
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Objective: To establish a rat model with respiratory and pulmonary responses caused by inhalation exposure to non-lethal concentrations of ammonia (NH3) that can be used for evaluation of new medical countermeasure strategies for NH3-induced acute lung injury (ALI). This is of great value since no specific antidotes of NH3-induced injuries exist and medical management relies on supportive and symptomatically relieving efforts.
Methods: Female Sprague-Dawley rats (8–9 weeks old, 213g ± 2g) were exposed to NH3 using two different exposure regimens; nose-only inhalation or intratracheal instillation. The experiment was terminated 5 h, 24 h, 14 and 28 days post-exposure.
Results: Nose-only inhalation of NH3 (9000–15 000 ppm) resulted in increased salivation and labored breathing directly post-exposure. Exposure did not increase inflammatory cells in bronchoalveolar lavage fluid but exposure to 12 000 ppm NH3 during 15 min reduced body weight and induced coagulation abnormalities by increasing serum fibrinogen levels. All animals were relatively recovered by 24 h. Intratracheal instillation of NH3 (1%) caused early symptoms of ALI including airway hyperresponsiveness, neutrophilic lung inflammation and altered levels of coagulation factors (increased fibrinogen and PAI-1) and early biomarkers of ALI (IL-18, MMP-9, TGFβ) which was followed by increased deposition of newly produced collagen 14 days later. Histopathology analysis at 5 h revealed epithelial desquamation and that most lesions were healed after 14 days.
Conclusions: This study demonstrates that intratracheal instillation can reproduce several early hallmarks of ALI. Our findings therefore support that the intratracheal instillation exposure regimen can be used for new medical countermeasure strategies for NH3-induced ALI.
Introduction
Ammonia (NH3) is a toxic, colorless, corrosive, alkaline gas with a sharp odor that is produced in large volumes because of its wide industrial application. Anhydrous NH3 is transported as pressurized liquid and if released into the air it expands to many times its original volume to form dangerous vapor clouds (AEGL Citation2007). It has been reported that NH3 concentrations 5000 ppm (3540 mg/m3) are rapidly fatal to humans (Henderson and Haggard Citation1943), causing acute respiratory failure due to severe damage to the lung and toxic pulmonary edema (AEGL Citation2007). Unintentional release as a result of incidents, e.g. at manufacturing facilities or during transportation, or intentional release, e.g. in terror attacks, could, therefore, result in mass exposure (Gorguner and Akgun Citation2010; UN Citation2014). Since mass casualty incidents after chemical release usually are unpredictable, an adequate preparedness is of great importance. The medical management after a toxic exposure to NH3 is mainly supportive, life-saving, and symptomatically relieving since no specific antidotes have been identified so far (Makarovsky et al. Citation2008).
Inhalation is the predominantly reported route of exposure and when inhaled at higher concentrations, NH3 severely affect the lower respiratory tract due to its high water solubility that allows it to quickly dissolve in the moist mucous membranes (Coon et al. Citation1970; Close et al. Citation1980; Makarovsky et al. Citation2008). Most of the toxic properties of NH3 relate to its alkaline pH in solution (EPA Citation2016). When dissolved in water, NH3 form ammonium (NH4+) and hydroxide ions (OH−) but increasing the pH above 9, the un-ionized form of NH3 becomes the predominant species (Stumm and Morgan Citation1981).
Human exposure to NH3 can lead to tissue break-down in the airways and destruction of cilia and the mucosal barrier, that causes inflammation and an increased risk of secondary infections (Arwood et al. Citation1985; Amshel et al. Citation2000). Major clinical manifestations and pathological findings in humans after exposure to NH3 include respiratory symptoms, such as hypoxia, secretions, sloughed epithelia, emphysema, edema, reactive smooth muscle contractions in the airways, hypovolemia and burns to the skin and eyes (Wiklund et al. Citation2001; Brautbar et al. Citation2003). The respiratory symptoms may lead to clinical impairment of respiratory function and damaged epithelia is often replaced by new connective tissue, which may be one of the causes of chronic lung disease following NH3 inhalation injury (Hatton et al. Citation1979; Flury et al. Citation1983; George et al. Citation2000). The chronic lung disease is similar to e.g. chronic obstructive lung disease (COPD), bronchiectasis, airway respiratory distress syndrome (ARDS), and bronchiolitis obliterans disease (BO). Complications after NH3 exposure could also progress into the need of prolonged ventilator support, or in the worst scenario, death (Kass et al. Citation1972; Sobonya Citation1977; Flury et al. Citation1983; Lessenger Citation2004; Makarovsky et al. Citation2008; Kerger and Fedoruk Citation2015).
Studies in animals have demonstrated both dose-effect and duration-effect relationships in the pathological changes of the respiratory tract after exposure to both non-lethal and lethal concentrations of NH3 (Appelman et al. Citation1982; AEGL Citation2007; Perkins et al. Citation2016). The LC50 for rats range from 7338 and 16 600 ppm for 60-min exposures up to 40 300 ppm for a 10-min exposure (MacEwen and Vernot Citation1972; Appelman et al. Citation1982). Due to ethical reasons, only non-lethal exposures of NH3 were utilized in this study, where the purpose was to follow both respiratory and vascular responses after short, high-level exposures to NH3. The overall aim of the study was to establish an animal model that could be used for evaluating new medical countermeasure strategies for NH3-induced ALI.
In this study, animals exposed to NH3 gas did not develop an inflammatory response at 24 h post-exposure regardless of NH3-concentration used, therefore this route of administration was not further analyzed. Instead, NH3 was directly administered in liquid form into the trachea with further distribution into the lung accordingly to a previously published method (Johansson et al. Citation2005; Wigenstam et al. Citation2012). Due to strain and species differences along with the great recovery, reversibility, and capability to repair fibrotic lesions of rodents, it is challenging to establish an in vivo model that mimics the progressive characters of human disease (Gauldie and Kolb Citation2008).
Materials and methods
Animals
Healthy female Sprague-Dawley rats (8–9 weeks old, Envigo RMS B.V, Netherlands) were used in this study. Animals were housed in plastic cages with absorbent bedding material and maintained on a 12 h daylight cycle, 22 °C, with a 30% relative humidity. Food (R36, Lantmännen, Sweden) and water were provided ad libitum. All rats were weighed before NH3-exposure (initial weight range of all rats was 213g ± 2g), and their health condition was monitored during the entire experimental period. Animals were terminated after the experiment with an overdose of pentobarbital sodium. The care of the animals and the experimental protocols were approved by the regional ethics committee on animal experiments in Umeå, Sweden.
NH3-exposure
Two different exposure regimens were used in the study: (1) NH3 via nose-only exposure (gas inhalation) and (2) NH3 via i.t. instillation (NH3 dissolved in water).
The rationale for dose selection, and exposure time of 10–20 min used in this study was considered to be relevant for a scenario of a large scale chemical disaster with NH3. Based on an estimation of the time period, an exposed individual could be located in a gas plume and survive long enough to get medical treatment. The i.t. administration of sub-lethal NH3 concentrations was performed as another alternative for establishing ALI when inhaled gas did not cause chemical lung damage.
