
Abstract
Objective
Ammonia (NH3) is a corrosive alkaline gas that can cause life-threatening injuries by inhalation. The aim was to establish a disease model for NH3-induced injuries similar to acute lung injury (ALI) described in exposed humans and investigate the progression of lung damage, respiratory dysfunction and evaluate biomarkers for ALI and inflammation over time.
Methods
Female BALB/c mice were exposed to an NH3 dose of 91.0 mg/kg·bw using intratracheal instillation and the pathological changes were followed for up to 7 days.
Results
NH3 instillation resulted in the loss of body weight along with a significant increase in pro-inflammatory mediators in both bronchoalveolar lavage fluid (e.g. IL-1β, IL-6, KC, MMP-9, SP-D) and blood (e.g. IL-6, Fibrinogen, PAI-1, PF4/CXCL4, SP-D), neutrophilic lung inflammation, alveolar damage, increased peripheral airway resistance and methacholine-induced airway hyperresponsiveness compared to controls at 20 h. On day 7 after exposure, deteriorating pathological changes such as increased macrophage lung infiltration, heart weights, lung hemorrhages and coagulation abnormalities (elevated plasma levels of PAI-1, fibrinogen, endothelin and thrombomodulin) were observed but no increase in lung collagen. Some of the analyzed blood biomarkers (e.g. RAGE, IL-1β) were unaffected despite severe ALI and may not be significant for NH3-induced damages.
Conclusions
NH3 induces severe acute lung injuries that deteriorate over time and biomarkers in lungs and blood that are similar to those found in humans. Therefore, this model has potential use for developing diagnostic tools for NH3-induced ALI and for finding new therapeutic treatments, since no specific antidote has been identified yet.
Introduction
Ammonia (NH3) is an industrial gas that is highly toxic and produced in large volumes due to its wide industrial application. Although often handled as a liquefied gas, a significant emission of NH3 would quickly generate a large gas cloud that could spread over vast areas. Exposure causes a wide range of injuries, documented both in humans and in animal models (AEGL Citation2007), and which need extensive and long-term medical care. The toxicity of NH3 primarily relates to its corrosive and alkaline properties that produce cytotoxic effects immediately upon contact with moist tissue (Agren et al. Citation2021).
The clinical manifestations after NH3-induced injuries show similarities to acute lung injury (ALI) or airway respiratory distress syndrome (ARDS) in humans, and the pathogenesis is very complex. Some of the pathophysiological findings after exposure to NH3 include hypoxia, mucus secretions, sloughed epithelia, emphysema, edema and reactive smooth muscle contractions in the airways, which may lead to impaired respiratory function and a need for ventilator support; or in the worst-case scenario, death (Kass et al. Citation1972; Sobonya Citation1977; Hatton et al. Citation1979; Flury et al. Citation1983; George et al. Citation2000; Wiklund et al. Citation2001; Brautbar et al. Citation2003; Lessenger Citation2004; Makarovsky et al. Citation2008; Kerger and Fedoruk Citation2015). In addition to pulmonary effects, several systemic and vascular measures are affected after inhalation exposure to NH3; such as increased heart rate and blood pressure, bradycardia, increased white blood cell count, thrombosis, hemorrhages and cardiac arrest accompanied by cyanosis (White Citation1971; Sobonya Citation1977; Montague and Macneil Citation1980; Ward et al. Citation1983; George et al. Citation2000).
In the hemostatic response to hemorrhage, the coagulation cascade plays an important role in promoting blood clotting and in orchestrating inflammatory and tissue repair responses. Increased coagulation and impaired or reduced fibrinolysis are associated with ALI/ARDS and have been shown to contribute to the exacerbation of symptoms, such as changes in fibrinogen, thrombomodulin, PAI-1 and endothelin expression (Idell et al. Citation1991; Ware et al. Citation2006; Wheeler and Bernard Citation2007; Sebag et al. Citation2011; Matthay et al. Citation2019; Whyte et al. Citation2020; Omiya et al. Citation2021). Several biomarkers have been proposed to predict morbidity and mortality of ALI/ARDS which include both pro-inflammatory cytokines: IL-1β, IL-2, IL-4, IL-6, IL-8, IL-18 and TNF-α and; other inflammatory mediators: receptor for advanced glycation end products (RAGE), angiopoietin-2 (Ang-2) and surfactant protein-D (SP-D) expressed during the exudative phase (Binnie et al. Citation2014; Mokra and Kosutova Citation2015; Butt et al. Citation2016; Spadaro et al. Citation2019).
Exposure to both sub-lethal and lethal concentrations of NH3 has previously been evaluated in animals (Appelman et al. Citation1982; Pauluhn Citation2013; Perkins et al. Citation2016; Citation2017; Elfsmark et al. Citation2019; Bein et al. Citation2021) but a limited number of studies have addressed the development of ALI/ARDS. There is a very steep concentration-mortality relationship for NH3, which complicates investigations into severe but not fatal injuries in animals. The aim of this study was to evaluate a set of biomarkers associated with inflammation, epithelial damage, endothelial damage and coagulation that have been identified in clinical studies as biomarkers for ALI/ARDS morbidity. In addition, the following fibroproliferative phase of ALI/ARDS including proliferation of fibroblasts, epithelial and endothelial cells was investigated after NH3 exposure by analyzing lung tissue collagen, endoglin (a TGF-β co-receptor) and platelet factor 4 (PF4/CXCL4). The release of PF4/CXCL4 from activated platelets induces neutrophil infiltration, pulmonary edema and lung tissue damage in sepsis-induced ALI/ARDS (Hwaiz et al. Citation2015) but has also been shown to promote lung fibrosis in vivo (Affandi et al. 2022).
Studies of combinations of biomarkers can provide a greater understanding of the processes that manifest as ALI/ARDS after chemical exposure. The present study was therefore designed to determine a dose of NH3 that causes injuries, which replicates the chemically induced lung damage in humans. However, there are some major challenges that need to be circumvented because mice are obligate nose breathers with a larger nasal cavity than humans, relative to body size. Moreover, rodents respond to NH3 inhalation exposures with reflex bradypnea which reduces minute ventilation to 30% of normal while oral breathing humans respond with hyperventilation (minute ventilation of 300%) (Silverman et al. Citation1949; Pauluhn Citation2013). In this study, the intratracheal instillation was used as an exposure method previously described for NH3 to study chemical-induced tracheobronchial-pulmonary injury in rodents (Elfsmark et al. Citation2019). The development of acute injuries was evaluated over time by analyzing changes in respiratory parameters (structural changes and airway hyperresponsiveness in small and large airways) and by histopathological evaluation of lung tissue. The differences in anatomy and respiratory responses in humans underline the importance of cautious interpretation of animal data. Since no specific antidote has been identified so far and medical management is primarily supportive, life-saving, and symptom-relieving (Makarovsky et al. Citation2008; Padappayil and Borger Citation2019), finding relevant animal models is highly motivated for the development of new treatment strategies for NH3-induced ALI/ARDS.
Materials and methods
Animals
All animal experiments were performed on female mice, as there are no indications that the chemical-induced injury differs between the sexes. BALB/c mice (9–10 weeks old, Envigo RMS B.V, Netherlands) were housed in a conventional animal facility; in plastic cages with absorbent bed material, and maintained on a 12-hour daylight cycle, 22 °C, with 30% relative humidity. Food (R36, Lantmännen, Sweden) and water were provided ad libitum. Mice were placed in a group of 6 in each cage and cages were enriched with nesting materials and plastic tubes. All mice were weighed before exposure to NH3 (average body weight (bw) from start: 20 ± 1.4 g) and their general health status was monitored daily after exposure. The animals were terminated after the experiment with an overdose of pentobarbital sodium. The care of the animals and experimental protocols were approved by the regional ethics committee for animal experiments in Umeå, Sweden; according to Directive 2010/64/EU.
Ammonia exposure and experimental design
NH3 (25% weight percent NH3, analytical grade, Merck Eurolab AB, Sweden) was diluted in water and administered by intratracheal (i.t.) instillation in a volume of 50 μL to anesthetized (enflurane) mice. The NH3 dose was determined from a small pilot study where different doses from 54.6 mg/kg·bw to 113.8 mg/kg·bw were tested via i.t. instillation and the toxic responses were evaluated at 20 h after exposure. Control mice, which received only the solvent, showed no response that differed from unexposed healthy mice.
