
Abstract
Background
Lysosomal ion channels are proposed therapeutic targets for a number of diseases, including those driven by NLRP3 inflammasome-mediated inflammation. Here, the specific role of the lysosomal big conductance Ca2+-activated K+ (BK) channel was evaluated in a silica model of inflammation in murine macrophages. A specific-inhibitor of BK channel function, paxilline (PAX), and activators NS11021 and NS1619 were utilized to evaluate the role of lysosomal BK channel activity in silica-induced lysosomal membrane permeabilization (LMP) and NLRP3 inflammasome activation resulting in IL-1β release.
Methods
Murine macrophages were exposed in vitro to crystalline silica following pretreatment with BK channel inhibitors or activators and LMP, cell death, and IL-1β release were assessed. In addition, the effect of PAX treatment on silica-induced cytosolic K+ decrease was measured. Finally, the effects of BK channel modifiers on lysosomal pH, proteolytic activity, and cholesterol transport were also evaluated.
Results
PAX pretreatment significantly attenuated silica-induced cell death and IL-1β release. PAX caused an increase in lysosomal pH and decrease in lysosomal proteolytic activity. PAX also caused a significant accumulation of lysosomal cholesterol. BK channel activators NS11021 and NS1619 increased silica-induced cell death and IL-1β release. BK channel activation also caused a decrease in lysosomal pH and increase in lysosomal proteolytic function as well as a decrease in cholesterol accumulation.
Conclusion
Taken together, these results demonstrate that inhibiting lysosomal BK channel activity with PAX effectively reduced silica-induced cell death and IL-1β release. Blocking cytosolic K+ entry into the lysosome prevented LMP through the decrease of lysosomal acidification and proteolytic function and increase in lysosomal cholesterol.
Introduction
Silicosis is an incurable progressive respiratory disease driven by chronic inflammation caused by inhalation of respirable crystalline silica (silica) particles <10 μm in size. Although silica related deaths have declined in recent decades, silicosis persists as an occupational hazard in many parts of the world (Leung et al. Citation2012) with cases of accelerated silicosis becoming increasingly predominant (Barnes et al. Citation2019; Leso et al. Citation2019; Burki Citation2021). Therefore, understanding the cellular mechanisms that perpetuate the onset of silicosis is paramount to the development of effective therapeutic strategies and can better inform understanding of chronic inflammation caused by a variety of exogenous and endogenous particles (i.e. asbestos, monosodium urate, cholesterol crystals, nanoparticles, PM2.5).
As inhaled silica enters the gas–air exchange region of the lungs, the alveoli, it is quickly cleared by the resident immune cells, the alveolar macrophages (AM). These sentinels are the first line of defense against particles and other inhaled exogenous material. Silica binds scavenger receptors on the surface of AM and is phagocytosed (Hamilton et al. Citation2006, Citation2008). Within the AM, the phagosome containing silica fuses with the lysosome to form a phagolysosome; there, the silica particle encounters lysosomal degradative enzymes. Fractured silica particles such as are encountered in environmental or occupational exposures are decorated with nearly free silanol (NFS) groups (Pavan et al. Citation2019, Citation2020). NFS bind preferentially with phosphatidylcholine over phosphatidylserine phospholipid headgroups (Pavan et al. Citation2022). It is likely that NFS binding to phosphatidylcholine headgroups is what drives the lysosomal membrane destabilization observed with silica exposure. Lysosomal membrane permeability (LMP) releases lysosomal proteases such as Cathepsin B into the cytosol and initiates the activation of the NLRP3 inflammasome and the release of IL-1β that perpetuates silica-induced inflammation (Cassel et al. Citation2008; Hornung et al. Citation2008; Hornung and Latz Citation2010; Katsnelson et al. Citation2015; Bunderson-Schelvan et al. Citation2016, Citation2017). Thus, furthering the understanding of the cellular mechanisms that contribute to particle-induced LMP is critical to preventing silica-caused inflammation and disease.
The lysosome is a specialized organelle responsible for degradation and recycling of intracellular and extracellular macromolecules and contains over 60 hydrolases in its low pH lumen (∼4.5) (De Duve et al. Citation1955; Ballabio and Bonifacino Citation2020). This pH is maintained by the proton pumping activity of the vacuolar ATPase (V-ATPase) (Forgac Citation2007). Fluxes of H+ and other ions, such as K+, are necessary to maintain lysosomal membrane potential and maintain the ionic homeostasis of the lysosome (Cang et al. Citation2015; Cao et al. Citation2015). For example, K+ influx into the lysosome has been demonstrated to cause hyperpolarization of lysosomal membrane potential (Xu and Ren Citation2015). Additionally, lysosomal membrane potential becomes hyperpolarized in the presence of α-crystalline silica Min-U-Sil 5 prior to onset of lysosomal membrane permeabilization (Ziglari et al. Citation2021). Ziglari et al. reported a particle-caused reduction in intracellular K+ at both physiological and high extracellular K+ concentrations, suggesting that particles induced K+ movement from the cytosol into the lysosome as well as the extracellular space (Ziglari et al. Citation2021). Currently, there are two known K+-selective channels in the lysosomal membrane, recently discovered transmembrane protein 175 (TMEM175) (Cang et al. Citation2015; Wie et al. Citation2021) and the large conductance Ca2+-activated K+ channel (BK) (Cao et al. Citation2015; Wang et al. Citation2017). TMEM175 is structurally distinct from other known K+ channels and thus far lacks a known specific inhibitor (Oh et al. Citation2020, Citation2022). BK channels, also known as MaxiK, Slo1 or KCa1.1 channels, are calcium-activated potassium channels that have a large single channel K+ conductance and were first described some 40 years ago (Marty Citation1981; Pallotta et al. Citation1981).
While the role of plasma membrane BK channels is often studied in excitable cells such as smooth muscle cells or neurons, the role of BK channel activity in lysosomal function is not well characterized. Previously, lysosomal BK channel activity has been reported to contribute to lysosomal cholesterol trafficking and proteolytic activity (Wang et al. Citation2017). Lysosomal cholesterol trafficking and lysosomal proteolytic function are integral in the development of LMP leading to silica induced cell death and IL-1β release (Kendall and Holian Citation2023; Sydor et al. Citation2023). Here, we assessed the role of lysosomal K+ channel activity in silica-induced LMP and subsequent IL-1β release. Specifically, we evaluated the effect of blocking BK channel-specific K+ entry into the lysosome to determine if preventing K+ entry into the lysosome can mitigate K+ efflux from silica exposed bone marrow derived macrophages (BMdM) and thereby reduce cell death and IL-1β release or if inhibition of lysosomal BK activity can reduce cell death and IL-1β release through modification of lysosomal function.