Nose-only inhalation
Animals were placed in individual nose-only containers (EMMS, UK) and simultaneous exposed in an inhalation tower (Battelle) providing an equal air flow to each animal of 3 l/min of the NH3 (AGA gas, Sweden; compressed gas in gas cylinders: 25 mol-% NH3, 75 mol-% nitrogen) gas mixture. Rats were subjected to a single exposure of NH3-gas mixture (sub-lethal concentrations in the range of 9000–15 000 ppm, during 10 or 20 min) (Perkins et al. Citation2016). The NH3-concentration in the inhalation tower was monitored, using the Fix View 7.0 control system (Intellution inc., Norwood, MA), throughout the exposure time (air flow: 20 l/min, temperature: 22 °C and relative humidity 11–12%) and the experiments were conducted in a designated fume hood for toxic gas exposures. While animals were inhaling NH3, control animals were breathing room air in their cages on the bench next to the fume hood (relative humidity 14% in room air). Data regarding body weight-loss, the levels of fibrinogen in serum, total cells in bronchoalveolar lavage fluid (BALF), neutrophils in BALF are shown for NH3-exposure via nose-only inhalation at 24 h. One group of animals exposed to 13 000 ppm was also analyzed on day-14 post-exposure for long-term effects (data not shown).
Intratracheal instillation
A sub-lethal concentration was decided on the basis of a small-scale pilot study where different concentrations were tested from 0.25% to 2.5% NH3 via i.t. instillation and the lung injury was evaluated at 24 h post-exposure. A final concentration of 1% NH3 (25% weight percent NH3, Merck Eurolab AB, Sweden) diluted in water, with a pH 11.75 at room temperature (approximately ≥90% un-ionized NH3 in the solution (Emerson et al. Citation1975)), was chosen for the study. The rats were anesthetized briefly using enflurane before NH3 was administered by i.t. instillation in a volume of 200 μl. Control rats, given only the solvent, did not show any response different from unexposed healthy rats. In the full-scale rat study, the concentration of 1% NH3 was used and its effects were evaluated at 5 h, 24 h, 14 days, or 28 days post-exposure. Data regarding body weight, cells in BALF, inflammatory cytokines, organ weights, respiratory mechanics, collagen deposition, and histopathology are shown for all animals.
Respiratory mechanics
Animals were weighed and anesthetized with pentobarbital sodium (50 mg/kg body weight (bw), i.p.). Rats were tracheostomized with a 15-gauge cannula and mechanically ventilated with a small animal ventilator (flexiVent™, SCIREQ®) at a frequency of 90 breaths/min and a tidal volume (VT) of 10 ml/kg bw. A positive end-expiratory pressure of 3 cmH2O was applied. Rats were paralyzed with pancuronium (0.1 mg/kg bw, i.p.) before four sigh maneuvers at 3 × VT were performed at the beginning of the experiment to establish stable baseline respiratory mechanics and to ensure a similar volume history before the experiments. The parameters recorded at baseline and after incremental doses of inhaled methacholine (MCh, acetyl-β-methylcholine chloride, Sigma-Aldrich (St. Louis, MO)) were: respiratory resistance (RRS), Newtonian resistance, (RN), tissue resistance (G), respiratory elastance (ERS), tissue elastance (H) and hysteresivity (η). With the flexiVent™ system, the respiratory properties were partitioned into central and peripheral airways and different tissue properties were distinguish in the lungs (Bates and Irvin Citation2003; Irvin and Bates Citation2003). In order to measure AHR, incremental doses of MCh were given at 5-min intervals. The MCh, diluted in saline to a volume of 20 µl, was given during 10 s as an aerosol (Aeroneb™ PRO, SCIREQ®). Each dose of MCh (0, 5 and 25 mg/ml) was aerosolized without any interference with the ventilation pattern.
Blood analyses and organ weights
Blood samples were taken from the aorta in the abdomen directly after terminal anesthesia. The blood was centrifuged (15 min, 20 °C, 3000 rpm) and the serum was saved in −70 °C until analysis of inflammatory mediators.
For the thrombocyte analyses, blood was withdrawn from the rats as described above and the blood was treated with K2EDTA (plasma) and then diluted (1:25) in 5 mL ISOTON® II diluent buffer (Beckman Coulter Inc.) and platelet-rich plasma (PRP) was prepared by centrifugation for 90 s at 600 × g. After a second dilution step (1:200) of 100 µL PRP supernatant, the number of thrombocytes was analyzed using an automatic cell counter, Z2 Coulter Particle count and Size Analyzer (Beckman Coulter Life Sciences, Indianapolis), with a 70 µm aperture tube. Thrombocytes above 1.5 µm in size were counted.
The rats in the NH3 i.t. instillation group were dissected and both heart and spleen were weighed at 24 h and 14 days post-exposure.
Analysis of cells in BALF
The lungs were lavaged five times via a tracheal tube with a total volume of 2 ml + 23 ml Ca2+/Mg2+ free Hanks’ balanced salt solution (HBSS, Sigma-Aldrich (St. Louis, MO)). The BALF was centrifuged (10 min, 4 °C, 1500 rpm). After removing the supernatant, the cell pellet was resuspended in 1 ml PBS. The supernatant (the first 2 ml recovered after lavage) was stored in −70 °C until analysis of inflammatory mediators. Leukocytes were counted manually in a hemacytometer and 20 000 cells were loaded onto slides using a Cytospin® centrifuge (Shandon© cytospin 3 cytocentrifuge, cell preparation system). Cytocentrifuged preparations were stained with May-Grünwald-Giemsa reagents (Merck Millipore, VWR International, Sweden) and differential cell counts of pulmonary inflammatory cells were performed in duplicates using standard morphological criteria and counting 200 cells per cytospin preparation.
Inflammatory mediators in BALF and serum
Inflammatory mediators in BALF and serum from animals exposed to NH3 via i.t. instillation were analyzed for the presence of interleukin (IL)-1β, CINC-1, MMP-9, and IL-6 using specific DuoSet assay kits from R&D Systems. MPO, PAI-1, and IL-18 were analyzed using specific assay kits from Nordic BioSite (ERK284, ERK2454, and EKR72, respectively) in Sweden and fibrinogen kit (E-25FIB) from Novakemi AB, Sweden. Fibrinogen was also analyzed in animals exposed to NH3 via the nose-only approach at 24 h. All analyses were performed according to the manufacturer’s instructions and analyzed using an ELISA reader (Thermo Scientific Mutilskan FC, Thermo Fischer Scientific Oy, Vantaa, Finland). ELISA data were analyzed using the software program for the ELISA reader (SkanIt for Multiskan FC 3.1. Ink), referring to the standards added to each plate. The three isoforms of TGFβ (TGF-β1-3) were analyzed simultaneously in BALF using a multiplex kit (Bio-Plex Pro TGF-β 3-Plex Immunoassay, Bio-Rad) and detected with a Bio-Plex™ system (Bio-Rad).
Lung tissue
Histopathological evaluation
After lavage and terminal anesthesia, the right lung lobe from animals exposed to NH3 via i.t. instillation was fixed in 4% paraformaldehyde until paraffin embedding. After embedding, the tissue was cut into 3 μm thick sections and mounted on positively charged slides. To assess inflammatory cell infiltration, the sections were deparaffinized, dehydrated, and stained with hematoxylin and eosin. Histopathological evaluations of stained sections were performed in a blinded manner using light microscopy for randomly selected animals in each experimental group (n = 4 rats/group). Specifically, scores from 0 to ++++ (0 = normal histology, ++++=very severe injury) were assigned to each of the following characteristics: (1) pulmonary congestion; (2) bronchi: mucosal damage, leukocyte infiltration in the bronchi space, and state of regeneration (mitosis in epithelium); (3) alveoli: leukocyte infiltration in the alveoli space; (4) interstitium: interstitial edema and leukocyte infiltration of the peribronchial and perivascular spaces, and interlobular septa.