For the time-course study, an NH3 dose of 91.0 mg/kg·bw was chosen based on the rationale that it caused a moderate effect on general health and lung toxicity (bodyweight loss and pulmonary inflammation) at 20 h, in line with the ALI described after chlorine exposure (Jonasson et al. Citation2013). Toxic responses in the mice were evaluated at 4 h, 20 h, 48 h or 7 days after exposure. However, the initially moderate injury induced by 91.0 mg/kg·bw at 20 h deteriorated over a 7-day follow-up period to lethal injuries and therefore, the collagen assessment experiment was executed (one week after exposure) using a lower NH3 dose of 72.8 mg/kg·bw that caused moderate injuries. At room temperature, the estimated pH of the solution was 11.75, hence approximately ≥90% of NH3 is in its un-ionized form (Emerson et al. Citation1975). A schedule of selected times for sampling of body weight, leukocytes in BALF, inflammatory mediators in BALF and plasma, whole blood analyzes, organ weights, respiratory mechanics, histopathology and collagen after instillation of NH3 are summarized in .
Figure 1. Schematic illustration of the experimental design of the study, including dose-response, collagen analysis and time course studies. The illustration shows the time axis after exposure to ammonia (NH3) via intratracheal instillation (i.t) and the analyses performed at the different time points; 4 h up to 7 days. (BALF: bronchoalveolar lavage fluid).
Respiratory mechanics
Measurement of respiratory mechanics was performed on anesthetized, paralyzed and tracheostomized mice as previously described (Hantos et al. Citation1992; Gomes et al. Citation2000; Jonasson et al. Citation2013), 20–24 h after NH3 exposure, using a small ventilator (FlexiVent; Scireq, Montreal, Quebec, Canada). The parameters recorded at baseline and after inhaled methacholine (MCh, acetyl-β-methylcholine chloride, Sigma-Aldrich (St. Louis, MO, USA)) were: respiratory resistance (RRS), Newtonian resistance, (RN), tissue resistance (G), respiratory elastance (ERS), and tissue elastance (H). To measure airway hyperresponsiveness (AHR), incremental doses of MCh were given at 5-min intervals. The MCh, diluted in saline to a volume of 20 µL, was given for 10 s as an aerosol (Aeroneb™ PRO, SCIREQ®). Each dose of MCh (0, 50 and 100 mg/mL) was aerosolized without affecting the ventilation pattern. The mice were sacrificed after the last recording of respiratory mechanics by a combination of chemical anesthesia, terminal retro-orbital bleeding and bilateral pneumothorax.
Blood sampling
After final anesthesia, blood was withdrawn by retro-orbital bleeding at 4, 20–24 and 48 h after exposure, collecting venous blood (Hoff Citation2000) that was then analyzed regarding; electrolytes, blood gases, blood chemistry, inflammatory mediators, cytokines and platelet numbers. On day 7, blood was prepared for analysis of inflammatory mediators and cytokines only.
Analysis of inflammatory mediators and cytokines was performed in plasma prepared from blood treated with K2EDTA. The plasma was retrieved after centrifugation (15 min, 20 °C, 3000 rpm) and stored at −70 °C until analysis. Analysis of hematology (hemoglobin (Hgb), Hematocrit (Hct)), chemistry (blood urea nitrogen (BUN), creatinine, ionized calcium (iCa) and glucose (Glu)), electrolytes (chloride (Cl−), sodium (Na+) and potassium (K+)), acid-base (pH, pCO2, HCO3, TCO2, anion gap and base excess), blood gases (pO2 and sO2) and lactate in whole blood (95 µL) was performed using a hand-held VetScan i-STAT analyzer (Abaxis via Triolab, Sweden).
Platelet-rich plasma (PRP) was prepared from K2EDTA-treated blood that was diluted (1:25) in 5 mL of ISOTON® II dilutent buffer (Beckman Coulter Inc.) and centrifuged for 90 seconds at 1720 rpm. After a second dilution step (1:200) of 100 µL in the PRP supernatant, the number of platelets (above 1.5 µm in size) was analyzed using an automatic cell counter, Z2 Coulter Particle Count and Size Analyzer (Beckman Coulter Life Sciences, Indianapolis, US), with a 70 µm aperture tube.
Bronchoalveolar lavage fluid (BALF) and organ weighing
BALF was collected for total and differential cell count analysis at 4 h, 20 h or 7 days after NH3 exposure by injecting and withdrawing 4 × 1 mL of ice-cold Hank’s balanced salt solution (HBSS, Sigma-Aldrich) through the endotracheal tube of euthanized mice. The BALF was centrifuged (1500 rpm, 10 min, 4 °C) and the supernatant was stored at −70 °C for later analysis. The number of leukocytes was counted in a Bürker chamber using trypan blue staining. From BALF for each animal, 20 000 cells were fixed on duplicate slides using a Cytospin® centrifuge (Shandon© cytospin 3 cyto-centrifuge, cell preparation system, Runcorn, UK) and stained with May-Grünwald-Giemsa reagents (Merck Millipore, VWR International, Sweden). The slides were blinded and a differential cell count of leukocyte subtypes (including macrophages, neutrophils, eosinophils and lymphocytes) was performed, counting 300 cells per slide. After collecting BALF from all mice, the heart, liver and spleen were dissected and weighed at all times after exposure.
Histopathology
After BALF was collected, the lung from animals both in the healthy control and in the 91.0 mg/kg·bw NH3 i.t. instillation group was fixed by placing the tissue in 4% paraformaldehyde until paraffin embedding. After embedding, the tissue was cut into 3 μm thick sections and mounted on positively charged slides. The sections were deparaffinized, dehydrated, and stained with hematoxylin and eosin before histopathological evaluations were performed in a blinded manner using light microscopy (20 and 40x magnification respectively) on animals from each experimental group (n = 3–4 mice/group, supplementary (S1) table). Specifically, scores (0 = normal histology; to 4= very severe injury) were assigned to each of the following characteristics: 1) pulmonary congestion; 2) bronchial mucosal damage, leukocyte infiltration and state of regeneration (mitosis in epithelium) in the bronchial space; 3) alveolar leukocyte infiltration and alveolar damage); 4) interstitial edema/congestion/hemorrhages and leukocyte infiltration of the peribronchial and perivascular spaces, and interlobular septa.
Analyses of collagen deposition
One week after exposure the left lung lobe was removed for assessment of deposited collagen, rinsed in HBSS, frozen in liquid nitrogen and then stored at −70 °C until analysis. For this analysis, a separate experiment was performed with a less toxic NH3 dose (72.8 mg/kg·bw) that did not cause lethal injuries at later times and time-matched healthy animals were used as controls. On the day of analysis, lungs were thawed on ice, cut into small pieces, and incubated in 10 mL of pepsin dissolved in 0.5 M acetic acid (1:10 ratio of pepsin/tissue wet weight) for 24 h at 4 °C with vigorous stirring. The solution was centrifuged at 2000 rpm for 15 min and the clear supernatant was used for collagen measurement. The collagen content was measured by a spectrophotometric method, the Sircol™ Collagen Assay kit for mice, according to the manufacturer’s description (Biocolor Ltd., Belfast, UK). Plates were analyzed with an ELISA reader (Thermo Fischer Scientific Oy) at 550 nm and data analysis was performed with the instrument software program provided. The calibration curve was set using the collagen standard included in the assay kit.
Determining mediators in BALF and blood
ELISA analyses
Inflammatory mediators were analyzed individually with specific ELISA kits for the presence of angiopoietin-2 (Ang-2), endoglin, endothelin, matrix metallopeptidase 9 (MMP-9), plasminogen activator inhibitor-1 (PAI-1), platelet factor 4 (PF4/CXCL4), receptor for advanced glycation end products (RAGE), surfactant protein D (SP-D), thrombomodulin (R&D Systems, Inc.) and murine fibrinogen (MFG-EIA, Enzyme Research Laboratories, Swansea, UK); according to the manufacturer’s instructions and analyzed with an ELISA reader (Thermo Scientific Mutilskan FC, Thermo Fischer Scientific Oy, Vantaa, Finland). Data analysis was performed using the ELISA reader software program (SkanIt for Multiskan FC 3.1. Ink), referring to the analyte-specific standards added to each plate.
Bio-Plex™ analyses
Inflammatory mediators in BALF and plasma in NH3-exposed mice were analyzed for the presence of interleukin (IL)-1α, IL-1β, IL-2, IL-3, IL-4, IL-5, IL-6, IL-9, IL-10, IL-12α, IL-12β, IL-13, IL-17, eotaxin, G-CSF, GM-CSF, IFNγ, KC, MCP-1, MIP-1α, MIP-1β, RANTES and TNFα. All cytokine analyses were performed simultaneously with a multiplex kit (Bio-Plex™ Pro Mouse Cytokine 23-plex panel, Group I) according to the manufacturer's instructions (Bio-Rad) and analyzed on a Bio-Plex™ system (Luminex Bio-Plex™ 200 System, Bio-Rad, Hercules, CA).