Materials and methods
Reagents
Iberiotoxin (IbTX; Abcam, ab120379), paxilline (Enzo Life Sciences, BML-KC155-0005), bafilomycin (BAF; Enzo Life Sciences, BML-CM110-0100), U18666A (Sigma, 662015-10MG), methyl-β-cyclodextrin (MβCD; Sigma, C4555), filipin (Sigma, F9765), digitonin (Sigma, 300410), 1,2-dioleoyl-sn-glycero-3-phospho-(1′-rac- glycerol) (18:1 Δ9-cis) (DOPG; Avanti, 840475), 4-Methylumbelliferyl-2-acetamido-2-deoxy-β-D-Glucopyranoside (Cayman Chemical, 26953), crystalline silica (Min-u-sil-5; Pennsylvania Glass Sand Corporation, Pittsburgh, PA), 1,3-dihydro-1-[2-hydroxy-5-(trifluoromethyl)phenyl]-5-(trifluoro-methyl)-2H-benzimidazole-2-one (NS1619; Abcam, ab141824), 1-(3,5-bis-trifluoromethyl-phenyl)-3-[4-bromo-2-(1H-tetrazol-5-yl)-phenyl]-thiourea (NS11021; Cayman Chemical, 15347), lipopolysaccharide (LPS; Sigma, L4516).
Mice
C57BL/6J were housed in the University of Montana’s specific-pathogen-free Laboratory Animal Resources facility with a 12 h light/dark cycle under controlled environmental conditions. Mice (8-12 weeks old) were euthanized by sodium pentobarbital (Euthasol™) overdose prior to collection of alveolar macrophages or legs. The animal procedures used in this study were approved by the Institutional Animal Care and Use Committee (IACUC) at the University of Montana.
Alveolar macrophage isolation and mexAM culture
AM were collected from C57BL/6 mice by whole lung lavage consisting of lung removal followed by repeated (x10) lavages with 1 mL of PBS (cold, sterile). Collected cells were centrifuged at 1500 RPM for 5 min and lavage fluid discarded. The cell pellet was resuspended in complete culture media (RPMI 1640, 10% fetal bovine serum, 1% sodium pyruvate, 1% penicillin/streptomycin). Primary AM were used immediately for in vitro experiments.
Murine ex vivo alveolar macrophages (mexAM) cells were generated by placing collected primary AM in T75 tissue-culture coated flasks at 4x106 cells in 10 mL media supplemented with hTGFβ1 (10 ng/mL, Peprotech, 100-21), murine GM-CSF (30 ng/mL, Peprotech 315-03), and rosiglitazone (1 μM, Peprotech, 1227342) and maintained in 5% CO2, 37 °C environment, as previously described (Gorki et al. Citation2022; Kendall et al. Citation2023). Media and growth factors were replaced every five days. mexAM were used for experimental needs or passaged every 7 days. For collection, cells were incubated with 5 mL of lidocaine (20 mM in PBS) at 37 °C for 5 min before scraping.
Crystalline silica
Crystalline silica (Min-U-Sil-5) was acid washed in 1 M HCl at 100 °C for 1 h, rinsed with sterile water multiple times, and dried at 200 °C. Silica was determined endotoxin-free by Limulus Amebocyte Lysate assay (Cambrex, Walkersville, MD). Min-u-sil-5 has an average diameter of 1.3 µm and has previously been characterized (Pavan et al. Citation2022). Prior to use, silica (5 mg/mL) was suspended in phosphate-buffered saline (PBS) and sonicated for 2 min (550 watts at 20 kHz) by Qsonica Q500 Sonicator with VWR circulating water bath. Unless noted, silica was applied to cells at a final concentration of 50 μg/mL in complete media.
Isolation and culture of bone marrow derived macrophages
BMdM were generated as described previously (Pfau et al. Citation2004; Migliaccio et al. Citation2008). Briefly, C57BL/6 mice were sacrificed, and hind legs collected. The femur and tibia bones were flushed with complete media (RPMI, 10% FBS, 1% Penicillin/Streptomycin, 1% Sodium Pyruvate, 0.1% β-mercaptoethanol) to collect bone marrow cells. Cells were incubated overnight (37 °C; 3.0x107 cells/T75 flask) to eliminate stromal cells by adherence. The following day, nonadherent cells were transferred to new T75 flasks (1.5 x 107) with macrophage colony stimulating growth factor (M-CSF) (20 ng/mL, R&D Systems). Cells were spiked with M-CSF (10 ng/mL) every 2-3 days. BMdM were collected by trypsin and gentle scraping and utilized for experimentation after day 10.
Toxicity and cytokine assays
For LDH & IL-1β assays, BMdM were plated in flat-bottom, tissue culture-treated 96-well plates at a density of 1x105 cells per well in 100 μL media and incubated with lipopolysaccharide (LPS) (20 ng/mL) for the course of the silica exposure. LPS promotes NLRP3 inflammasome priming through activation of the NFκB pathway and upregulation of pro-IL-1β transcription. Cells were treated with 10 μM PAX or 100 nM IbTX for 30 min prior to addition of silica (50 μg/mL) and remained in the media for the duration of the 24 h silica exposure. NS11021 (1 μM) and NS1619 (5 μM) for 1 h prior to addition of silica. MβCD (10 μg/mL) and DOPG (50 μM) experiments required 1 h treatment before addition of PAX for 30 min. Following silica addition, BMdM were incubated (37 °C, 5% CO2) for 24 h. Cytotoxicity was determined by lactate dehydrogenase (LDH) assay (Promega G1780) per the manufacturer’s instructions and absorbance at 490 nm measured by Molecular Devices SpectraMax M4 plate reader. NLRP3 inflammasome activation was assayed by measuring the release of IL-1β in cell supernatants using an IL-1β ELISA kit (R&D Systems, DY401) according to the manufacturer’s instructions and absorbance measured at 450 nm.
Lysosomal membrane permeabilization (LMP) assay
LMP results in release of lysosomal enzymes into the cellular cytosol where they contribute to NLRP3 inflammasome activation. LMP was determined by measuring lysosomal-specific enzyme β-N-acetylglucosaminidase (NAG) activity in the cytosol as previously described (Aits et al. Citation2015; Jessop et al. Citation2017). BMdM were plated in 96-well plates at a density of 1x105 cells per well and pretreated with PAX (10 μM) for 30 min prior to addition of silica (50 μg/mL) for 24 h. BMdM were permeabilized with 100 μL per well cytosolic extraction buffer (250 mM sucrose, 20 mM HEPES, 10 mM KCl, 1.5 mM MgCl2, 1 mM EDTA, 1 mM EGTA, 0.5 mM pefabloc, pH 7.5) with an optimized dose of digitonin (12.5 μg/mL) for 15 min on ice with rocking. Digitonin was optimized by titration (data not shown) to determine a concentration that effectively permeabilized the plasma membrane, as indicated by LDH released into the extracellular space, while minimally disrupting the lysosomal membrane, as indicated by NAG values. NAG activity in the cytosol was measured by reaction of fluorogenic substrate 4-methylumbelliferyl-2-acetamido-2-deoxy-B-D-glucopyranoside (300 μg/mL) (Cayman, 26953) with cytosolic extracts (30 μL) in reaction buffer (100 μL, 0.2 M sodium citrate, pH 4.5) and assessed on a plate reader (20 min kinetic read, 45 s intervals, 356 nmex, 444 nmem, 30 °C). NAG activity for each condition was compared to NAG values for complete lysis (200 μg/mL digitonin) of the same condition (NAG12.5/NAG200).