Analyses of collagen deposition
After lavage and terminal anesthesia, the left lung lobe from rats exposed by i.t. instillation was removed, rinsed in HBSS, and frozen in liquid nitrogen and then stored in −70 °C, until analysis. They were thawed on ice, 500 mg of tissue from the same part of lung from each animal was cut into small pieces, and incubated in 10 ml of pepsin dissolved in 0.5 M acetic acid (1:10 ratio of pepsin/tissue wet weight) for 24 h at 4 °C with vigorous stirring. The solution was then centrifuged at 2000 rpm for 15 min and the clear solution was used for collagen measurement. Collagen content was measured by a spectrophotometric method, Sircol™ Collagen Assay kit for rats, according to the manufacturer’s description (Biocolor Ltd., Belfast, UK). Plates were read using an ELISA reader (Thermo Fischer Scientific Oy) at 550 nm. Analysis of data was performed with the software program for the ELISA reader. The calibration curve was set up on the basis of the collagen standard provided by the manufacturer. The assay was performed in duplicate and the mean was determined for each individual sample.
Deposition of collagen and fibrin was also analyzed histologically in the right lung lobe from animals exposed to NH3 via i.t. instillation following fixation of tissue in 4% paraformaldehyde and paraffin embedding. After embedding, the tissue was cut into 3 μm thick sections and mounted on positively charged slides and stained using a Masson’s Trichrome (Sigma-Aldrich) staining protocol according to the manufacturer’s description and photos were taken at 100× magnification using light microscopy. Histological evaluations of Masson’s Trichrome stained sections were performed in a blinded manner using light microscopy for randomly selected animals in each experimental group (n = 4 rats/NH3 group).
Statistical analysis
Data are presented as means ± standard error of means (SEM). Statistical significance was assessed by parametric methods using a two-way analysis of variance (ANOVA) or when appropriate, Student’s unpaired t-test or one-way ANOVA followed by Bonferroni post-hoc test to determine differences between NH3-exposed groups and age-matched control groups. A statistical result of p < 0.05 was considered significant. For correlation analysis, Pearson’s correlation analysis was used. The GraphPad Prism program (version 6.07 GraphPad Software Inc., San Diego, CA) was used for statistical analyses and preparing graphs.
Results
Health effects after NH3-exposure via nose-only exposure
Study design
A dose-response study was performed in rats exposed to NH3 via nose-only exposure where the following non-lethal concentrations were used: Group (Gr) 1: 9000 ppm during 10 min (n = 8), Gr. 2: 9000 ppm during 20 min (n = 5), Gr. 3: 12 000 ppm during 10 min (n = 9), Gr. 4: 12 000 ppm during 15 min (n = 6), Gr. 5: 13 000 ppm during 10 min (n = 7) and Gr. 6: 15 000 ppm during 10 min (n = 3) (, data are not shown for Gr. 6 and fibrinogen analyses were not analyzed for all concentrations). Effects on these animals were evaluated up to 24 h after exposure. One group of animals exposed to 13 000 ppm group (Gr. 5) was also evaluated 14 days post-exposure (data not shown, n = 7).
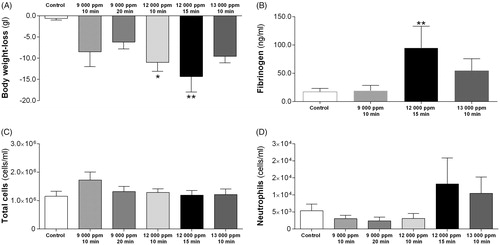
Figure 1. (A–D) Acute effects in rats, 24 h post-exposure, of anhydrous ammonia (NH3) exposure via nose-only inhalation (9000–13 000 ppm during 10–20 min); (A) recordings of body weight loss (weight before and after exposure using registered weight); (B) the level of fibrinogen in serum; (C) total cell counts (cells/ml) in bronchoalveolar lavage fluid (BALF); (D) the number of neutrophils (cells/ml) in BALF. The figure bars show increasing concentrations (ppm) of NH3 regardless of exposure time and the values indicate means ± SEM, n = 5–9 rats per group. Statistical significances of NH3-exposed compared to age-matched control rats are indicated (*p < 0.05 and **p < 0.01).
General health condition
Directly after nose-only inhalation exposure, animals showed discomfort with labored breathing but returned to normal breathing patterns within 5 h. In Gr. 3 and Gr. 4, nose-only inhalation resulted in increased salivation, nasal hemorrhagic secretions, labored breathing directly after exposure and body weight-loss at 24 h post-exposure (). Visual appearance at 5 h of all exposed rats was similar to that of control animals. The exception to this was the animals of Gr. 6, exposed to 10-min inhalation of 15 000 ppm NH3, that after 5 h suffered from reduced body circulation and showed progressive discomfort. The animals in that group were hypersensitive to anesthesia and when dissected, the GI-tract was filled with trapped air at 5 h post-exposure. Therefore, 15 000 ppm or higher concentrations of NH3 were not examined further. Animals in the 14-day group were similar to that of age-matched control animals (data not shown).
Leukocytes in BALF and fibrinogen levels in serum
At 24 h post-exposure, rats in Gr. 4 displayed elevated fibrinogen levels in serum (). Regardless of NH3-concentration, analyses of BALF showed no elevated number of inflammatory cells ().
Since animals did not have an inflammatory response at 24 h post-exposure regardless of NH3-concentration, this route of administration was not further analyzed.
Health effects after NH3-exposure via i.t. instillation
Study design
A dose-response study was performed in a limited number of rats where doses from 0.25% to 1.5% (n = 11) were tested by administrating NH3 dissolved in water via i.t. instillation (data not shown). After 24 h, NH3-exposed rats subjected to concentrations of 1.5% and higher showed discomfort, were hypersensitive to anesthesia and when dissected, the GI-tract was filled with trapped air. There was no further investigation using higher concentrations of NH3 (above 1%) due to ethical considerations, and avoiding exposing animals to fatal concentrations of NH3. The rationale for excluding higher concentration was that animals would show too much discomfort according to the permission given by the regional ethics committee on animal experiments in accordance with Swedish law. The permission admits studies in animals that induce lung damage that could only be reconstituted by careful physiological measurements, avoid evoking lung damage that could be life-threatening and avoid deterioration in visible general functions. Symptoms such as body weight loss, reduced mobility are expected to occur after exposure within the first days but to be of a transitory nature.
General health condition
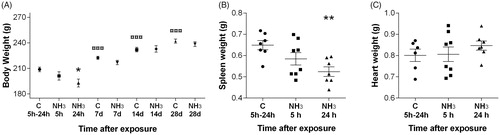
Rats exposed to 1% NH3 i.t. showed body weight loss and were affected in their general health condition up to 24 h post-exposure (). Exposed rats showed breathing obstructions, i.e. their breathing became labored with noticeable expiratory wheezing. All affected animals were recovered within a few days and at day seven the body weight was similar to that of age-matched control animals (). Rats had significantly decreased spleen weight at 24 h after NH3-exposure in comparison to control rats and the heart weight was unchanged (). At day 14, the spleen weight was back to normal (data not shown).