Statistical analysis
Data are shown as a scatter plot with a line indicating the mean or as mean ± standard error of the mean (SEM) in the graphs. Statistical significance was assessed by parametric methods using Student’s unpaired t-test, one-way analysis of variance (ANOVA) or two-way analysis of variance (ANOVA) to determine differences between groups. ANOVA tests were followed by the Bonferroni post hoc test. A statistical result of p < 0.05 was considered significant. The statistical analyzes were performed and graphs were prepared using the GraphPad Prism program (version 8.0 GraphPad Software Inc., San Diego, CA, USA).
Results
Evaluation of dose-response
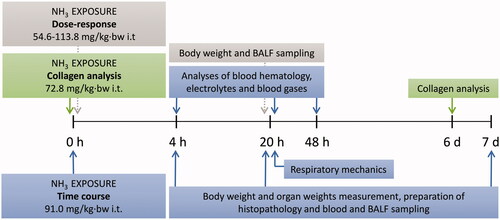
A dose-response study was performed in mice exposed to NH3 i.t. where the following doses were used: Group (Grp.) 1: 54.6 mg/kg·bw (n = 7), Grp. 2: 72.8 mg/kg·bw (n = 7), Grp. 3: 91.0 mg/kg·bw (n = 7) and Grp. 4: 113.8 mg/kg·bw (n = 7). The effects of NH3 on these animals were evaluated 20 h after exposure (). All exposed groups showed significant body weight loss () and increased infiltration of neutrophils in BALF. Compared to controls, neutrophilia was statistically significant at the highest dose (), but the animals in this group also showed lethargy and severe wheezing at 20 h. Based on these results, NH3 doses of 113.8 mg/kg·bw or higher were not used in further experiments due to ethical considerations. The NH3 dose of 91.0 mg/kg·bw was therefore chosen for the time course study. However, transient symptoms such as loss of body weight due to changes in eating behavior and reduced mobility are expected to occur during the first days after exposure.
Figure 2. Results from the dose-response study show (A) body weight (BW)-loss, (B) the total number of leukocytes in bronchoalveolar lavage fluid (BALF), and (C) the percentage of neutrophils in BALF at 20 h after exposure to increasing doses of ammonia (NH3) via intratracheal instillation. Scatter plots with means are indicated for each group. One-way analysis of variance (ANOVA), ***p < 0.001 vs. control group.
Time course of toxic responses
Observed health effects after four hours
Initially, 4 h after exposure, the animals were lethargic and showed labored breathing, but no significant body weight loss was observed and organ weights did not differ from control animals (). Mice showed no histopathologic manifestations in lung tissue compared to control mice (data not shown) but upregulation of cytokines in BALF was seen (IL-1α, IL-1β, IL-5, IL-6, G-CSF, KC, TNFα, MIP-1β and MMP-9) (). The total number of leukocytes was not elevated but a small significant increase in neutrophils was detected in BALF at 4 h (). In plasma, there was an increase in IL-6, G-CSF, KC, Fibrinogen, SP-D, MMP-9 and a decrease in Ang-2, IL-12 and IL-13 ( and ). In addition, NH3-exposed mice showed significantly lower levels of Cl-, BUN, pO2 and sO2; and higher levels of pCO2 and HCO3 in venous blood, compared to control animals (). Platelet counts increased significantly in whole blood after NH3 exposure ().
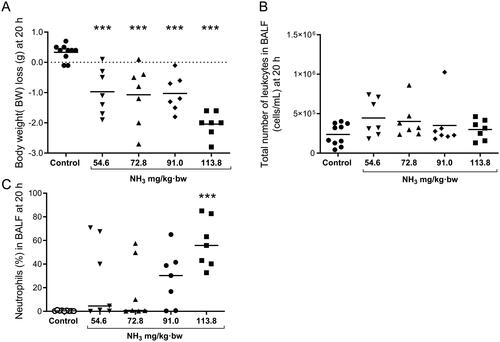
Figure 3. Results from the time course study show (A) body weight (BW)-loss, (B) spleen weights (C) liver weights, and (D) heart weights at 4 h, 20 h and 7 days (% of BW) after exposure to an ammonia (NH3) dose of 91.0 mg/kg·bw via intratracheal instillation and in time-matched controls. Scatter plots with means are indicated for each group. Student’s unpaired t-test, *p < 0.05, **p < 0.01 and ***p < 0.001 between NH3-group and the time-matched control group.
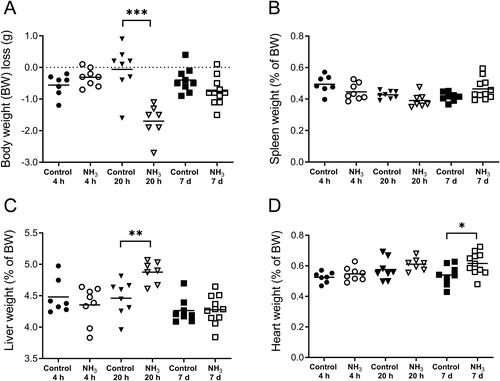
Figure 4. Results from the full-scale study show the total number of (A) leukocytes, (B) macrophages, and (C) neutrophils in bronchoalveolar lavage fluid (BALF) at 4 h, 20 h and 7 days after an ammonia (NH3) dose of 91.0 mg/kg·bw via intratracheal instillation and controls. Scatter plots with means are indicated for each group. Student’s unpaired t-test, *p < 0.05 and **p < 0.01 between NH3-group and the time-matched control group.
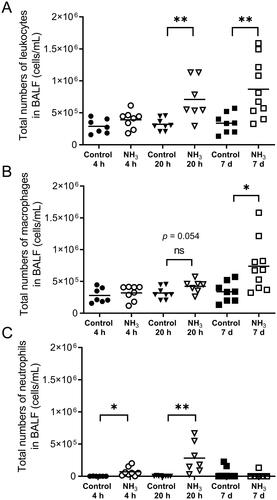
Figure 5. Analyzes of the level of (A) fibrinogen (B) plasminogen activator inhibitor-1 (PAI-1) (C) platelet factor 4 (PF4/CXCL4) (D) endothelin, (E) thrombomodulin, (F) endoglin (G) the receptor for advanced glycation end products (RAGE), and (H) angiopoietin-2 (Ang-2) at 4 h, 20 h and 7 days after exposure to an ammonia (NH3) dose of 91.0 mg/kg·bw via intratracheal instillation and in time-matched controls. Scatter plots with means are indicated for each group. Student’s unpaired t-test, *p < 0.05, **p < 0.01 and ***p < 0.001 between NH3-group and the time-matched control group.
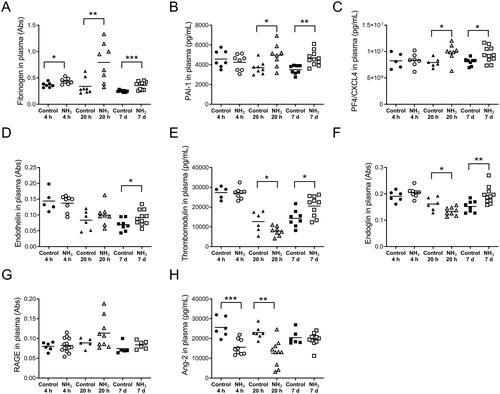
Table 1. Inflammatory mediators in bronchoalveolar lavage fluid (BALF) were measured at 4 hours, 20 hours and 7 days after ammonia (91.0 mg/kg·bw) i.t. instillation exposure.
Table 2. Inflammatory mediators in plasma were measured at 4 hours, 20 hours and 7 days after ammonia (91.0 mg/kg·bw) i.t. instillation exposure.
Table 3. Venous blood is characterized by hematology, chemistry, electrolytes, acid-base, blood gases, lactate and thrombocyte numbers; at 4 hours, 20 hours and 48 hours following exposure to ammonia (91.0 mg/kg·bw) i.t. instillation.