Lysosomal pH
LysoTracker Red DND-99 (LTR; Invitrogen, L7528) was used to measure lysosomal pH. BMdM were plated at 50,000 cells/well overnight. Cells were treated with PAX, NS11021, NS1619, or bafilomycin A1 (BAF) for 1 h. Cells were washed and 100 μL/well media containing LysoTracker Red (100 nM) was added. Cells with LTR were incubated (37 °C; 30 min), washed with PBS, and fluorescence measured by BioTek Cytation 5 Imaging Reader or SpectraMax Microplate Reader at 575 nmex/595 nmem. A decrease in LTR fluorescence compared to control cells indicated a reduction of lysosomal acidification.
Lysosomal proteolytic activity
To assess lysosomal proteolytic activity in BMdM, fluorescence analysis using the DQ Green BSA probe was performed. This albumin conjugated fluorochrome is taken into the cell via endocytosis and degraded in the lysosome where the resulting small, unquenched fragments fluoresce brightly. BMdM were plated in 96-well plates at 50,000 or 100,000 cells per well with PAX, NS11021, NS1619 or BAF treatment for 1 h. After incubation with drugs, cells were washed and incubated with 100 μL/well DQ Green BSA (10 μg/mL in complete media (37 °C, 1 h). Cells were washed with PBS and the fluorescence measured with a Molecular Devices SpectraMax M4 Microplate Reader or BioTek Cytation 5 Imaging Reader (488 nmex/525 nmem). A decrease in DQ Green BSA fluorescence compared to control cells corresponded to a reduction in lysosomal degradative activity.
Filipin imaging
BMdM were plated at 1.5x105 cells in 4-well Greiner Cellview cell culture dish with glass bottom and treated with PAX, NS11021, U18666A, or U18666A + NS11021 for 24 h. Plated BMdM were washed twice with PBS prior to fixation with 4% paraformaldehyde for 10 min at room temperature. Fixed cells were washed twice with PBS and stained with filipin staining solution (2.5 mg/ml stock of filipin in DMSO diluted 1:3 with 2% BSA/PBS) for 2 h at 37 °C in the dark. The staining solution was removed and replaced with phenol-red free complete RPMI 1640 media. All fluorescence images were collected on a Zeiss 880 LSM (Carl Zeiss Microscopy, LLC., White Plaines, NY) at the UM Center for Biomolecular Structure and Dynamics with a 63x Plan Apo 1.4 NA oil immersion objective. Filipin was excited with a 405 nm laser. The laser power and detector gain were held constant across all conditions. The Analyze Particles function of ImageJ was used with a set particle threshold size of 10-500 pixel to quantify filipin puncta per cell.
Particle uptake
Silica particle uptake was determined by flow cytometry side scatter values as previously described (Kendall et al. Citation2023). In short, BMdM were incubated with silica (50 μg/mL) for 1 h, washed, and resuspended in PBS for analysis by Life Technologies Attune NxT Accoustic Focusing Cytometer in the Fluorescence Cytometry Core at the University of Montana. Analysis was performed using FlowJo v10 software.
Statistical analysis
Statistical significance was defined as p < 0.05. An unpaired-student’s t-test was used to compare the statistical difference of two means. For the comparison of more than two means, one or two-way ANOVA were used. For comparisons between all groups, a Holm-Sidak’s post-test was used. A minimum of three experimental replicates were performed for each data set. All statistical tests and graphs were generated with Prism 9.0 by GraphPad Software (San Diego, CA, USA).
Results
PAX prevents silica-induced cell death and IL-1β release across multiple macrophage models
To evaluate if blocking K+ movement through the lysosomal BK channel could mitigate silica-induced cell death and IL-1β release, the effect of two specific BK channel inhibitors, iberiotoxin (IbTX) and paxilline (PAX), on silica induced cell death and IL-1β release was examined. IbTX is a toxin derived from Indian Red (Bathus tamulus) scorpion venom that is cell impermeable and binds the external face of the BK channel (Galvez et al. Citation1990; Candia et al. Citation1992). PAX is an indole diterpene biosynthetic product of the filamentous fungal species Penicillium paxilli. In contrast to IbTX, PAX is cell permeable and binds the closed state of the BK channel at a ratio of one PAX molecule per channel (Saikia et al. Citation2007; Zhou and Lingle Citation2014; Zhou et al. Citation2020). IbTX can block BK channel activity at nanomolar concentrations and abolished lysosomal hyperpolarization induced by BK channel activity at a dose of 100 nM (Yamamura et al. Citation2001), while PAX applied at 10 μM abolished BK channel activity (Yang et al. Citation2019).
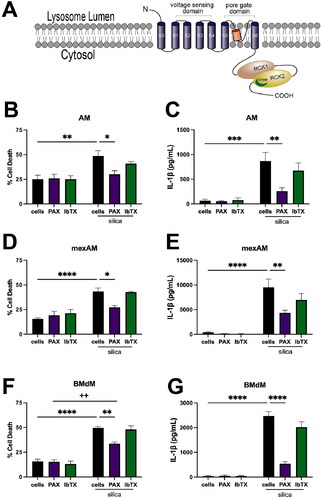
The effect of PAX and IbTX was evaluated in three murine macrophage models, primary AM, cultured murine ex vivo alveolar macrophages (mexAM) (Gorki et al. Citation2022; Kendall et al. Citation2023), and BMdM. Macrophages were treated with lipopolysaccharide (LPS; 20 ng/mL) for the duration of the silica exposure to prime NLRP3 activation. AM were treated with the respective PAX (10 μM) or IbTX (100 nM) for 30 min prior to silica (50 μg/mL) application. After 24 h, cell death and IL-1β release were assessed. PAX significantly reduced silica-induced cell death () and IL-1β release () in AM. In contrast, the cell-impermeable IbTX had negligible impacts on cell death () and IL-1β release (). These same effects were observed in both mexAM () and BMdM (). The effect of PAX on decreasing silica-induced cell death and inflammation suggests that specific inhibition of the lysosomal BK channel contributes to the decrease, in contrast to IbTX which failed to reduce cell death or IL-1β release. Based on the consistent response to PAX observed in AM, mexAM, and BMdM, targeting lysosomal BK channel activity demonstrates potential as a mechanistic target in the prevention of silica-induced cell death and IL-1β release. Due to the similarity of responses in all three macrophage models, the remaining experiments were conducted in BMdM.