Figure 2. (A–C) Effects in rats subjected to ammonia (1% NH3) via intratracheal instillation on (A) body weight at 5 h–28 days after exposure, (B) spleen weight (g) and (C) heart weight (g) at 5 h and 24 h after exposure. Values indicate means ± SEM, n = 6–8 rats per group. Statistical significances of NH3-exposed animal compared to age-matched control rats (*p < 0.05 and **p < 0.01), and control groups at 14 and 28 days compared to control rats at 5 h–24 h are indicated (![]() p < 0.001).
p < 0.001).
Leukocytes in BALF and thrombocytes in blood
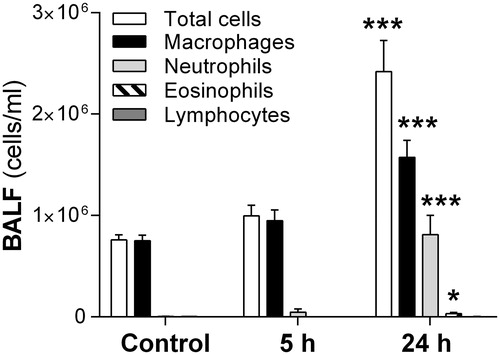
NH3-exposed (1%) rats had increased number of total leukocytes, macrophages, neutrophils (32 ± 4% of total cells), and eosinophils in BALF compared to the control group 24 h post-exposure (). At day 14, the inflammatory response in BALF was comparable with control animals (data not shown).
Figure 3. Total cell counts in bronchoalveolar lavage fluid (BALF) in rats at 5 h and 24 h after intratracheal instillation of ammonia (1% NH3). Values indicate means ± SEM, n = 5–8 rats per group. Statistical significances of NH3-exposed compared to age-matched control rats are indicated (*p < 0.05 and ***p < 0.001).
At 24 h, there was a slight increase in thrombocyte numbers in blood from rats exposed to 1% NH3 when compared to controls (p = 0.07, ).
Table 1. In female rats exposed to i.t. instillation of ammonia (1% NH3) (A) the number of thrombocytes (×109/L) in blood was measured at 24 h, (B) inflammatory mediators in bronchoalveolar lavage fluid (BALF) and serum were measured at 5 h and 24 h after exposure and (C) the amount of collagen was quantified at 14 and 28 days after exposure.
Respiratory mechanics after 1% NH3
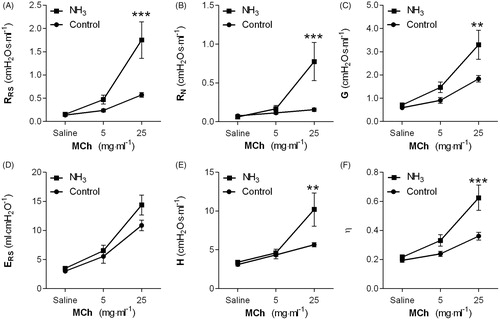
The baseline parameters, respiratory elastance (ERS: 3.0 ± 0.1 vs. 3.5 ± 0.1 ml·cmH2O−1, p < 0.05), and tissue resistance (G: 0.7 ± 0.0 vs. 0.6 ± 0.0 cmH2O·s·ml−1, p < 0.05) were the only baseline parameters significantly increased after NH3-exposure.
Recordings of the MCh-induced changes in respiratory response at 24 h post-exposure showed significant differences in respiratory resistance (RRS), Newtonian resistance (RN), tissue resistance (G), tissue elastance (H), and hysteresivity (η) between exposed and control animals (). There was no correlation between the MCh-induced respiratory responses and histopathological findings in the 24 h-group. At day 14, the respiratory physiology parameters were normalized and remained normal at 28 days post-exposure (data not shown).
Figure 4. (A–F) Respiratory mechanics in rats at 24 h after exposure intratracheal instillation of ammonia (1% NH3). Measurements of methacholine (MCh)-induced (A) RRS: respiratory resistance, (B) RN: Newtonian resistance, (C) G: tissue resistance, (D) ERS: respiratory elastance, (E) H: tissue elastance, and (F) η: hysteresivity were performed using the Flexivent™. Values indicate means ± SEM, n = 6 rats per group. Statistical significances of NH3-exposed compared to age-matched control rats are indicated (**p < 0.01 and ***p < 0.001).
Inflammatory mediators in serum and in BALF
At 5 h post-exposure (1% NH3) in serum, the level of MMP-9 was reduced and at 24 h IL-1β, MMP-9, PAI-1, and fibrinogen were significantly elevated whereas CINC-1 was significantly decreased in serum (). In BALF, only IL-18 was increased at 5 h, and IL-1β, TGFβ1, and TGFβ3 were significantly decreased at 5 h. At 24 h, MMP-9 and TGFβ1-2 were significantly increased, and IL-6 and TGFβ3 were significantly reduced when compared to controls in BALF.
At day 14, a six-fold increase of fibrinogen was measured in serum whereas levels of the other quantified mediators were comparable with control animals (). Inflammatory mediators in serum and in BALF were not analyzed at day 28.
Histopathology and collagen deposition in lung
Histopathological analysis at 5 h and 24 h post-exposure (1% NH3) revealed mild to severe epithelial desquamation characterized by degeneration, apoptotic figures, and necrosis in parts of the epithelium. There were also findings of fibrin effusions in the bronchial lumen and infiltrated neutrophils in large and small-sized bronchi at 5 h and mild to severe edema in the interstitium of both peribronchial and perivascular spaces, and interlobular septa. The amount of fibrinous exudates correlated to the extent of epithelial desquamation in the lung at 5 h ( and , ). In the 24 h group, the peribronchial space around large bronchi and the perivascular space displayed numerous polymorphs and macrophages together with mild to severe inflammation in a few small-sized bronchi. Alveoli displayed small hemorrhages with focal effusion of neutrophils and macrophages in the alveolar lumens. At 24 h there was more pulmonary congestion and less edema in the interstitium as compared to that shown in animals in the 5 h group. Shared for all animals, was that the main damage was centered in the terminal bronchioles, alveolar ducts, and adjacent alveoli. The severe lesions included damage of the epithelial cell lining, exfoliation of damaged cells and infiltrates of polymorphs and macrophages (i.e. acute inflammatory cells) which at sites filled the bronchioloalveolar lumens. Animals with more severe lesions displayed more neutrophils in bronchioloalveolar lumens (, ). Furthermore, no abnormal signs were observed in vehicle-exposed animals, suggesting that the i.t. instillation of the vehicle did not cause toxicity in the animals (, ). After 14-days, there was an increase in collagen deposition in the left lung lobe which was confirmed by quantitative measurement () but the histopathological analysis did not reveal an on-going lung tissue fibrosis at 14 or 28 days post-exposure (, ).
Figure 5. (A–F) Lung tissue sections stained with Masson’s Trichrome in order to visualize fibrin (red stain) and collagen deposition (blue stain) in small-bronchial regions (top panel C and E) and alveolar regions (lower panel, D and F) of rats subjected to ammonia (1% NH3) via intratracheal instillation evaluated in comparison to (A,B) healthy controls. Representative sections are shown from (C,D) 5 h and (E,F) 24 h post-exposure. Photos were taken at 100× magnification using light microscopy. The figure shows representative tissue sections from each group of 4 animals.