Observed health effects after 20 hours
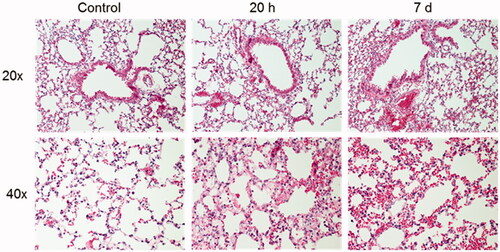
Health deteriorated over time and several of the animals showed clear signs of anorexia, lethargy, dyspnea, wheezing and hypothermia after exposure. Compared to the initial starting weight, all NH3-exposed mice showed significant body weight loss (). There was a significant increase of total leukocytes, mainly neutrophils, in the airways and alveoli compared to control animals (). Baseline parameters for both respiratory resistance (RRS) and tissue resistance (G) increased significantly after NH3 exposure (, baseline parameters). All measured respiratory parameters in NH3-exposed lungs changed significantly when exposed to methacholine: including respiratory (RRS), Newtonian (RN) and tissue (G) resistance; as well as respiratory (ERS) and tissue (H) elastance (). Histopathological evaluation of hematoxylin and eosin-stained lung tissue sections showed that injuries following exposure to NH3 were apparent after 20 h and were confined to peripheral parts (appearing as alveolar regions were more prone to be damaged) in the lungs, in line with the results of respiratory mechanics on peripheral airways. Damage to the alveolar region could have been induced directly by NH3 or indirectly by making the walls of the alveoli more fragile and sensitive to the mechanical forces of bronchoalveolar lavage. The larger airways (bronchi) appeared mainly unaffected regarding mucosal damage, regeneration (mitosis of the epithelium) and inflammatory infiltrates. Pulmonary congestion and mild interstitial edema could be noted along with an inflammatory cell reaction in the peribronchial and perivascular areas (). Inflammatory profiling of BALF and plasma revealed significantly altered inflammatory patterns ( and , ) in exposed animals. In BALF, the mediators IL-1α, IL-1β, IL-5, IL-6, IL-12p40, G-CSF, MCP-1, MIP-1α, MIP-1β, MMP-9, SP-D and RANTES increased; in plasma, corresponding biomarkers increased were IL-6, IL-10, G-CSF, KC, fibrinogen, PAI-1, PF4/CXCL4 and SP-D. At the same time, Ang-2, IL-9, IL-12, thrombomodulin and endoglin were significantly down-regulated in plasma together with IL-9 in BALF. The other mediators analyzed in BALF and plasma were not affected by the exposure. At 20 h, the mice also showed significantly lower levels of Cl-, BUN, pO2 and sO2; and higher levels of pCO2, TCO2, Hct and HCO3 in venous blood, compared to control animals. The platelet count in whole blood was not increased at 20 h after NH3 exposure ().
Figure 6. Respiratory mechanics in mice at 20 h after exposure to an ammonia (NH3) dose of 91.0 mg/kg·bw compared to healthy controls. Measurements of methacholine (MCh)-induced (A) respiratory resistance, RRS, (B) Newtonian resistance, RN, (C) tissue resistance, G, (D) respiratory elastance, ERS, and (E) tissue elastance, H, was performed using the Flexivent™. Baseline parameters show data from NH3 exposed animals and control animals recorded without MCh provocation. Values indicate means ± SEM. Two-way analysis of variance (ANOVA) to determine differences between groups followed by Bonferroni post hoc test, *p < 0.05, **p < 0.01 and ***p < 0.001 vs. control group.
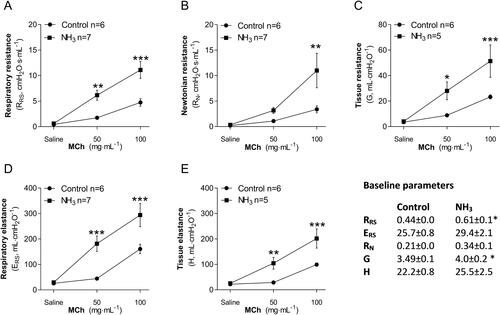
Figure 7. Histopathological evaluation of lung sections from mice exposed to an ammonia dose of 91.0 mg/kg·bw at two different time-points after exposure, 20 h and 7 days, and healthy controls. Photographs show representative bronchial (upper panels) and alveolar areas (lower panels) of the lungs at each time. Sections stained with hematoxylin and eosin and evaluated using light microscopy using 20 and 40x magnification.
Electrolytes and blood gases in venous blood were also monitored on day 2 (48 h) after exposure (). Exposed mice showed significantly lower levels of Cl-, glucose, pO2 and sO2; and higher levels of pCO2, Na+, TCO2, Hct, Hb and HCO3 in venous blood, compared to time-matched control animals. The number of platelets remained within the normal range at this time.
Observed health effects after seven days
The health of the exposed animals deteriorated over time and by day 7 after exposure, many of the mice had not recovered, showing decreased body weight and labored breathing and wheezing. The survival rate after 7 days was estimated at 69% (11/16 animals survived). Animals in this group were not healthy enough to survive respiratory mechanics measurements; many animals were excluded from this analysis since they died during anesthesia, but all other biological samples were taken from these individuals. Analysis revealed an increase in the ratio of heart weight to body weight in mice exposed to NH3 that was not evident in control animals ().
On day 7, the animals had a significantly higher number of BALF leukocytes than control animals and cell differentiation confirmed an inflammatory profile with a marked increase in macrophages (). Histopathological evaluation of the lung showed that the findings observed at 20 h were still present but had deteriorated into more severe lesions in bronchial mucosa, alveoli and interstitium. Interstitial hemorrhages increased at day 7 compared to both the control animals and the 20-h-group. The inflammatory cell response in the peribronchial alveoli and perivascular areas had returned to control levels in all but one individual (), although there were still increased concentrations of IL-1α, IL-1β, IL-12, IL-13, MCP-1, TNFα, and RANTES in the BALF from surviving animals. Some of the mediators that were significantly upregulated in BALF at 20 h had returned to control levels (IL-6, G-CSF, KC, MIP-1α, MIP-1β, SP-D, MMP-9) at day 7, whereas the cytokine IL-13 were upregulated in BALF compared to control animals only at this time (). In plasma, there were increased levels of fibrinogen, PAI-1, PF4/CXCL4, endothelin, thrombomodulin and endoglin. Some of the biomarkers (IL-6, IL-10, G-CSF, KC and SP-D) that were significantly upregulated after 20 h in plasma were now equal to the levels in control animals. One week after exposure, levels of IL-9, IL-12, IL-13, and RANTES were significantly down-regulated in plasma compared to control animals ( and ). To avoid life-threatening symptoms shown on day 7, another group of animals were exposed to a lower dose of NH3 (72.8 mg/kg·bw) and analyzed one week later for lung collagen deposition, but the amount of collagen did not differ from the control animals (data not shown).
Discussion
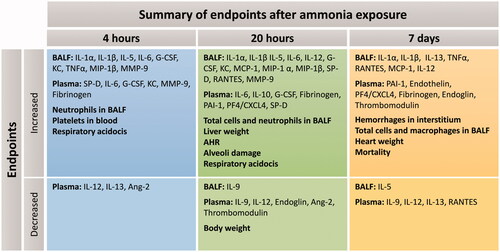
In this murine study, we show that exposure to inhaled NH3 induces a markedly toxic response, not only locally but also systemically, damages that deteriorate over time. Several of the symptoms and biomarkers identified after NH3 exposure are also significant for ALI/ARDS in humans. There are currently no effective medical treatments to improve the health outcome after exposure to NH3 and the treatment given is mainly supportive and symptom-relieving. One aim of this study was therefore to find a dose of NH3, which induces non-lethal damage in mice and that can be used as a disease model for evaluation of diagnostic tools and therapeutic treatments. Another aim was to improve the understanding of NH3-induced ALI, focusing on the manifestations of symptoms and systemic changes after exposure (), for that reason, it was important to conduct a wide range of pathological examinations over time.
Figure 8. Summary of all results at each time point after exposure to an ammonia dose of 91.0 mg/kg·bw via intratracheal instillation (at 4 h, 20 h and 7 days). The top row (increased) shows induced biomarkers and pathophysiological conditions, and the bottom row (decreased) biomarkers decrease in relation to time-matched control groups. (BALF: bronchoalveolar lavage fluid. AHR: airway hyperresponsiveness).