Figure 1. Inhibiting lysosomal BK function mitigates silica-induced cell death and IL-1β release in multiple murine macrophage models. Respective macrophage models were primed with LPS (20 ng/mL) and then treated with selective BK channel inhibitors PAX (10 μM) or IbTX (100 nM) for 30 min prior to silica (50 μg/mL) application for 24 h. Cell death and IL-1β release were assessed in (A) & (B) primary AM, (C) & (D) mexAM, and (E) & (F) BMdM. Data expressed as mean ± SEM. n= 4. * p < 0.05, ** p < 0.01, *** p < 0.001, and **** p < 0.0001 compared to silica-treated macrophages, as determined by two-way ANOVA with Tukey’s multiple comparisons test. ++ p < 0.05 compared to PAX-treated BMdM.
PAX prevents silica-induced decrease in intracellular K+
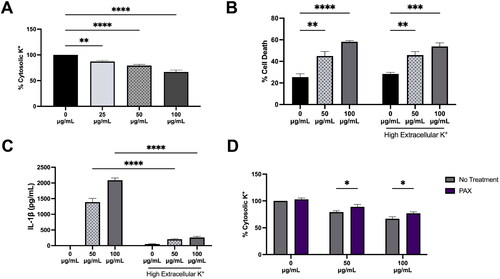
Many NLRP3 inflammasome activators such as nigericin, ATP, or crystalline particles are known to demonstrate a decrease in cytosolic K+ prior to IL-1β release suggesting cytosolic K+ efflux is an upstream event of NLRP3 inflammasome activity (Munoz-Planillo et al. 2013). Because the lysosomal BK channel facilitates the movement of K+ out of the cytosol into the lysosome, it was necessary to examine the impact of BK channel activity on cytosolic K+ in silica exposure. Others have noted a decrease in cytosolic potassium with silica exposure (Cassel et al. Citation2008; Ziglari et al. Citation2021) that was confirmed here with silica demonstrating a dose dependent decrease in cytosolic K+ in BMdM after a 1 h exposure (). Preventing K+ efflux from the cell has been proposed as a way to prevent NLRP3-mediated IL-1β release, therefore K+ efflux from the cell was prevented by disrupting the K+ gradient with a high extracellular K+ (45 mM) environment. High extracellular K+ effectively reduced silica-induced IL-1β release for both 50 μg/mL and 100 μg/mL (); however, it failed to prevent silica-induced cell death () suggesting LMP-caused cell death is not dependent on activation of the NLPR3 inflammasome. The impact of PAX on silica-induced loss in cytosolic K+ was assessed. PAX pretreatment for 30 min prior to 1 h silica exposure significantly reduced the particle-induced decrease in cytosolic K+ (). These results demonstrate that preventing cytosolic K+ decrease with high extracellular K+ prevents silica-induced IL-1β release but fails to prevent silica-induced cell death. In contrast, PAX treatment effectively offset the decrease in cytosolic K+, prevented IL-1β release, and prevented silica-induced cell death suggesting that blocking the lysosomal BK channel-mediated movement of K+ out of the cytosol prevents silica-induced cell death.
Figure 2. Silica causes dose-dependent cytosolic K+ decrease in BMdM. Data expressed as mean ± SEM. (A) Cytosolic K+ as determined by IPG 2AM fluorescent intensity after 1 h silica treatment. n = 6. ** p < 0.01 or *** p < 0.001 compared to untreated BMdM as determined by one-way ANOVA with Dunnett’s multiple comparisons test. (B) Cell death in supernatants of LPS-primed BMdM treated with silica (50 μg/mL or 100 μg/mL) for 24 h in normal K+ media (5 mM) compared to high K+ media (45 mM). n = 4. ** p < 0.01, *** p < 0.001 or **** p < 0.0001 compared to untreated BMdM in each group as determined by two-way ANOVA with Tukey’s multiple comparisons test. (C) IL-1β release measured in supernatants of LPS-primed BMdM treated with silica (50 μg/mL or 100 μg/mL) for 24 h in normal K+ media (5 mM) compared to high K+ media (45 mM). n = 4. **** p < 0.0001 compared to same silica dose in normal K+ media as determined by two-way ANOVA with Tukey’s multiple comparisons test. (D) Cytosolic K+ in BMdM pretreated with PAX for 30 min prior to 1 h silica (50 μg/mL or 100 μg/mL). n = 6. * p < 0.05 compared to silica dose without PAX treatment as determined by two-way ANOVA with Tukey’s multiple comparison’s test.
PAX prevents silica-induced lysosomal membrane permeability
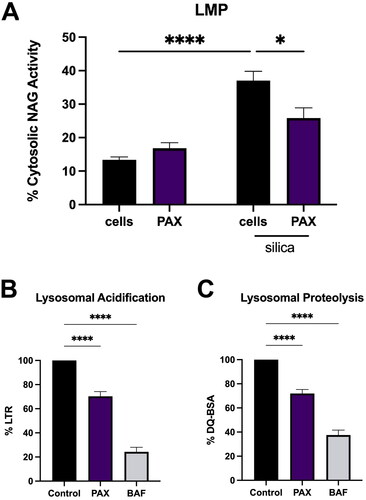
LMP has been reported to be a key event in NLRP3 inflammasome activation by particles such as aluminum, multiwalled carbon nanotubes, silica, and monosodium urate crystals (MSU) (Hornung et al. Citation2008; Munoz-Planillo et al. 2013; Bunderson-Schelvan et al. Citation2017). Preventing LMP has been demonstrated to prevent both particle-induced cell death and IL-1β release (Biswas et al. Citation2017; Jessop et al. Citation2017; Burmeister et al. Citation2019; Fletcher et al. Citation2021; Kendall and Holian Citation2023). As PAX treatment effectively reduced both silica-induced IL-1β and cell death () without altering silica particle uptake (Supplemental Figure 1), PAX’s effect on LMP was examined. PAX treatment for 30 min was followed by 24 h silica (50 μg/mL) exposure. LMP was measured by assessing activity of lysosomal specific enzyme β-N-acetylglucosaminidase (NAG) in cytosolic extracts as described in Methods. PAX pretreatment significantly reduced cytosolic NAG activity due to silica () demonstrating that blocking lysosomal BK ion channel activity prevents LMP.