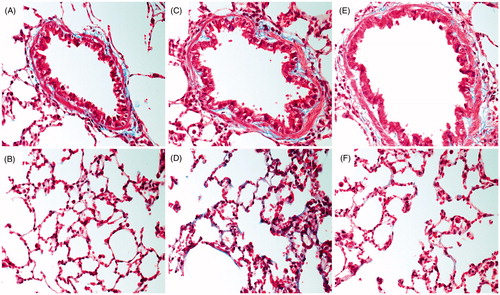
Figure 6. (A–H) Histopathological changes in alveolar (top panel C, E, and G) and bronchial regions (lower panel, D, F, and H) of rats subjected to ammonia (1% NH3) via intratracheal instillation evaluated in comparison to (A,B) healthy controls. Representative sections are shown from (C,D) 5 h, (E,F) 24 h, and (G,H) 14 days and 28 days post-exposure. Sections were stained with hematoxylin-eosin and evaluated with light microscopy using 40× magnification (n = 4 rats per group).
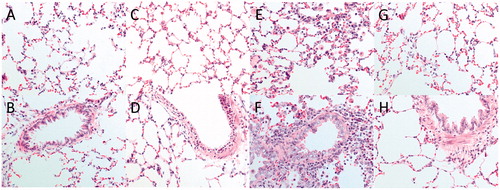
Table 2. Histopathology, scoring the main features of lung lesions at different time-points after ammonia (1% NH3) i.t. instillation exposure.
Discussion
Limited studies have addressed the effects of a single inhaled exposure to high concentrations of NH3 and the concentration of NH3 used in this study was considered to be relevant for a scenario of a large scale chemical disaster at an industrial plant or during transportation, which can result in life-threatening lung injuries. This study addresses lung effects after a single exposure to NH3 employing two experimental exposure regimens, nose-only gas inhalation, and i.t. instillation in female Sprague-Dawley rats. The exposures of NH3 were non-lethal but with detectable effects on the general health and acute responses in airways. Timing and magnitude of adverse reactions, such as the inflammatory response, involvement of the immune system both in the lung and systemically, organ weight changes, and late pulmonary abnormalities, such as pulmonary fibrosis were compared with non-exposed healthy animals.
All animals exposed to NH3 gas via the nose-only system exhibited clinical signs including nasal secretions and labored breathing which intensified with increased NH3-concentrations, during and directly after the inhalation exposure, suggesting that inhalation to NH3 gas contributes to an upper respiratory toxicity. Rats are obligate nose breathers and the nasal cavity of rats is larger relative to body size and has more inner windings than those of humans. Presumably, the inhaled NH3 gas was effectively dissolved in mucus and retained in the upper airway passages consistent with a wash-in/wash-out phenomenon (Silverman et al. Citation1949; Pauluhn Citation2013). This means that the inspired NH3 continued to be removed by the upper passages, which then returned progressively larger amounts to expired air as it passed over the mucosal surfaces in the nose preventing the inspired NH3 to go gradually deeper into the respiratory tract (Silverman et al. Citation1949) which would explain the findings in rats exposed to NH3 gas. Equivalent health effects were observed after nose-only exposure to both anhydrous and aerosolized NH3 which most likely excludes the possibility that the observed mild response was due to reaction with humid exposure air (data not shown).
Body weight loss, an indication of acute toxicity, was recorded in animals exposed to inhalation of higher concentrations than 9000 ppm of NH3 gas in comparison to controls but, in contrast to other studies (Coon et al. Citation1970; Close et al. Citation1980; Perkins et al. Citation2017), inhalation at the highest concentrations of NH3 did not result in measurable ALI, i.e. there was no evidence of lung injury, inflammatory response or physiological dysfunction. This is consistent with a study in mice, where exposure to three times the RD50 (RD: A 50% reduction in the respiration rate, 711 ppm) in mice only resulted in a slight to a moderate injury of the nasal cavity but no lower respiratory tract lesions (Zissu Citation1995). However, in our study, higher concentrations of inhaled NH3 (12 000 ppm) resulted in an upregulation of fibrinogen in serum at 24 h after exposure. A weakness in the present study was that the blood was not further analyzed with respect to e.g. elevated white blood cell count, blood gases, or pulmonary artery thrombosis which is a part of the clinical findings after NH3-exposure. In a study by Sjoblom et al. (Citation1999), using New Zealand White rabbits, NH3 exposure as an aerosol decreased PaO2, increased peak airway pressure and changed hemodynamics. However, during the six-hour observation period, the blood gas parameters improved gradually in all rabbits, suggesting that the rabbits were recovering from the NH3-induced injury. Also, it has recently been reported that exposure of male Sprague-Dawley rats via the inhalation route (head-only exposure system, 20 000 ppm NH3 during 20 min) lead to vascular effects such as increase in total blood cell counts of white blood cells, neutrophils, and thrombocytes 24 h post-exposure (Perkins et al. Citation2017).
It has previously been demonstrated that male rats are markedly more susceptible to NH3 than female rats (Appelman et al. Citation1982), however, for comparison to studies of other lung irritants in our laboratory, the present study addresses the effects of NH3 in female rats (Wigenstam, Elfsmark, Koch, et al. Citation2016). The effects after NH3 gas exposure was shown to differ between other lung-toxic chemicals which are less soluble in water such as Cl2 (Wigenstam, Elfsmark, Koch, et al. Citation2016), SO2 (Wigenstam, Elfsmark, Bucht, et al. Citation2016) and the poorly water-soluble compound, phosgene (Grainge and Rice Citation2010), which all give rise to a number of pathological changes both short and long-term. With respect to the likely presumable wash-in and wash-out phenomenon where the inhaled gas retained in the upper airways, another method to better mimic the exposure situation in humans was to expose the lung directly by i.t. instillation to circumvent possible effects of the upper airways, if they are of a protective nature. In comparison to Cl2 and SO2, NH3-exposure appears to induce another inflammatory pattern. Differences that may be coupled to different: (1) pathology of lung injury, (2) concentrations, and (3) magnitude of lung damage after exposure to the toxic chemicals. Another finding, supporting that the injury after NH3-exposure is different from Cl2 and SO2, is that animals instilled with NH3 are less hypersensitive to MCh-challenge 24 h post-exposure. Animals exposed to Cl2 and SO2 responded to the lowest concentration of MCh (5 mg/ml) probably because of another type or magnitude of lung injury such as epithelial damage at this time-point (Wigenstam, Elfsmark, Bucht, et al. Citation2016; Wigenstam, Elfsmark, Koch, et al. Citation2016). Even though, the NH3-induced inflammation in the lung was milder in comparison to the other toxic gases at 24 h, the inflammation in the terminal bronchioles contributed to the increasing forces of the baseline tissue resistance and small airway wall stiffness. At the same time-point, the highest MCh-dose induced AHR and closure or narrowing of larger and smaller airways, based on the large effects of MCh on resistance (RN and RRS), tissue resistance (G) and tissue elastance (H). The changes in respiratory mechanics, e.g. MCh-induced changes in increased hysteresivity (η), most likely reflected remodeling of the lung. Further studies are needed to determine the mechanism behind why there was no correlation between the MCh-induced changes in respiratory mechanics and histopathological findings in the 24 h-group and one hypothesis for this is that NH3 trigger afferent nerve endings that can lead to the release of physiologically active neuropeptides (Barnes Citation1995). This may, in turn, lead to increased sensitivity to exogenous provocations such as MCh, resulting in increased AHR without increased inflammation and massive damage to the lung tissue.
Similar to the other toxic lung irritants, the rats instilled with NH3 were not hypersensitive to MCh-challenge at day 14 and the respiratory physiological parameters were normalized and remained normal at subsequent time-points. This NH3-exposure model lacks the long-term hypersensitive airways (symptoms of RADS) which has previously been reported in humans exposed to lung irritants (Brooks et al. Citation1985; Aslan et al. Citation2006; Brooks and Bernstein Citation2011).