To establish a model for NH3-induced lung injury, an intratracheal instillation method was used as previously described in rats (Elfsmark et al. Citation2019). The rationale for selecting this administration route was based on a pilot study in which mice exposed by nose-only inhalation to anhydrous NH3 gas (750–6750 ppm · h, data not shown) showed no developing signs of lung injuries, lung inflammation or AHR, not even at the highest concentration that was fatal to most individuals. This is consistent with other findings in mice, where exposures to three times the concentration that induced a 50% reduction in respiratory rate (3 × RD50) caused only mild to moderate injuries in the nasal cavity but no lesions in the lower respiratory tract (Zissu Citation1995). The inhaled NH3 gas is probably retained in the upper respiratory tract by a wash-in/wash-out phenomenon, typically for highly water-soluble chemicals, where gas dissolved in the mucosa gradually returns to the exhaled air in large quantities, preventing it from gradually reaching the deeper respiratory tract (Silverman et al. Citation1949; Pauluhn Citation2013). To expose the lungs directly through i.t. instillation bypasses the nasal region and has possible protective effects on the upper respiratory tract of rodents, recreating an exposure situation more similar to oral breathing. In line with the selected doses for the present study (91.0 mg/kg·bw or 1.82 mg/mouse), MacEwan et al. reported a 1-hour LC50 value of 3385 mg/m3 (4837 ppm) in mice exposed to NH3 (MacEwen and Vernot Citation1972). Assuming a maximal respiratory depression of 30% (Li and Pauluhn Citation2010) and a minute volume of 2 L/kg·min (20 g mouse) (Mailhot-Larouche et al. Citation2018) in these mice throughout the exposure, this corresponds to a 50% lethal dose of 2.7 mg in mice. Exposing mice to 91.0 mg/kg·bw NH3 intratracheally resulted in labored breathing, wheezing and signs of dyspnea, acute neutrophilic airway inflammation, increased pro-inflammatory mediators and pro-coagulant factors and signs of pulmonary edema; indications of developing ALI at 20 h after exposure showing many similarities to ALI/ARDS described in humans (Matthay et al. Citation2019). NH3 exposure induces a biphasic injury in mice, an acute phase that gradually worsens over the following days/weeks to a fatal or sub-chronic phase, which has also been observed in humans exposed to moderate NH3 concentrations over a long period (30 min or more). The late phase in these patients has been associated with obstructive and restrictive lung diseases and pathological findings of bronchiectasis, tracheobronchial ulcerations, pneumonia, small airway lesions, edema and patchy bronchiolitis obliterans (Sobonya Citation1977; Flury et al. Citation1983; Leduc et al. Citation1992; Leung and Foo Citation1992; de la Hoz et al. Citation1996; Pirjavec et al. Citation2009; Ortiz-Pujols et al. Citation2014). In some patients, the late lung injuries developed without initial obstruction in the upper respiratory tract (Close et al. Citation1980). There are also case reports describing persistent hyperreactive bronchoconstriction in patients recovering from a single high-level exposure to ammonia (Flury et al. Citation1983; de la Hoz et al. Citation1996), a condition known as reactive airway dysfunction syndrome (RADS) or acute irritant-induced asthma that is diagnosed by positive methacholine challenge test and persistent respiratory symptoms (Brooks et al. Citation1985). Hyperreactive airways after single inhalation exposure to high levels of an irritating chemical develop within 24 h and may persist for up to several years after the incident (Brooks Citation2018). In line with this, the present study show methacholine-induced hyperresponsiveness in both central and peripheral airways in all NH3-exposed animals at 20 h, but since far from all case studies include methacholine testing, the disorder may be underestimated in NH3-exposed humans.
ALI/ARDS is challenging to study, partly because the patient group is heterogeneous in terms of comorbidities, etiology and severity of the disease. There are different clinical sub-phenotypes of ALI/ARDS, which are characterized by direct lung epithelial damage or by indirect lung injury with pronounced damage to the vascular endothelium. Direct lung injury, mainly caused by pneumonia or gastric acid aspiration, has been characterized by more severe lung epithelial injury and, as also shown in the NH3-exposed mice, by higher levels of plasma surfactant protein (SP)-D, a biomarker for lung epithelial injury and type II alveolar cell injury. At the same time, the levels of Ang-2, a pro-inflammatory mediator for endothelial injury, are lower compared to indirect ALI/ARDS (Calfee et al. Citation2015; Koyama et al. Citation2019). When the lung epithelium barrier is disrupted by inflammation or acute injury, it causes leakage of lung-specific proteins into the circulation and SP-D can be found peripherally in plasma from patients with lung disease (Hartl and Griese Citation2006). Increased plasma SP-D within 48 h has been validated in patients as one of the top diagnostic biomarkers of ALI/ARDS (Atochina-Vasserman Citation2012; Ware et al. Citation2013; Binnie et al. Citation2014; Park et al. Citation2017; Sunil et al. Citation2018). In this study, NH3 induced SP-D leakage into plasma within 4 h that was still present after 20 h, and at the same time detectable in BALF. SP-D regulates both pro-inflammatory/cytotoxic M1 and anti-inflammatory/wound repair M2 macrophage activation and lack of SP-D leads to more lung damage e.g. inflammation and oxidative stress (Sunil et al. Citation2018). The inflammatory response in the alveoli and interstitium can reduce the production of surfactants and inactivate the remaining surfactant, promoting widespread atelectasis and respiratory acidosis (Wheeler and Bernard Citation2007; Albert Citation2012). The NH3-induced ALI in mice shows signs of a hyper-inflammatory response in the airways with many pro-inflammatory mediators upregulated within one day of exposure, e.g. IL-5, IL-6, G-CSF, KC (IL-8 analogue), MIP-1α, and MIP-1β, some of which were still present in BALF on day seven (IL-1β, IL-1α, IL-12p40, MCP-1, TNFα and RANTES). In addition to increased inflammation in the airways, exposure to NH3 causes changes in baseline parameters for both respiratory resistance (RRS) and tissue resistance (G), effects that may be due to the development of atelectasis, which is known to decrease blood flow and increase resistance and elastance in the lung (Woodson et al. Citation1963; Albert Citation2012).
The results of this study show increased levels of biomarkers in blood from day 1 (IL-6, IL-10, G-CSF, fibrinogen, PAI-1, PF4/CXCL4, SP-D, MMP-9) up to 7 days (PAI-1, endothelin, PF4/CXCL4, fibrinogen, thrombomodulin) after exposure, which were associated with either epithelial damage, endothelial damage, coagulation or angiogenesis. These biomarkers have been shown to be correlated with increased mortality in patients with ALI/ARDS (Aisiku et al. Citation2016; Matthay et al. Citation2019; Spadaro et al. Citation2019). Soluble RAGE, a biomarker of type I alveolar epithelial cell injury (Briot et al. Citation2009) known to be increased in plasma from ALI/ARDS patients (Matthay et al. Citation2019) was slightly increased but not significantly in NH3 exposed mice. However, a study in which exogenous RAGE was administered to mice with LPS-induced lung injury suggests that the receptor was released as an anti-inflammatory response rather than a consequence of parenchymal lung injury (Griffiths and McAuley Citation2008; Zhang et al. Citation2008). An important early prognostic biomarker for systemic inflammation, organ failure and ALI/ARDS is MMP-9 (Hsu et al. Citation2015; Jordakieva et al. Citation2021), this protein was detected in plasma at 4 h and could still be detected in BALF at 20 h. The effect of NH3 in the blood is a rapid process and at 4 h after exposure, the number of platelets increased significantly. Activated platelets play a key role during hemostasis, but they can also modulate the immune response by releasing pro-inflammatory mediators such as PF4/CXCL4 and by direct interaction with neutrophils and vascular endothelial cells, facilitating neutrophil migration and contributing to microvascular permeability and organ damage (Liverani et al. Citation2018). In this study, PF4/CXCL4 was significantly elevated after both 20 h and 7 days. Previous studies have shown that PF4/CXCL4 expression is elevated following trauma and its proposed main physiological function is to increase blood clot formation but a putative pro-fibrotic role cannot be ruled out (Kowalska et al. Citation2010; Cai et al. Citation2020; Affandi et al. 2022). Circulating endoglin, endothelin and thrombomodulin that were noted at day 7 in exposed mice, are all known to promote inflammation and endothelial dysfunction and correlate with poorer outcomes and organ failure (Langleben et al. Citation1993; Ware et al. Citation2003; Nakano et al. Citation2007; Kapur et al. Citation2012). In addition, BALF pro-inflammatory mediators such as TNFα and IL-1β that are expressed in NH3-exposed mice have also been pointed out as the cause of the endothelial disruption observed in ALI/ARDS (Matthay et al. Citation2019).