Figure 3. PAX prevents LMP caused by silica and disrupts lysosomal acidification. Data expressed as mean ± SEM. (A) LMP assessed by cytosolic NAG activity in BMdM pretreated with PAX (10 μM) for 30 min before silica (50 μg/mL) exposure for 24 h. n = 5. * p < 0.05 or **** p < 0.0001 compared to silica treated BMdM as determined by two-way ANOVA with Tukey’s multiple comparison test. (B) Lysosomal acidification as assessed by lysotracker Red (LTR) or (C) lysosomal proteolytic activity as assessed by DQ-BSA after 1 h treatment with PAX (10 μM) or BAF (100 nM). n = 6. **** p < 0.0001 compared to untreated BMdM as determined by one-way ANOVA with Dunnett’s multiple comparisons test.
PAX reduces lysosomal acidification and proteolytic function
The function of lysosomal ion channels is critical to maintaining lysosomal homeostasis, not the least of which is the acidification of the lysosome. Ion channel activity largely contributes to lysosomal membrane potential facilitating the H+ pumping activity of the V-ATPase (Mindell Citation2012). Lysosomal acidification has been demonstrated as necessary for LMP and IL-1β release following silica exposure while inhibiting V-ATPase driven acidification with bafilomycin A1 (BAF), reducing lysosomal acidification with imipramine (IMP) or fluoxetine (FLX) contributes to increased lysosomal cholesterol and can reduce LMP, cell death, and IL-1β release (Jessop et al. Citation2017; Kendall and Holian Citation2023). Hypothesizing that blocking lysosomal BK ion channel activity could alter lysosomal acidification and proteolytic function, lysosomal pH and general proteolytic function of PAX-treated BMdM were assessed. BMdM were treated with PAX (10 μM) or BAF (100 nM) for 1 h then incubated with LysoTracker Red (LTR; 100 nM) for 30 min. LTR fluorescence demonstrated a significant decrease with PAX (). DQ-Green BSA (DQ-BSA) was used to measure lysosomal proteolytic activity. DQ-BSA fluoresces brightly when cleaved by lysosomal proteases. BMdM were treated with PAX (10 μM) or BAF (100 nM) for 1 h, then loaded with DQ-BSA (10 μg/mL) for 1 h. PAX treatment significantly reduced DQ-BSA fluorescence (). These results demonstrate that blocking lysosomal BK channel activity with PAX significantly decreases lysosomal acidification and proteolytic activity.
PAX causes lysosomal cholesterol accumulation
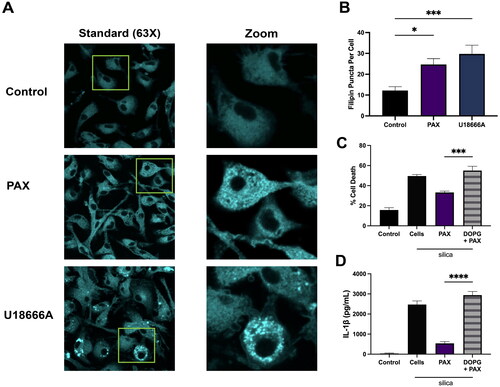
Alkalinization of lysosomal pH contributes to reduced NPC1 and NPC2 activity and can cause an increase in lysosomal free cholesterol (Friedland et al. Citation2003; Qian et al. Citation2020). Previously, disrupting lysosomal acidification with the V-ATPase inhibitor BAF or with cationic amphiphilic drugs (CADs) imipramine (IMP) or fluoxetine (FLX) was demonstrated to contribute to cholesterol accumulation (Kendall and Holian Citation2023). Since disruption of lysosomal pH and proteolytic function can alter lysosomal cholesterol homeostasis, lysosomal free cholesterol accumulation was examined in PAX-treated BMdM with fluorescent filipin staining. Filipin specifically binds free cholesterol and has been used to monitor increased lysosomal-associated accumulation of cholesterol by the NPC1 inhibitor U18666A (Reiners et al. Citation2011; Lu et al. Citation2015). BMdM were treated with PAX (10 μM) or U18666A (10 μg/mL) for 24 h then fixed and stained with filipin. Filipin-stained cells were imaged by laser-scanning microscopy () and free cholesterol-stained puncta quantified using Image J (). PAX treatment demonstrated a significant increase in filipin puncta per cell similar to U18666A (). These results demonstrate that inhibiting lysosomal BK channel activity increases lysosomal cholesterol.
Figure 4. Lysosomal cholesterol is necessary for PAX’s reduction of silica-induced IL-1β release. Data expressed as mean ± SEM. (A) Representative images BMdM treated with PAX (10 μM) or U18666A (10 μg/mL) for 24 h prior to filipin staining. (B) Quantification of filipin-stained free cholesterol puncta per cell using Image J. n = 7. * p < 0.05 and *** p < 0.001 compared to untreated BMdM as determined by one way ANOVA with Dunnett’s multiple comparisons test. (C) LDH and (D) IL-1β in supernatants of BMdM treated with DOPG for 1 h prior to PAX and silica for 24 h. n = 4. *** p < 0.001 and **** p < 0.0001 compared to PAX + silica treated as determined by one-way ANOVA with Dunnett’s multiple comparisons test.
PAX effect on cell death and IL-1β release is reversed by removal of lysosomal cholesterol
To further confirm that cholesterol accumulation was necessary for PAX’s ability to reduce silica-induced LMP and IL-1β release, BMdM were depleted of lysosomal cholesterol prior to PAX treatment. Phosphatidyl glycerol (DOPG) is the biosynthetic precursor of lysobisphosphatidic acid (LBPA) that interacts with NPC2 cholesterol transporter and promotes free cholesterol transport out of the lysosome (McCauliff et al. Citation2019). Both DOPG and LBPA exogenous treatments have demonstrated reduced intracellular cholesterol accumulation in an NPC1 deficient cell model (McCauliff et al. Citation2019; Ilnytska et al. Citation2021). Furthermore, DOPG decreases lysosomal free cholesterol and increases LMP and IL-1β release in silica-treated BMdM (Sydor et al. Citation2023). Here, LPS-primed BMdM were treated with DOPG for 1 h prior to 30 min treatment with PAX and silica application for 24 h. DOPG treatment completely negated PAX’s reduction of both silica-induced cell death () and IL-1β release (). These results demonstrate the necessity of lysosomal cholesterol accumulation to the ability of PAX to reduce silica-induced LMP, cell death, and IL-1β release.