Following lung-damaging exposure, lung repair mechanisms are often initiated immediately, involving cytokine release and cell recruitment and activation of the coagulation cascade. However, differently, from other toxic lung irritants such as Cl2 and SO2, NH3-exposure did not induce a quantifiable burst of the pro-inflammatory cytokines IL-1β, IL-6, MPO, and CINC-1 in BALF at 5-24 h post-exposure preceding the recruitment of inflammatory cells into the airways. Instead, during the acute inflammatory phase, at 5 h, a significant increase of IL-18 was observed in BALF but not at the later time-point 24 h. This may indicate that IL-18 plays a critical role during the very acute stage of the NH3-induced lung injury and contributes to the systemic pro-inflammatory cytokine response (Dolinay et al. Citation2012). This is in line with the histopathological findings that the lung damage was already established at 5 h after exposure, with slightly more epithelial desquamation and interstitial edema than after 24 h post-exposure. The lung lesions had not progressed further at 24 h post-exposure. Previous studies have shown that increased levels of IL-18 correlate with disease severity, lung injury, and mortality in patients with ALI/ARDS (Dolinay et al. Citation2012; Makabe et al. Citation2012; Butt et al. Citation2016). Another specific indicator of early systemic inflammation that portends subsequent lung inflammation, injury, and ALI/ARDS is MMP-9 which was significantly increased at 24 h both in BALF and serum (Fligiel et al. Citation2006; Craig et al. Citation2015; Hsu et al. Citation2015). MMP-9 is stored in neutrophil granules, is rapidly released after cellular activation and has been shown to facilitate inflammation-driven leukocyte extravasation (macrophages migration) and endothelial permeability. In addition to being essential for an adequate inflammatory response, MMP-9 is also associated with on-going fibrotic processes and is involved in tissue repair (Santos et al. Citation2006; Albaiceta et al. Citation2008). Another cytokine, important for the acute-phase reaction, that was elevated in serum 24 h after exposure was IL-1β. This cytokine has also been shown to play an immediate and direct role in the repair of injured pulmonary tissue (Kolb et al. Citation2001). Previous studies have shown that IL-1β is a potent inducer of TGF-β, and that at least a part of its pro-fibrotic effects may be ascribed to this growth factor (Pan et al. Citation1996; Kolb et al. Citation2001; Lappalainen et al. Citation2005). In the present study, the maximum increase of TGF-β1 and TGF-β2 were seen at 24 h in BALF compared to age-matched control animals at this time-point. In humans, especially during the later stages of ALI that involves tissue repair and remodeling, TGF-β has been considered a critical factor with a central role in lung fibrosis (Fahy et al. Citation2003). Tissue fibrosis is generally considered to arise due to a failure of normal wound healing responses, i.e. if the tissue is severely damaged fibroblasts enter the location of tissue injury and produce collagen to replace lost tissue (Kovacs and DiPietro Citation1994). It has also been shown that TGF-β1 can increase alveolar epithelial permeability and act as a central mediator of ALI (Pittet et al. Citation2001; Fahy et al. Citation2003; Peters et al. Citation2014). Previous studies have demonstrated that both TGF-β1 and TGF-β2 similarly increase fibroblast collagen production. TGF-β2 may be secreted in response to injury and is involved in the host defense and wound repair by promoting mucin expression (Chu et al. Citation2004; Thompson et al. Citation2006). In comparison to age-matched control group, and divergent to data from Sprague-Dawley rats exposed to SO2 and Cl2 (Wigenstam, Elfsmark, Bucht, et al. Citation2016; Wigenstam, Elfsmark, Koch, et al. Citation2016), there was no further increase of TGF-β levels in BALF and, together with the lung histology findings, the injury was almost totally recovered 14 days post-exposure. However, even if the difference was subtle between exposed and unexposed rats, there was a significant new production or plasma leakage of collagen in the lung tissue which was confirmed by quantitative measurement of collagen deposition in lung tissue at day 14.
In contrast to most of the acute-phase inducing pro-inflammatory cytokines analyzed in BALF, concentrations of the acute-phase proteins fibrinogen and PAI-1 were significantly increased in serum after NH3-exposure, displaying a similar pattern as mice and rats exposed to Cl2 (Wigenstam et al. Citation2015; Wigenstam, Elfsmark, Koch, et al. Citation2016). The occurrence of these biomarkers is consistent with other studies, supporting the theory that the inflammatory response after a chemical exposure is not solely limited to the lung. Fibrinogen, that can leak into injured and inflamed tissues, is one of the principal plasma proteins together with thrombin, and is involved in blood clotting (e.g. homeostasis) (Mosesson et al. Citation2001). Fibrinogen is converted into fibrin (Laurens et al. Citation2006) which is a potent deactivator of pulmonary surfactants in the lung (Wygrecka et al. Citation2008). The deactivation raises the surface tension in the lung, making the smaller airspaces more prone to collapse. Fibrin breakdown products have also been shown to have the capacity to increase vascular smooth muscle contractility and directly be involved in the AHR (Wagers et al. Citation2004). The fibrinogen levels in serum correlated significantly to both the number of neutrophils (p = 0.04, r = 0.71) and the total number of cells in BALF (p = 0.04, r = 0.70) suggesting that fibrinogen increases with more extensive airway inflammation. The amount of fibrinous effusions in peripheral lung at the early time-point also correlated with increased epithelial desquamation. There was also a correlation between PAI-1 and fibrinogen in serum at 24 h (p = 0.02, r = 0.77). PAI-1 is an inhibitor of the plasminogen activator and therefore, elevation of PAI-1 may lead to an unfavorable shift in the balance between fibrin deposition and fibrinolysis. It has been shown that there is an increased risk for developing severe ALI with higher mortality rates if PAI-1 is released into the circulation (McClintock et al. Citation2008). Within few hours after exposure, there was enhanced fibrin deposition in the bronchial lumen probably due to altered bronchoalveolar fibrin turnover and coagulopathy, both markers of ALI (Allen et al. Citation2007; Bhargava et al. Citation2014; Li and Pauluhn Citation2017). Another blood cell type that has a role in homeostasis, is the thrombocyte that when activated orchestrates a sequence of events designed to promote hemostasis. Additionally, the effects of thrombocytes are pro-inflammatory and may contain large quantities of TGF-β (Yadav and Kor Citation2015). The heart of the NH3-exposed rat was not enlarged after exposure but the results point towards a NH3-induced vascular toxicity because of increased levels of fibrin in tissue and elevated thrombocyte numbers and fibrinogen/PAI-1 in blood. Further studies are needed to determine the role of the thrombocytes in this model and what set of events that trigger the thrombocytes to become activated. The elevated inflammatory biomarkers in the vascular system may also be of interest to use as an early noninvasive specific diagnostic tool for chemical-induced ALI that enables monitoring the prognosis of delayed effects after exposure and predicting the individual need of early medical treatment to avoid long-term injuries.
Conclusion
The purpose was to follow both respiratory and vascular responses after short, high-level exposures to NH3 with the overall aim to establish an animal model that can be used for new medical countermeasure strategies for NH3-induced ALI. The i.t. instillation exposure regimen using non-lethal concentrations of NH3 caused an ALI with symptoms such as acute neutrophilic lung inflammation and AHR which were followed by fibrinolytic abnormalities.