Here, exposure to NH3 induced acute symptoms that worsened over time, which led to fatalities and a seven-day lethality of 31% in exposed animals. NH3-exposed mice showed significantly higher pCO2 and HCO3 blood levels than control animals, which may be a sign of respiratory acidosis caused by poor alveolar ventilation and/or airway obstruction (Epstein and Singh Citation2001). Respiratory acidosis, acidosis, hypercarbia and increased levels of pCO2 in patients with moderate to severe NH3-induced injuries are also described in human case reports (de la Hoz et al. Citation1996, Pirjavec et al. Citation2009, Flury et al. Citation1983, O’Kane Citation1983, Leung and Foo Citation1992). Sudden increases in pulmonary vascular resistance due to e.g. pulmonary edema, pneumonia, hypoxemia-driven vasoconstriction, or micro-thrombi, may lead to increased heart weight and acute right heart failure in ARDS patients (Pinsky Citation2016; Bhattacharya and Ellison Citation2021; Sato et al. Citation2021). The direct cause of NH3 induced death could not be ascertained, but the findings of increased heart weight and affected lung function (e.g. stiffer lung) in the exposed group suggest heart failure as a possible cause of death. On day 7, there were increased levels of plasma endoglin from NH3-exposed animals compared to control animals. Clinically, increased plasma levels of soluble endoglin that reflect endothelial activation and vascular remodeling can be used to predict pulmonary arterial hypertension (PAH) morbidity and complications such as acute heart failure and cardiovascular changes, such as cardiac fibrosis (Malhotra et al. Citation2013; Berezin Citation2016).
Histopathological evaluation of the lungs of animals that survived up to 7 days did not provide any further information on the possible cause of death; however, one individual hade severe interstitial hemorrhages and another had extensive mucosal damage in the bronchi, showing necrosis and erosion but also areas of granulation tissues. These changes extended into the peribronchial alveoli and interstitium. Evaluation of histopathological changes at 20 h showed great variation within the group; mild to severe damage to the alveoli and mild to moderate infiltration of leukocytes in the interstitium. This pattern reflects the autopsy findings of NH3 lesions in the respiratory tract of human cases, with a broad spatial distribution of the lesions from the pharynx to small airways consisting of denudation of mucous membranes, edema, inflammatory infiltrates and hemorrhages and large individual variations in the severity of the lesions (Sobonya Citation1977; Close et al. Citation1980). One plausible explanation for the variations within a group may also be the method of exposing NH3. The intratracheal instillation method may lead to local high concentration exposure and more uneven distribution in the lung compared to inhalation exposure (Driscoll et al. Citation2000). Nevertheless, the lung tissues were severely infiltrated by leukocytes, mainly macrophages that are known to drive the production of several pro-inflammatory cytokines e.g. TNFα, IL-1β, IL-6, KC and IL-12 (Duque and Descoteaux Citation2014; Laskin et al. Citation2019) that were also detected in our NH3-injury model. On day 7, an increased level of IL-13 was detected in BALF, indicating an activated TH type 2 (TH2) response in the lung; both activated macrophages (M2 cells) and epithelial cells can produce IL-13, which has been reported to be involved in the pathogenesis of pulmonary fibrosis (Hancock et al. Citation1998; Byers and Holtzman Citation2011; Passalacqua et al. Citation2017; Laskin et al. Citation2019). High levels of plasma PF4/CXCL4 and endoglin also indicate pro-fibrotic responses (Gerrits et al. Citation2020; Affandi et al. 2022); however, the regenerative and repair processes in the lung tissue were probably in an early stage at day 7 because the collagen deposition had not changed significantly.
Many of the biomarkers examined in this present study are recognized as important mechanisms orchestrating ALI/ARDS, but they have not been described in NH3 induced acute lung injury previously. Our studies indicate, however, that some of the well-known ALI/ARDS biomarkers e.g. plasma levels of IL-1β and RAGE are not significantly increased despite severe lung injury, which could be due to species differences, at different times kinetics of the markers or differences in ALI/ARDS etiology. It has been shown that the combination of clinical risk factors and biomarkers can better predict the severity of the disease compared to either clinical risk factors or biomarkers alone (Ware et al. Citation2010; Spadaro et al. Citation2019). There are limitations to the present study such as the lack of an i.t. control of the alkalinity properties of NH3 in combination with the use of a solvent (water) vehicle. Sodium hydroxide (NaOH, 20 mM, pH 9.0) was added to rat precision-cut lung slices (PCLS) and had no effect on the airway contraction while NH3 with the same pH significantly increased the contractility. NaOH had no effect on toxicity or viability in PCLS (Agren et al. Citation2021). Another limitation is that individual animals could not be sampled repeatedly over time, which would have been helpful in identifying and time-correlating biomarkers that can predict injury outcomes after exposure to NH3. Nevertheless, we believe that the mouse model is better suited for testing and validation of effective treatment protocols than in even larger animals because there are more measurable endpoints/targets to evaluate and counteract in this species (Dodd and Gross Citation1980; Sjoblom et al. Citation1999; Elfsmark et al. Citation2019).
Conclusion
The pathogenesis following an NH3 exposure is very complex and the most important conclusions of this study are that: 1) NH3 i.t. instillation induces an acute lung injury affecting the immune system, hemostasis, lung tissue and lung function which deteriorated into fatal injuries within one week; and 2) many of the biomarkers significant of ALI/ARDS in humans are expressed in NH3-induced lung injuries but not all. These induced biomarkers may be potential targets for new therapeutic treatments or diagnostic tools to predict the risk of severe NH3-induced ALI, but additional studies are needed to address this further.
Acknowledgements
Barbro Ekstrand-Hammarström, Åsa Gustafsson, Elisabeth Wigenstam and Karin Wallgren are gratefully acknowledged for invaluable help with the animal experiments and while writing the manuscript.
Disclosure statement
The authors report no declarations of interest.
Additional information
Funding
References
- AEGL. 2007. Acute exposure guideline levels for selected airborne chemicals. Ammonia prepared by the national research council of the national academies. Washington D.C, US: National Academies Press; p. 58–114.
- Affandi AJ, Carvalheiro T, Ottria A, de Haan JJ, Brans M, Brandt MM, Tieland RG, Lopes AP, Fernandez BM, Bekker C, et al. 2022. CXCL4 drives fibrosis by promoting several key cellular and molecular processes. Cell Rep. 38(1):110189.
- Agren L, Elfsmark L, Akfur C, Jonasson S. 2021. High concentrations of ammonia induced cytotoxicity and bronchoconstriction in a precision-cut lung slices rat model. Toxicol Lett. 349:51–60.
- Aisiku IP, Yamal JM, Doshi P, Benoit JS, Gopinath S, Goodman JC, Robertson CS. 2016. Plasma cytokines IL-6, IL-8, and IL-10 are associated with the development of acute respiratory distress syndrome in patients with severe traumatic brain injury. Crit Care. 20:288.
- Albert RK. 2012. The role of ventilation-induced surfactant dysfunction and atelectasis in causing acute respiratory distress syndrome. Am J Respir Crit Care Med. 185(7):702–708.
- Appelman LM, ten Berge WF, Reuzel PG. 1982. Acute inhalation toxicity study of ammonia in rats with variable exposure periods. Am Ind Hyg Assoc J. 43(9):662–665.
- Atochina-Vasserman EN. 2012. S-nitrosylation of surfactant protein D as a modulator of pulmonary inflammation. Biochim Biophys Acta. 1820(6):763–769.
- Bein K, Ganguly K, Martin TM, Concel VJ, Brant KA, Di Y, Upadhyay S, Fabisiak JP, Vuga LJ, Kaminski N, et al. 2021. Genetic determinants of ammonia-induced acute lung injury in mice. Am J Physiol Lung Cell Mol Physiol. 320(1):L41–L62.
- Berezin AE. 2016. Prognostication in different heart failure phenotypes: the role of circulating biomarkers. J Circ Biomark. 5:6.
- Bhattacharya PT, Ellison MB. 2021. Right ventricular hypertrophy., StatPearls. Treasure Island (FL): StatPearls Publishing.
- Binnie A, Tsang JL, dos Santos CC. 2014. Biomarkers in acute respiratory distress syndrome. Curr Opin Crit Care. 20(1):47–55.
- Brautbar N, Wu MP, Richter ED. 2003. Chronic ammonia inhalation and interstitial pulmonary fibrosis: a case report and review of the literature. Arch Environ Health. 58(9):592–596.
- Briot R, Frank JA, Uchida T, Lee JW, Calfee CS, Matthay MA. 2009. Elevated levels of the receptor for advanced glycation end products, a marker of alveolar epithelial type I cell injury, predict impaired alveolar fluid clearance in isolated perfused human lungs. Chest. 135(2):269–275.