Activation of lysosomal BK channel exacerbates silica-induced cell death and IL-1β release
As inhibition of lysosomal BK activity was effective in reducing LMP, cell death, and inflammation due to silica, studies to evaluate the impact of activating the lysosomal BK channel were conducted. NS1619 and NS11021 are each reported selective BK channel activators (Olesen et al. Citation1994; Bentzen et al. Citation2007). NS1619 is a specific activator that targets the α subunit of the BK channel, likely by binding the S6-RCK1 linker () (Gessner et al. Citation2012) while NS11021 works to slow overall BK channel closing rate thereby stabilizing the open conformation and increasing channel open probability (Rockman et al. Citation2020). BMdM were pretreated with NS11021 (1 μM) or NS1619 (5 μM) for 1 h prior to silica application for 24 h. Silica particle uptake was unchanged with NS11021 or NS1619 treatment (Supplemental Figure 1). Silica-induced cell death was increased with BK activation as NS11021 increased cell death to near significance (p = 0.08) and NS1619 increased cell death significantly (). Both NS11021 and NS1619 significantly increased IL-1β release compared to silica-only BMdM (). These results demonstrate that in contrast to the effects of inhibiting lysosomal BK channel activity, activation of the lysosomal BK channel promotes silica-induced cell death and IL-1β release in BMdM.
Figure 5. Activating lysosomal BK channel increases silica-induced cell death and IL-1β release by increasing lysosome acidification and cholesterol trafficking. Data expressed as mean ± SEM. (A) Cell death and (B) IL-1β release as measured in supernatants of LPS-primed BMdM treated with NS11021 (1 μM) or NS1619 (5 μM) for 1 h prior to silica (50 μg/mL) for 24 h. n = 4. * p < 0.05, ** p < 0.01 and **** p < 0.0001 compared to silica treated BMdM as determined by two-way ANOVA with Tukey’s multiple comparisons test. (C) Lysotracker Red or (D) DQ-BSA relative fluorescent intensity percentage (compared to untreated BMdM) of BMdM treated with NS11021 (1 μM), NS1619 (5 μM), or BAF (100 nM) for 1 h. n = 4-6. * p < 0.05 or **** p < 0.0001 compared to untreated BMdM as determined by one-way ANOVA with Dunnett’s multiple comparison’s test. (E) Filipin puncta per cell as quantified by Image J following NS11021, U18666A, or U18666A + NS11021 treatment for 24 h. n = 5-10 images (∼50 cells). *** p < 0.001 compared to U18666A treated as determined by one-way ANOVA with Dunnett’s multiple comparison’s test.
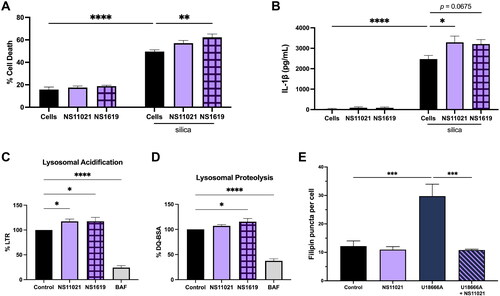
Activation of lysosomal BK channel promotes lysosomal acidification and cholesterol trafficking
Activation of the BK channel has been previously reported to promote lysosomal acidification and cholesterol efflux (Zhong et al. Citation2016; Khan et al. Citation2019). Here, the impact on lysosomal function was assessed with BK activators NS11021 and NS1619. BMdM were treated with NS11021 (1 μM) or NS1619 (5 μM) for 1 h prior to loading with Lysotracker Red or DQ-BSA and compared to BAF (100 nM). NS11021 and NS1619 each significantly increased the LTR fluorescent intensity compared to untreated BMdM (). NS11021 treatment slightly increased lysosomal proteolytic activity, while NS1619 increased DQ-BSA fluorescent intensity significantly compared to control cells (). Finally, the impact of BK activation on cholesterol trafficking out of the lysosome was assessed in U18666A-loaded cells. U18666A directly inhibits NPC1 transporter activity and causes a significant increase in lysosomal cholesterol (Liscum and Faust Citation1989; Lu et al. Citation2015). Here, BMdM were treated with NS11021 (1 μM), U18666A (10 μg/mL), or U18666A + NS11021 for 24 h, then fixed and stained with filipin. NS11021 alone showed no demonstratable change in filipin puncta compared to untreated BMdM; however, NS11021 treatment rescued U18666A-induced filipin puncta formation (). The increased LTR and DQ-BSA fluorescent output and decreased filipin puncta staining indicated that BK activation with NS11021 or NS1619 enhances lysosomal acidification and proteolytic activity and contributes to enhanced cholesterol trafficking from the lysosome thus reducing lysosomal cholesterol.
Discussion
Silicosis and other particle-induced fibrotic diseases are driven by NLRP3-mediated inflammation. In order to understand the mechanisms that perpetuate particle-induced inflammation in alveolar macrophages it is necessary to understand the key rate-limiting step, namely, LMP. Previously, Ziglari et al. demonstrated a hyperpolarization of the lysosomal membrane that preceded the onset of silica-induced LMP and suggested lysosomal hyperpolarization triggers LMP (Ziglari et al. Citation2021). High extracellular K+ failed to abrogate the lysosomal membrane hyperpolarization due to silica nor did it completely offset the decrease in cytosolic K+ (Ziglari et al. Citation2021). Those observations suggest that lysosomal K+ movement is important to the mechanisms of silica-induced inflammation in macrophages. As LMP and decreased cytosolic K+ are both known activators of the NLRP3 inflammasome and promote IL-1β release in particle exposure scenarios, the current study was designed to elucidate the role of lysosomal BK channel-mediated K+ movement in silica-induced inflammation.
Cell permeable BK inhibitor paxilline (PAX) effectively reduced silica-induced cell death and IL-1β release in three murine macrophage models () demonstrating that inhibiting intracellular BK ion channel activity reduces silica-induced inflammation. Furthermore, PAX significantly offset the dose dependent decrease in cytosolic K+ observed with silica treatment (). Interestingly, high extracellular K+ effectively reduced IL-1β release caused by silica () but it failed to prevent cell death (), suggesting that while cytosolic K+ efflux to the extracellular space may contribute to IL-1β release in silica-induced inflammation, cell death is independent of NLRP3 activity and subsequent IL-1β release. This is in agreement with other reports of silica-induced cell death in macrophages being inflammasome-independent (Cassel et al. Citation2008; Wierenga et al. Citation2019). Thus, blocking movement of cytosolic K+ into the lysosome though the BK channel with PAX was effective in preventing both silica-induced cell death and IL-1β release () in contrast to high extracellular K+ which only prevented silica-induced IL-1β release. The entry of cytosolic K+ into the lysosome contributes a small part of the total decrease in cytosolic K+ movement following silica exposure (), yet even the smallest perturbations in ion flux across the lysosomal membrane can have profound effects on lysosomal homeostasis as evidenced by PAX’s ability to prevent silica-induced LMP (). The impact of PAX on silica-induced cell death, LMP, and IL-1β release highlights the importance of LMP in silica’s role in cell death and inflammation.