Acknowledgements
Karin Wallgren and Bo Koch are gratefully acknowledged for invaluable help with the animal experiments. Anders Bucht for thankful suggestions while conducting the experiments. This work was supported by grants from the Swedish Center for Disaster Toxicology at the National Board of Health and Welfare and the Swedish Ministry of Defense.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- AEGL. 2007. Acute exposure guideline levels for selected airborne chemicals. Vol. 6. Washington D.C, US: National Academies Press. Chapters 2–8, Ammonia prepared by the National Research Council of the National Academies; pp. 58–114.
- Albaiceta GM, Gutierrez-Fernandez A, Parra D, Astudillo A, Garcia-Prieto E, Taboada F, Fueyo A. 2008. Lack of matrix metalloproteinase-9 worsens ventilator-induced lung injury. Am J Physiol Lung Cell Mol Physiol. 294(3):L535–L543.
- Allen GB, Leclair T, Cloutier M, Thompson-Figueroa J, Bates JH. 2007. The response to recruitment worsens with progression of lung injury and fibrin accumulation in a mouse model of acid aspiration. Am J Physiol Lung Cell Mol Physiol. 292(6):L1580–L1589.
- Amshel CE, Fealk MH, Phillips BJ, Caruso DM. 2000. Anhydrous ammonia burns case report and review of the literature. Burns. 26(5):493–497.
- Appelman LM, ten Berge WF, Reuzel PG. 1982. Acute inhalation toxicity study of ammonia in rats with variable exposure periods. Am Ind Hyg Assoc J. 43(9):662–665.
- Arwood R, Hammond J, Ward GG. 1985. Ammonia inhalation. J Trauma. 25(5):444–447.
- Aslan S, Kandis H, Akgun M, Cakir Z, Inandi T, Gorguner M. 2006. The effect of nebulized NaHCO3 treatment on "RADS" due to chlorine gas inhalation. Inhal Toxicol. 18(11):895–900.
- Barnes PJ. 1995. Overview of neural mechanisms in asthma. Pulm Pharmacol. 8(4–5):151–159.
- Bates JH, Irvin CG. 2003. Measuring lung function in mice: the phenotyping uncertainty principle. J Appl Physiol. 94(4):1297–1306.
- Bhargava M, Becker TL, Viken KJ, Jagtap PD, Dey S, Steinbach MS, Wu B, Kumar V, Bitterman PB, Ingbar DH, et al. 2014. Proteomic profiles in acute respiratory distress syndrome differentiates survivors from non-survivors. PLoS One. 9(10):e109713.
- Brautbar N, Wu MP, Richter ED. 2003. Chronic ammonia inhalation and interstitial pulmonary fibrosis: a case report and review of the literature. Arch Environ Health. 58(9):592–596.
- Brooks SM, Bernstein IL. 2011. Irritant-induced airway disorders. Immunol Allergy Clin North Am. 31(4):747–768, vi.
- Brooks SM, Weiss MA, Bernstein IL. 1985. Reactive airways dysfunction syndrome (RADS). Persistent asthma syndrome after high level irritant exposures. Chest. 88(3):376–384.
- Butt Y, Kurdowska A, Allen TC. 2016. Acute lung injury: a clinical and molecular review. Arch Pathol Lab Med. 140(4):345–350.
- Chu HW, Balzar S, Seedorf GJ, Westcott JY, Trudeau JB, Silkoff P, Wenzel SE. 2004. Transforming growth factor-beta2 induces bronchial epithelial mucin expression in asthma. Am J Pathol. 165(4):1097–1106.
- Close LG, Catlin FI, Cohn AM. 1980. Acute and chronic effects of ammonia burns on the respiratory tract. Arch Otolaryngol. 106(3):151–158.
- Coon RA, Jones RA, Jenkins LJ, Jr., Siegel J. 1970. Animal inhalation studies on ammonia, ethylene glycol, formaldehyde, dimethylamine, and ethanol. Toxicol Appl Pharmacol. 16(3):646–655.
- Craig VJ, Zhang L, Hagood JS, Owen CA. 2015. Matrix metalloproteinases as therapeutic targets for idiopathic pulmonary fibrosis. Am J Respir Cell Mol Biol. 53(5):585–600.
- Dolinay T, Kim YS, Howrylak J, Hunninghake GM, An CH, Fredenburgh L, Massaro AF, Rogers A, Gazourian L, Nakahira K, et al. 2012. Inflammasome-regulated cytokines are critical mediators of acute lung injury. Am J Respir Crit Care Med. 185(11):1225–1234.
- Emerson K, Russo RC, Lund RE, Thurston RV. 1975. Aqueous ammonia equilibrium calculations; effect of pH and temperature. J Fish Res Board Can. 32:2378–2383.
- EPA. 2016. Toxicological Review of Ammonia Noncancer Inhalation - Supplemental Information―Ammonia. In: Agency USEP, editor. Washington, DC, U.S. p. 1–139.
- Fahy RJ, Lichtenberger F, McKeegan CB, Nuovo GJ, Marsh CB, Wewers MD. 2003. The acute respiratory distress syndrome: a role for transforming growth factor-beta 1. Am J Respir Cell Mol Biol. 28(4):499–503.
- Fligiel SE, Standiford T, Fligiel HM, Tashkin D, Strieter RM, Warner RL, Johnson KJ, Varani J. 2006. Matrix metalloproteinases and matrix metalloproteinase inhibitors in acute lung injury. Hum Pathol. 37(4):422–430.
- Flury KE, Dines DE, Rodarte JR, Rodgers R. 1983. Airway obstruction due to inhalation of ammonia. Mayo Clin Proc. 58(6):389–393.
- Gauldie J, Kolb M. 2008. Animal models of pulmonary fibrosis: how far from effective reality? Am J Physiol Lung Cell Mol Physiol. 294(2):L151.
- George A, Bang RL, Lari AR, Gang RK, Kanjoor JR. 2000. Liquid ammonia injury. Burns. 26(4):409–413.
- Gorguner M, Akgun M. 2010. Acute inhalation injury. Eurasian J Med. 42(1):28–35.
- Grainge C, Rice P. 2010. Management of phosgene-induced acute lung injury. Clin Toxicol (Phila). 48(6):497–508.
- Hatton DV, Leach CS, Beaudet AL, Dillman RO, Di Ferrante N. 1979. Collagen breakdown and ammonia inhalation. Arch Environ Health. 34(2):83–87.
- Henderson Y, Haggard H. 1943. Noxious gases. 2nd ed. New York: Reinhold Publishing Corporation.
- Hsu AT, Barrett CD, DeBusk GM, Ellson CD, Gautam S, Talmor DS, Gallagher DC, Yaffe MB. 2015. Kinetics and role of plasma matrix metalloproteinase-9 expression in acute lung injury and the acute respiratory distress syndrome. Shock. 44(2):128–136.
- Irvin CG, Bates JH. 2003. Measuring the lung function in the mouse: the challenge of size. Respir Res. 4(1):4.
- Johansson L, Svensson L, Bergstrom U, Jacobsson-Ekman G, Arner ES, van Hage M, Bucht A, Gafvelin G. 2005. A mouse model for in vivo tracking of the major dust mite allergen Der p 2 after inhalation. FEBS J. 272(13):3449–3460.
- Kass I, Zamel N, Dobry CA, Holzer M. 1972. Bronchiectasis following ammonia burns of the respiratory tract. A review of two cases. Chest. 62(3):282–285.
- Kerger BD, Fedoruk MJ. 2015. Pathology, toxicology, and latency of irritant gases known to cause bronchiolitis obliterans disease: does diacetyl fit the pattern?. Toxicol Rep. 2:1463–1472.