- Brooks SM. 2018. Irritants, irritancy and irritant induced asthma. J Allergy Ther. 09(01):273.
- Brooks SM, Weiss MA, Bernstein IL. 1985. Reactive airways dysfunction syndrome (RADS). Persistent asthma syndrome after high level irritant exposures. Chest. 88(3):376–384.
- Butt Y, Kurdowska A, Allen TC. 2016. Acute lung injury: a clinical and molecular review. Arch Pathol Lab Med. 140(4):345–350.
- Byers DE, Holtzman MJ. 2011. Alternatively activated macrophages and airway disease. Chest. 140(3):768–774.
- Cai Z, Greene MI, Zhu Z, Zhang H. 2020. Structural features and PF4 functions that occur in Heparin-Induced Thrombocytopenia (HIT) complicated by COVID-19. Antibodies. 9(4):52.
- Calfee CS, Janz DR, Bernard GR, May AK, Kangelaris KN, Matthay MA, Ware LB. 2015. Distinct molecular phenotypes of direct vs indirect ARDS in single-center and multicenter studies. Chest. 147(6):1539–1548.
- Close LG, Catlin FI, Cohn AM. 1980. Acute and chronic effects of ammonia burns on the respiratory tract. Arch Otolaryngol. 106(3):151–158.
- de la Hoz RE, Schlueter DP, Rom WN. 1996. Chronic lung disease secondary to ammonia inhalation injury: a report on three cases. Am J Ind Med. 29(2):209–214.
- Dodd KT, Gross DR. 1980. Ammonia inhalation toxicity in cats: a study of acute and chronic respiratory dysfunction. Arch Environ Health. 35(1):6–14.
- Driscoll KE, Costa DL, Hatch G, Henderson R, Oberdorster G, Salem H, Schlesinger RB. 2000. Intratracheal instillation as an exposure technique for the evaluation of respiratory tract toxicity: uses and limitations. Toxicol Sci. 55(1):24–35.
- Duque GA, Descoteaux A. 2014. Macrophage cytokines: involvement in immunity and infectious diseases. Front Immunol. 5:491–412.
- Elfsmark L, Agren L, Akfur C, Wigenstam E, Bergstrom U, Jonasson S. 2019. Comparisons of acute inflammatory responses of nose-only inhalation and intratracheal instillation of ammonia in rats. Inhal Toxicol. 31(3):107–118.
- Emerson K, R RC, Lund RE, Thurston RV. 1975. Aqueous ammonia equilibrium calculations; effect of pH and temperature. J Fish Res Board Can. 32:2378–2383.
- Epstein SK, Singh N. 2001. Respiratory acidosis. Respir Care. 46(4):366–383.
- Flury KE, Dines DE, Rodarte JR, Rodgers R. 1983. Airway obstruction due to inhalation of ammonia. Mayo Clin Proc. 58(6):389–393.
- George A, Bang RL, Lari AR, Gang RK, Kanjoor JR. 2000. Liquid ammonia injury. Burns. 26(4):409–413.
- Gerrits T, Zandbergen M, Wolterbeek R, Bruijn JA, Baelde HJ, Scharpfenecker M. 2020. Endoglin promotes myofibroblast differentiation and extracellular matrix production in diabetic nephropathy. IJMS. 21(20):7713.
- Gomes RF, Shen X, Ramchandani R, Tepper RS, Bates JH. 2000. Comparative respiratory system mechanics in rodents. J Appl Physiol. 89(3):908–916.
- Griffiths MJ, McAuley DF. 2008. RAGE: a biomarker for acute lung injury. Thorax. 63(12):1034–1036.
- Hancock A, Armstrong L, Gama R, Millar A. 1998. Production of interleukin 13 by alveolar macrophages from normal and fibrotic lung. Am J Respir Cell Mol Biol. 18(1):60–65.
- Hantos Z, Daroczy B, Suki B, Nagy S, Fredberg JJ. 1992. Input impedance and peripheral inhomogeneity of dog lungs. J Appl Physiol. 72(1):168–178.
- Hartl D, Griese M. 2006. Surfactant protein D in human lung diseases. Eur J Clin Invest. 36(6):423–435.
- Hatton DV, Leach CS, Beaudet AL, Dillman RO, Di Ferrante N. 1979. Collagen breakdown and ammonia inhalation. Arch Environ Health. 34(2):83–87.
- Hoff J. 2000. Methods of blood collection in the mouse. Lab Animal. 29:47–53.
- Hsu AT, Barrett CD, DeBusk GM, Ellson CD, Gautam S, Talmor DS, Gallagher DC, Yaffe MB. 2015. Kinetics and role of plasma matrix metalloproteinase-9 expression in acute lung injury and the acute respiratory distress syndrome. Shock. 44(2):128–136.
- Hwaiz R, Rahman M, Zhang E, Thorlacius H. 2015. Platelet secretion of CXCL4 is Rac1-dependent and regulates neutrophil infiltration and tissue damage in septic lung damage. Br J Pharmacol. 172(22):5347–5359.
- Idell S, Koenig KB, Fair DS, Martin TR, McLarty J, Maunder RJ. 1991. Serial abnormalities of fibrin turnover in evolving adult respiratory distress syndrome. Am J Physiol. 261(4 Pt 1):L240–248.
- Jonasson S, Koch B, Bucht A. 2013. Inhalation of chlorine causes long-standing lung inflammation and airway hyperresponsiveness in a murine model of chemical-induced lung injury. Toxicology. 303:34–42.
- Jordakieva G, Budge-Wolfram RM, Budinsky AC, Nikfardjam M, Delle-Karth G, Girard A, Godnic-Cvar J, Crevenna R, Heinz G. 2021. Plasma MMP-9 and TIMP-1 levels on ICU admission are associated with 30-day survival. Wien Klin Wochenschr. 133(3–4):86–95.
- Kapur NK, Wilson S, Yunis AA, Qiao X, Mackey E, Paruchuri V, Baker C, Aronovitz MJ, Karumanchi SA, Letarte M, et al. 2012. Reduced endoglin activity limits cardiac fibrosis and improves survival in heart failure. Circulation. 125(22):2728–2738.
- Kass I, Zamel N, Dobry CA, Holzer M. 1972. Bronchiectasis following ammonia burns of the respiratory tract. A review of two cases. Chest. 62(3):282–285.
- Kerger BD, Fedoruk MJ. 2015. Pathology, toxicology, and latency of irritant gases known to cause bronchiolitis obliterans disease: does diacetyl fit the pattern? Toxicol Rep. 2:1463–1472.
- Kowalska MA, Rauova L, Poncz M. 2010. Role of the platelet chemokine platelet factor 4 (PF4) in hemostasis and thrombosis. Thromb Res. 125(4):292–296.
- Koyama K, Katayama S, Tonai K, Shima J, Koinuma T, Nunomiya S. 2019. Biomarker profiles of coagulopathy and alveolar epithelial injury in acute respiratory distress syndrome with idiopathic/immune-related disease or common direct risk factors. Crit Care. 23(1):283.
- Langleben D, DeMarchie M, Laporta D, Spanier AH, Schlesinger RD, Stewart DJ. 1993. Endothelin-1 in acute lung injury and the adult respiratory distress syndrome. Am Rev Respir Dis. 148(6 Pt 1):1646–1650.
- Laskin DL, Malaviya R, Laskin JD. 2019. Role of macrophages in acute lung injury and chronic fibrosis induced by pulmonary toxicants. Toxicol Sci. 168(2):287–301.
- Leduc D, Gris P, Lheureux P, Gevenois PA, De Vuyst P, Yernault JC. 1992. Acute and long term respiratory damage following inhalation of ammonia. Thorax. 47(9):755–757.
- Lessenger JE. 2004. Anhydrous ammonia injuries. J Agromedicine. 9(2):191–203.
- Leung CM, Foo CL. 1992. Mass ammonia inhalational burns–experience in the management of 12 patients. Ann Acad Med Singap. 21(5):624–629.
- Li WL, Pauluhn J. 2010. Comparative assessment of the sensory irritation potency in mice and rats nose-only exposed to ammonia in dry and humidified atmospheres. Toxicology. 276(2):135–142.
- Liverani E, Mondrinos MJ, Sun S, Kunapuli SP, Kilpatrick LE. 2018. Role of protein kinase C-delta in regulating platelet activation and platelet-leukocyte interaction during sepsis. PLOS One. 13(4):e0195379.