Lysosomal pH and cholesterol content play a significant role in silica-induced LMP and IL-1β release (Jessop et al. Citation2017; Kendall and Holian Citation2023; Sydor et al. Citation2023). Decreased lysosomal pH and increased lysosomal cholesterol are protective of silica-induced LMP and IL-1β release (Kendall and Holian Citation2023). PAX was previously reported to decrease lysosomal proteolytic activity while NS1619 reduced cholesterol accumulation suggesting that lysosomal BK activity contributes to both lysosomal pH homeostasis and cholesterol transport (Zhong et al. Citation2016; Wang et al. Citation2017). Here, inhibition of lysosomal BK activity caused a decrease in lysosomal acidification and proteolytic function () while BK activation increased lysosomal acidification and proteolytic function (). Filipin staining revealed a significant accumulation of lysosomal cholesterol after 24 h treatment with PAX (), while activation of the lysosomal BK channel prevented accumulation of lysosomal cholesterol (). The importance of lysosomal cholesterol to PAX’s ability to reduce silica-induced LMP was further highlighted as cholesterol trafficking promoter DOPG completely abrogated the PAX decrease of IL-1β release. (). These results confirm that blocking lysosomal BK channel activity prevents silica-induced LMP, cell death, and inflammation by increasing lysosomal pH and diminishing proteolytic function thereby promoting an accumulation of lysosomal cholesterol (). Future studies should examine silica-induced LMP in BK null macrophages as embryonic fibroblasts from BK null C57/BL6 mice demonstrated a slight increase in lysosomal cholesterol (Wang et al. Citation2017).
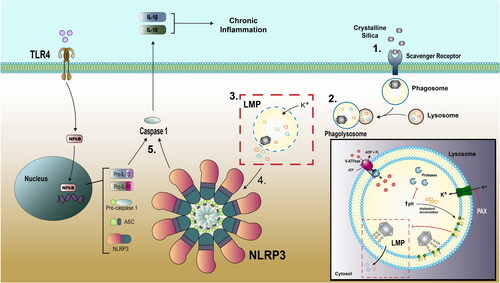
Figure 6. Silica causes LMP in macrophages. (1) Silica is recognized by a scavenger receptor and internalized by macrophage in phagosome. (2) Phagosome fuses with lysosome creating phagolysosome where particle encounters acid pH and hydrolyzing enzymes. (3) Silica particle interacts with lysosomal membrane via nearly free silanol attachments to phospholipid head groups and initiates K+ influx and LMP. (4) Release of hydrolyzing enzymes (such as cathepsin B) initiate NLRP3 inflammasome activity. (5) NLRP3 inflammasome activity includes activated caspase 1 and pro-inflammatory cytokine IL-1β.
In addition to its contribution to lysosomal pH and cholesterol movement, BK activity can regulate Ca2+ movement by providing a counter ion flux to Ca2+ release from the lysosome through both transient receptor potential mucolipin 1 (TRPML1) and two-pore channels (TPC) Ca2+ channels and contribute to large shifts in lysosome membrane potential that further contribute to lysosomal Ca2+ ion channel activity (Cao et al. Citation2015; Kendall and Holian Citation2021). Ca2+ acts as a second messenger and is necessary to many aspects of cellular function. Apilimod, a PIKfyve kinase inhibitor that has been used in the treatment of B-cell non-Hodgkin lymphoma (Gayle et al. Citation2017), was reported to cause a TRPML1-mediated lysosomal Ca2+ release that initiated downstream mitochondrial damage and subsequent NLRP3 activation (Hou et al. Citation2023). Furthermore, lysosomal Ca2+ signaling was reported as necessary for pro-IL-1β generation following NLRP3 priming and, in fact, the decrease of lysosomal Ca2+ signaling prevented the effective priming of the NLRP3 inflammasome (Platt et al. Citation2022). While release of lysosomal Ca2+ contributed to the stabilization of IL-1β mRNA and increased IL-1β (Weber & Schilling, Citation2014). Thus, lysosomal Ca2+ release could contribute to effective priming of the NLRP3 inflammasome as well as enhanced activation, resulting in enhanced pro- IL-1β expression and increased IL-1β production. Therefore, further examination should be given to the role of lysosomal Ca2+ movement in silica-induced LMP and IL-1β release.
In conclusion, blocking lysosomal BK activity prevents LMP, cell death, and IL-1β release following silica exposure in multiple models of murine alveolar macrophages. Inhibited lysosomal BK channel activity further decreases lysosomal proteolytic function and causes an increase of lysosomal cholesterol that has been demonstrated to be effective in reducing LMP, cell death, and inflammation. Slowing the onset of silica-induced cell death allows macrophages to clear the silica from the lung microenvironment and can delay or slow the progression of fibrosis in a silicosis disease scenario. This examination of lysosomal K+ ion channel activity provides insight into the mechanisms that contribute to silica-induced LMP, cell death, and NLRP3-initiated inflammation, supporting the notion that lysosomal ion channels can be a therapeutic target in disease. Further research is necessary to fully understand the molecular mechanisms whereby lysosomal ion channel function and cholesterol accumulation prevent silica-induced inflammation and how these mechanisms can be translated to other particle interactions with lysosomes such as endogenous uric acid crystals in gout.
Supplemental Material
Download MS Word (14.6 KB)Supplemental Material
Download JPEG Image (183.8 KB){kind=link}
Supplemental Material
Download MS Word (12.5 KB)Acknowledgements
The authors thank Britten Postma of the University of Montana’s Inhalation and Pulmonary Physiology Core for assistance in alveolar macrophage isolation and bone marrow collection and Dr. Jessica Ray of the University of Montana’s Fluorescence Cytometry Core for assistance and training on the use of the Attune fluorescence cytometer.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
Data available upon request.
Additional information
Funding
References
- Aits S, Jaattela M, Nylandsted J. 2015. Methods for the quantification of lysosomal membrane permeabilization: a hallmark of lysosomal cell death. Methods Cell Biol,. 126:261–285. doi: 10.1016/bs.mcb.2014.10.032.
- Ballabio A, Bonifacino JS. 2020. Lysosomes as dynamic regulators of cell and organismal homeostasis. Nat Rev Mol Cell Biol. 21(2):101–118. doi: 10.1038/s41580-019-0185-4.
- Barnes H, Goh NSL, Leong TL, Hoy R. 2019. Silica-associated lung disease: an old-world exposure in modern industries. Respirology. 24(12):1165–1175. doi: 10.1111/resp.13695.
- Bentzen BH, Nardi A, Calloe K, Madsen LS, Olesen SP, Grunnet M. 2007. The small molecule NS11021 is a potent and specific activator of Ca2+-activated big-conductance K + channels. Mol Pharmacol. 72(4):1033–1044. doi: 10.1124/mol.107.038331.
- Biswas R, Trout KL, Jessop F, Harkema JR, Holian A. 2017. Imipramine blocks acute silicosis in a mouse model. Part Fibre Toxicol. 14(1):36. doi: 10.1186/s12989-017-0217-1.