- Kolb M, Margetts PJ, Anthony DC, Pitossi F, Gauldie J. 2001. Transient expression of IL-1beta induces acute lung injury and chronic repair leading to pulmonary fibrosis. J Clin Invest. 107(12):1529–1536.
- Kovacs EJ, DiPietro LA. 1994. Fibrogenic cytokines and connective tissue production. FASEB J. 8(11):854–861.
- Lappalainen U, Whitsett JA, Wert SE, Tichelaar JW, Bry K. 2005. Interleukin-1beta causes pulmonary inflammation, emphysema, and airway remodeling in the adult murine lung. Am J Respir Cell Mol Biol. 32(4):311–318.
- Laurens N, Koolwijk P, de Maat MP. 2006. Fibrin structure and wound healing. J Thromb Haemost. 4(5):932–939.
- Lessenger JE. 2004. Anhydrous ammonia injuries. J Agromed. 9(2):191–203.
- Li W, Pauluhn J. 2017. Phosgene-induced acute lung injury (ALI): differences from chlorine-induced ALI and attempts to translate toxicology to clinical medicine. Clin Transl Med. 6(1):19.
- MacEwen J, Vernot E. 1972. Toxic hazards research unit annual report: 1972 Wright-Patterson Air Force Base, OH AMRL-TR-72-62.
- Makabe H, Kojika M, Takahashi G, Matsumoto N, Shibata S, Suzuki Y, Inoue Y, Endo S. 2012. Interleukin-18 levels reflect the long-term prognosis of acute lung injury and acute respiratory distress syndrome. J Anesth. 26(5):658–663.
- Makarovsky I, Markel G, Dushnitsky T, Eisenkraft A. 2008. Ammonia–when something smells wrong. Isr Med Assoc J. 10(7):537–543.
- McClintock D, Zhuo H, Wickersham N, Matthay MA, Ware LB. 2008. Biomarkers of inflammation, coagulation and fibrinolysis predict mortality in acute lung injury. Crit Care. 12(2):R41.
- Mosesson MW, Siebenlist KR, Meh DA. 2001. The structure and biological features of fibrinogen and fibrin. Ann N Y Acad Sci. 936:11–30.
- Pan LH, Ohtani H, Yamauchi K, Nagura H. 1996. Co-expression of TNF alpha and IL-1 beta in human acute pulmonary fibrotic diseases: an immunohistochemical analysis. Pathol Int. 46(2):91–99.
- Pauluhn J. 2013. Acute inhalation toxicity of ammonia: revisiting the importance of RD50 and LCT01/50 relationships for setting emergency response guideline values. Regul Toxicol Pharmacol. 66(3):315–325.
- Perkins MW, Wong B, Tressler J, Coggins A, Rodriguez A, Devorak J, Sciuto AM. 2016. Assessment of inhaled acute ammonia-induced lung injury in rats. Inhal Toxicol. 28(2):71–79.
- Perkins MW, Wong B, Tressler J, Rodriguez A, Sherman K, Andres J, Devorak J, Wilkins LW, Sciuto AM. 2017. Adverse respiratory effects in rats following inhalation exposure to ammonia: respiratory dynamics and histopathology. Inhal Toxicol. 29(1):32–41.
- Peters DM, Vadasz I, Wujak L, Wygrecka M, Olschewski A, Becker C, Herold S, Papp R, Mayer K, Rummel S, et al. 2014. TGF-beta directs trafficking of the epithelial sodium channel ENaC which has implications for ion and fluid transport in acute lung injury. Proc Natl Acad Sci USA. 111(3):E374–E383.
- Pittet JF, Griffiths MJ, Geiser T, Kaminski N, Dalton SL, Huang X, Brown LA, Gotwals PJ, Koteliansky VE, Matthay MA, et al. 2001. TGF-beta is a critical mediator of acute lung injury. J Clin Invest. 107(12):1537–1544.
- Santos FB, Nagato LK, Boechem NM, Negri EM, Guimaraes A, Capelozzi VL, Faffe DS, Zin WA, Rocco PR. 2006. Time course of lung parenchyma remodeling in pulmonary and extrapulmonary acute lung injury. J Appl Physiol (1985). 100(1):98–106.
- Silverman L, Whittenberger JL, Muller J. 1949. Physiological response of man to ammonia in low concentrations. J Industr Hyg Toxicol. 31(2):74–78.
- Sjoblom E, Hojer J, Kulling PE, Stauffer K, Suneson A, Ludwigs U. 1999. A placebo-controlled experimental study of steroid inhalation therapy in ammonia-induced lung injury. J Toxicol Clin Toxicol. 37(1):59–67.
- Sobonya R. 1977. Fatal anhydrous ammonia inhalation. Hum Pathol. 8(3):293–299.
- Stumm W, Morgan JJ. 1981. Aquatic chemistry: an introduction emphasizing chemical equilibria in natural waters. 2nd ed. New York: John Wiley & Sons Ltd.
- Thompson HG, Mih JD, Krasieva TB, Tromberg BJ, George SC. 2006. Epithelial-derived TGF-beta2 modulates basal and wound-healing subepithelial matrix homeostasis. Am J Physiol Lung Cell Mol Physiol. 291(6):L1277–L1285.
- UN. 2014. Report of the independent international commission of inquiry on the Syria Arab Republic. United Nations General Assembly, Human Rights Counsil A/HRC/27/60.
- Wagers SS, Norton RJ, Rinaldi LM, Bates JH, Sobel BE, Irvin CG. 2004. Extravascular fibrin, plasminogen activator, plasminogen activator inhibitors, and airway hyperresponsiveness. J Clin Invest. 114(1):104–111.
- Wigenstam E, Elfsmark L, Bucht A, Jonasson S. 2016. Inhaled sulfur dioxide causes pulmonary and systemic inflammation leading to fibrotic respiratory disease in a rat model of chemical-induced lung injury. Toxicology. 368–369:28–36.
- Wigenstam E, Elfsmark L, Koch B, Bucht A, Jonasson S. 2016. Acute respiratory changes and pulmonary inflammation involving a pathway of TGF-beta1 induction in a rat model of chlorine-induced lung injury. Toxicol Appl Pharmacol. 309:44–54.
- Wigenstam E, Jonasson S, Koch B, Bucht A. 2012. Corticosteroid treatment inhibits airway hyperresponsiveness and lung injury in a murine model of chemical-induced airway inflammation. Toxicology. 301(1–3):66–71.
- Wigenstam E, Koch B, Bucht A, Jonasson S. 2015. N-acetyl cysteine improves the effects of corticosteroids in a mouse model of chlorine-induced acute lung injury. Toxicology. 328:40–47.
- Wiklund L, Ronquist G, George M. 2001. Ammonia–not the culprit. Arch Dis Child. 84(6):525.
- Wygrecka M, Jablonska E, Guenther A, Preissner KT, Markart P. 2008. Current view on alveolar coagulation and fibrinolysis in acute inflammatory and chronic interstitial lung diseases. Thromb Haemost. 99(3):494–501.
- Yadav H, Kor DJ. 2015. Platelets in the pathogenesis of acute respiratory distress syndrome. Am J Physiol Lung Cell Mol Physiol. 309(9):L915–L923.
- Zissu D. 1995. Histopathological changes in the respiratory tract of mice exposed to ten families of airborne chemicals. J Appl Toxicol. 15(3):207–213.