198. doi: 10.1016/S0025-5408(01)00715-2
- Dewaele A, Torrent M, Loubeyre P, et al. Compression curves of transition metals in the Mbar range: experiments and projector augmented-wave calculations. Phys Rev B. 2008;78:104102. doi: 10.1103/PhysRevB.78.104102
- Ming LC, Manghnani MH. Isothermal compression of bcc transition metals to 100 kbar. J Appl Phys. 1978;49:208–212. doi: 10.1063/1.324325
- Dewaele A, Loubeyre P, Mezouar M. Equations of state of six metals above 94 GPa. Phys Rev B. 2004;70:094112. doi: 10.1103/PhysRevB.70.094112
- Hu Q, Guo S, Wang JM, et al. Parametric study of amorphous high-entropy alloys formation from two new perspectives: atomic radius modification and crystalline structure of alloying elements. Sci Rep. 2017;7:39917. doi: 10.1038/srep39917
- Takeuchi A, Amiya K, Wada T, et al. High-entropy alloys with a hexagonal close-packed structure designed by equi-atomic alloy strategy and binary phase diagrams. JOM. 2014;66:1984–1992. doi: 10.1007/s11837-014-1085-x
- Cynn H, Yoo CS, Baer B, et al. Martensitic fcc-to-hcp transformation observed in xenon at high pressure. Phys Rev Lett. 2001;86:4552–4555. doi: 10.1103/PhysRevLett.86.4552
- Lucas MS, Wilks GB, Mauger L, et al. Absence of long-range chemical ordering in equimolar FeCoCrNi. Appl Phys Lett. 2012;100:251907. doi: 10.1063/1.4730327
- Maiti S, Steurer W. arXiv:1601.06015.
- Santodonato LJ, Zhang Y, Feygenson M, et al. Deviation from high-entropy configurations in the atomic distributions of a multi-principal-element alloy. Nat Comm. 2015;6:5964. doi: 10.1038/ncomms6964
- Zhang YW, Stocks GM, Jin K, et al. Influence of chemical disorder on energy dissipation and defect evolution in concentrated solid solution alloys. Nat Comm. 2015;6:8736. doi: 10.1038/ncomms9736
- Tsai KY, Tsai MH, Yeh JW. Sluggish diffusion in Co–Cr–Fe–Mn–Ni high-entropy alloys. Acta Mater. 2013;61:4887–4897. doi: 10.1016/j.actamat.2013.04.058
- Sheng HW, Liu HZ, Cheng YQ, et al. Polyamorphism in a metallic glass. Nat Mat. 2007;6:192–197. doi: 10.1038/nmat1839
- Zeng QS, Li YC, Feng CM, et al. Anomalous compression behavior in lanthanum/cerium-based metallic glass under high pressure. Proc Natl Acad Sci USA. 2007;104:13565–13568. doi: 10.1073/pnas.0705999104
- Zeng QS, Ding Y, Mao WL, et al. Origin of pressure-induced Polyamorphism in Ce(75)Al(25) metallic Glass. Phys Rev Lett. 2010;104:105702. doi: 10.1103/PhysRevLett.104.105702
- Decremps F, Morad G, Garbarino G, et al. Polyamorphism of a Ce-based bulk metallic glass by high-pressure and high-temperature density measurements. Phys Rev B. 2016;93:054209. doi: 10.1103/PhysRevB.93.054209
- West AR. Solid state chemistry and its application. New Delhi: Wiley; 1984, p. 367.
- Owen LR, Pickering EJ, Playford HY, et al. An assessment of the lattice strain in the CrMnFeCoNi high-entropy alloy. Acta Mater. 2017;122:11–18. doi: 10.1016/j.actamat.2016.09.032
- Oh HS, Ma D, Leyson GP, et al. Lattice distortions in the FeCoNiCrMn high entropy alloy studied by theory and experiment. Entropy. 2016;18:321. doi: 10.3390/e18090321
- Song H, Tian F, Hu QM, et al. Local lattice distortion in high-entropy alloys. Phys Rev Mater. 2017;1:023404. doi: 10.1103/PhysRevMaterials.1.023404
- Klug HP, Alexander LE. X-ray diffraction procedures for polycrystalline and amorphous materials. New York: Wiley; 1954, p. 126.
- Zhou YJ, Zhang Y, Wang FJ, et al. Phase transformation induced by lattice distortion in multiprincipal component CoCrFeNiCuxAl1–x solid-solution alloys. Appl Phys Lett. 2008;92:241917. doi: 10.1063/1.2938690
- Lindemann FA. The calculation of molecular vibration frequencies. Physik Z. 1910;11:609–612.
- Ye YF, Liu CT, Yang Y. A geometric model for intrinsic residual strain and phase stability in high entropy alloys. Acta Mater. 2015;94:152–161. doi: 10.1016/j.actamat.2015.04.051
- Egami T. Atomic level stresses. Prog Mater Sci. 2011;56:637–653. doi: 10.1016/j.pmatsci.2011.01.004
- Lu K, Li Y. Homogeneous nucleation catastrophe as a kinetic stability limit for superheated crystal. Phy Rev Lett. 1998;80:4474–4477. doi: 10.1103/PhysRevLett.80.4474
- Allen GL, Gile WW, Jesser WA. The melting temperature of microcrystals embedded in a matrix. Acta Metall. 1980;28:1695–1701. doi: 10.1016/0001-6160(80)90022-X