- MacEwen J, Vernot E. 1972. Toxic hazards research unit annual report: 1972 Health & Environmental Research Online (HERO). Wright-Patterson Air Force Base, OH: Aerospace Medical Research Laboratory.
- Mailhot-Larouche S, Deschenes L, Lortie K, Gazzola M, Marsolais D, Brunet D, Robichaud A, Bosse Y. 2018. Assessment of respiratory function in conscious mice by double-chamber plethysmography. J Vis Exp. doi: https://doi.org/10.3791/57778.
- Makarovsky I, Markel G, Dushnitsky T, Eisenkraft A. 2008. Ammonia–when something smells wrong. Isr Med Assoc J. 10(7):537–543.
- Malhotra R, Paskin-Flerlage S, Zamanian RT, Zimmerman P, Schmidt JW, Deng DY, Southwood M, Spencer R, Lai CS, Parker W, et al. 2013. Circulating angiogenic modulatory factors predict survival and functional class in pulmonary arterial hypertension. Pulm Circ. 3(2):369–380.
- Matthay MA, Zemans RL, Zimmerman GA, Arabi YM, Beitler JR, Mercat A, Herridge M, Randolph AG, Calfee CS. 2019. Acute respiratory distress syndrome. Nat Rev Dis Primers. 5(1):18.
- Mokra D, Kosutova P. 2015. Biomarkers in acute lung injury. Respir Physiol Neurobiol. 209:52–58.
- Montague TJ, Macneil AR. 1980. Mass ammonia inhalation. Chest. 77(4):496–498.
- Nakano Y, Tasaka S, Saito F, Yamada W, Shiraishi Y, Ogawa Y, Koh H, Hasegawa N, Fujishima S, Hashimoto S, et al. 2007. Endothelin-1 level in epithelial lining fluid of patients with acute respiratory distress syndrome. Respirology. 12(5):740–743.
- O'Kane GJ, 1983. Inhalation of ammonia vapour. A report on the management of eight patients during the acute stages. Anaesthesia 38:1208–1213.
- Omiya K, Sato H, Sato T, Wykes L, Hong M, Hatzakorzian R, Kristof AS, Schricker T. 2021. Albumin and fibrinogen kinetics in sepsis: a prospective observational study. Crit Care. 25(1):436.
- Ortiz-Pujols S, Jones SW, Short KA, Morrell MR, Bermudez CA, Tilley SL, Cairns BA. 2014. Management and sequelae of a 41-year-old Jehovah's witness with severe anhydrous ammonia inhalation injury. J Burn Care Res. 35(3):e180-183–e183.
- Padappayil RP, Borger J. 2019. Ammonia toxicity. Treasure Island (FL): StatPearls.
- Park J, Pabon M, Choi A, Siempos II, Fredenburgh LE, Baron RM, Jeon K, Chung CR, Yang JH, Park CM, et al. 2017. Plasma surfactant protein-D as a diagnostic biomarker for acute respiratory distress syndrome: validation in US and Korean cohorts. BMC Pulm Med. 17(1):204.
- Passalacqua G, Mincarini M, Colombo D, Troisi G, Ferrari M, Bagnasco D, Balbi F, Riccio A, Canonica GW. 2017. IL-13 and idiopathic pulmonary fibrosis: possible links and new therapeutic strategies. Pulm Pharmacol Ther. 45:95–100.
- Pauluhn J. 2013. Acute inhalation toxicity of ammonia: revisiting the importance of RD50 and LCT01/50 relationships for setting emergency response guideline values. Regul Toxicol Pharmacol. 66(3):315–325.
- Perkins MW, Wong B, Tressler J, Coggins A, Rodriguez A, Devorak J, Sciuto AM. 2016. Assessment of inhaled acute ammonia-induced lung injury in rats. Inhal Toxicol. 28(2):71–79.
- Perkins MW, Wong B, Tressler J, Rodriguez A, Sherman K, Andres J, Devorak J, W LW, Sciuto AM. 2017. Adverse respiratory effects in rats following inhalation exposure to ammonia: respiratory dynamics and histopathology. Inhal Toxicol. 29(1):32–41.
- Pinsky MR. 2016. The right ventricle: interaction with the pulmonary circulation. Crit Care. 20:266.
- Pirjavec A, Kovic I, Lulic I, Zupan Z. 2009. Massive anhydrous ammonia injury leading to lung transplantation. J Trauma. 67(4):E93–97.
- Sato R, Dugar S, Cheungpasitporn W, Schleicher M, Collier P, Vallabhajosyula S, Duggal A. 2021. The impact of right ventricular injury on the mortality in patients with acute respiratory distress syndrome: a systematic review and meta-analysis. Crit Care. 25(1):172.
- Sebag SC, Bastarache JA, Ware LB. 2011. Therapeutic modulation of coagulation and fibrinolysis in acute lung injury and the acute respiratory distress syndrome. Curr Pharm Biotechnol. 12(9):1481–1496.
- Silverman L, Whittenberger JL, Muller J. 1949. Physiological response of man to ammonia in low concentrations. J Ind Hyg Toxicol. 31:74–78.
- Sjoblom E, Hojer J, Kulling PE, Stauffer K, Suneson A, Ludwigs U. 1999. A placebo-controlled experimental study of steroid inhalation therapy in ammonia-induced lung injury. J Toxicol Clin Toxicol. 37(1):59–67.
- Sobonya R. 1977. Fatal anhydrous ammonia inhalation. Hum Pathol. 8(3):293–299.
- Spadaro S, Park M, Turrini C, Tunstall T, Thwaites R, Mauri T, Ragazzi R, Ruggeri P, Hansel TT, Caramori G, et al. 2019. Biomarkers for acute respiratory distress syndrome and prospects for personalised medicine. J Inflamm. 16:1.
- Sunil VR, Vayas KN, Cervelli JA, Ebramova EV, Gow AJ, Goedken M, Malaviya R, Laskin JD, Laskin DL. 2018. Protective role of surfactant protein-D against lung injury and oxidative stress induced by nitrogen mustard. Toxicol Sci. 166(1):108–122.
- Ward K, Murray B, Costello GP. 1983. Acute and long-term pulmonary sequelae of acute ammonia inhalation. Ir Med J. 76(6):279–281.
- Ware LB, Camerer E, Welty-Wolf K, Schultz MJ, Matthay MA. 2006. Bench to bedside: targeting coagulation and fibrinolysis in acute lung injury. Am J Physiol Lung Cell Mol Physiol. 291(3):L307–311.
- Ware LB, Fang X, Matthay MA. 2003. Protein C and thrombomodulin in human acute lung injury. Am J Physiol Lung Cell Mol Physiol. 285(3):L514–521.
- Ware LB, Koyama T, Billheimer DD, Wu W, Bernard GR, Thompson BT, Brower RG, Standiford TJ, Martin TR, Matthay MA, et al. 2010. Prognostic and pathogenetic value of combining clinical and biochemical indices in patients with acute lung injury. Chest. 137(2):288–296.
- Ware LB, Koyama T, Zhao Z, Janz DR, Wickersham N, Bernard GR, May AK, Calfee CS, Matthay MA. 2013. Biomarkers of lung epithelial injury and inflammation distinguish severe sepsis patients with acute respiratory distress syndrome. Crit Care. 17(5):R253.
- Wheeler AP, Bernard GR. 2007. Acute lung injury and the acute respiratory distress syndrome: a clinical review. Lancet. 369(9572):1553–1564.
- White ES. 1971. A case of near fatal ammonia gas poisoning. J Occup Med. 13:549–550.
- Whyte CS, Morrow GB, Mitchell JL, Chowdary P, Mutch NJ. 2020. Fibrinolytic abnormalities in acute respiratory distress syndrome (ARDS) and versatility of thrombolytic drugs to treat COVID-19. J Thromb Haemost. 18(7):1548–1555.
- Wiklund L, Ronquist G, George M. 2001. Ammonia–not the culprit. Arch Dis Child. 84(6):525.
- Woodson RD, Raab DE, Ferguson DJ. 1963. Pulmonary hemodynamics following acute atelectasis. Am J Physiol. 205:53–56.
- Zhang H, Tasaka S, Shiraishi Y, Fukunaga K, Yamada W, Seki H, Ogawa Y, Miyamoto K, Nakano Y, Hasegawa N, et al. 2008. Role of soluble receptor for advanced glycation end products on endotoxin-induced lung injury. Am J Respir Crit Care Med. 178(4):356–362.
- Zissu D. 1995. Histopathological changes in the respiratory tract of mice exposed to ten families of airborne chemicals. J Appl Toxicol. 15(3):207–213.