- Bunderson-Schelvan M, Hamilton RF, Trout KL, Jessop F, Gulumian M, Holian A. 2016. Approaching a unified theory for particle-induced inflammation. InL T. Otsuki, Yoshioka, Y., Holian, A., editors, Biological effects of fibrous and particulate substances (p. 51–76). Japan: Springer. doi: 10.1007/978-4-431-55732-6_3.
- Bunderson-Schelvan M, Holian A, Hamilton RF. Jr 2017. Engineered nanomaterial-induced lysosomal membrane permeabilization and anti-cathepsin agents. J Toxicol Environ Health B Crit Rev. 20(4):230–248. doi: 10.1080/10937404.2017.1305924.
- Burki TK. 2021. Silicosis and the countertop industry. Lancet Respir Med. 9(11):1217. doi: 10.1016/S2213-2600(21)00452-5.
- Burmeister R, Rhoderick JF, Holian A. 2019. Prevention of crystalline silica-induced inflammation by the anti-malarial hydroxychloroquine. Inhal Toxicol. 31(7):274–284. doi: 10.1080/08958378.2019.1668091.
- Candia S, Garcia ML, Latorre R. 1992. Mode of action of iberiotoxin, a potent blocker of the large conductance Ca(2+)-activated K + channel. Biophys J. 63(2):583–590. doi: 10.1016/S0006-3495(92)81630-2.
- Cang C, Aranda K, Seo YJ, Gasnier B, Ren D. 2015. TMEM175 is an organelle K(+) channel regulating lysosomal function. Cell. 162(5):1101–1112. doi: 10.1016/j.cell.2015.08.002.
- Cao Q, Zhong XZ, Zou Y, Zhang Z, Toro L, Dong XP. 2015. BK channels alleviate lysosomal storage diseases by providing positive feedback regulation of lysosomal Ca2+ release. Dev Cell. 33(4):427–441. doi: 10.1016/j.devcel.2015.04.010.
- Cassel SL, Eisenbarth SC, Iyer SS, Sadler JJ, Colegio OR, Tephly LA, Carter AB, Rothman PB, Flavell RA, Sutterwala FS. 2008. The Nalp3 inflammasome is essential for the development of silicosis. Proc Natl Acad Sci U S A. 105(26):9035–9040. doi: 10.1073/pnas.0803933105.
- De Duve C, Pressman BC, Gianetto R, Wattiaux R, Appelmans F. 1955. Tissue fractionation studies. 6. Intracellular distribution patterns of enzymes in rat-liver tissue. Biochem J. 60(4):604–617. doi: 10.1042/bj0600604.
- Fletcher P, Hamilton RF, Jr., Rhoderick JF, Pestka JJ, Holian A. 2021. Docosahexaenoic acid impacts macrophage phenotype subsets and phagolysosomal membrane permeability with particle exposure. J Toxicol Environ Health A. 84(4):152–172. doi: 10.1080/15287394.2020.1842826.
- Forgac M. 2007. Vacuolar ATPases: rotary proton pumps in physiology and pathophysiology. Nat Rev Mol Cell Biol. 8(11):917–929. doi: 10.1038/nrm2272.
- Friedland N, Liou HL, Lobel P, Stock AM. 2003. Structure of a cholesterol-binding protein deficient in Niemann-Pick type C2 disease. Proc Natl Acad Sci U S A. 100(5):2512–2517. doi: 10.1073/pnas.0437840100.
- Galvez A, Gimenez-Gallego G, Reuben JP, Roy-Contancin L, Feigenbaum P, Kaczorowski GJ, Garcia ML. 1990. Purification and characterization of a unique, potent, peptidyl probe for the high conductance calcium-activated potassium channel from venom of the scorpion Buthus tamulus. J Biol Chem. 265(19):11083–11090. doi: 10.1016/s0021-9258(19)38560-6.
- Gayle S, Landrette S, Beeharry N, Conrad C, Hernandez M, Beckett P, Ferguson SM, Mandelkern T, Zheng M, Xu T, et al. 2017. Identification of apilimod as a first-in-class PIKfyve kinase inhibitor for treatment of B-cell non-Hodgkin lymphoma. Blood. 129(13):1768–1778. doi: 10.1182/blood-2016-09-736892.
- Gessner G, Cui YM, Otani Y, Ohwada T, Soom M, Hoshi T, Heinemann SH. 2012. Molecular mechanism of pharmacological activation of BK channels. Proc Natl Acad Sci U S A. 109(9):3552–3557. doi: 10.1073/pnas.1114321109.
- Gorki AD, Symmank D, Zahalka S, Lakovits K, Hladik A, Langer B, Maurer B, Sexl V, Kain R, Knapp S. 2022. Murine ex vivo cultured alveolar macrophages provide a novel tool to study tissue-resident macrophage behavior and function. Am J Respir Cell Mol Biol. 66(1):64–75. doi: 10.1165/rcmb.2021-0190OC.
- Hamilton RF, Jr., Thakur SA, Holian A. 2008. Silica binding and toxicity in alveolar macrophages. Free Radic Biol Med. 44(7):1246–1258. doi: 10.1016/j.freeradbiomed.2007.12.027.
- Hamilton RF, Jr., Thakur SA, Mayfair JK, Holian A. 2006. MARCO mediates silica uptake and toxicity in alveolar macrophages from C57BL/6 mice. J Biol Chem. 281(45):34218–34226. doi: 10.1074/jbc.M605229200.
- Hornung V, Bauernfeind F, Halle A, Samstad EO, Kono H, Rock KL, Fitzgerald KA, Latz E. 2008. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat Immunol. 9(8):847–856. doi: 10.1038/ni.1631.
- Hornung V, Latz E. 2010. Critical functions of priming and lysosomal damage for NLRP3 activation. Eur J Immunol. 40(3):620–623. doi: 10.1002/eji.200940185.
- Hou Y, He H, Ma M, Zhou R. 2023. Apilimod activates the NLRP3 inflammasome through lysosome-mediated mitochondrial damage. Front Immunol. 14:1128700. doi: 10.3389/fimmu.2023.1128700.
- Ilnytska O, Lai K, Gorshkov K, Schultz ML, Tran BN, Jeziorek M, Kunkel TJ, Azaria RD, McLoughlin HS, Waghalter M, et al. 2021. Enrichment of NPC1-deficient cells with the lipid LBPA stimulates autophagy, improves lysosomal function, and reduces cholesterol storage. J Biol Chem. 297(1):100813. doi: 10.1016/j.jbc.2021.100813.
- Jessop F, Hamilton RF, Jr., Rhoderick JF, Fletcher P, Holian A. 2017. Phagolysosome acidification is required for silica and engineered nanoparticle-induced lysosome membrane permeabilization and resultant NLRP3 inflammasome activity. Toxicol Appl Pharmacol. 318:58–68. doi: 10.1016/j.taap.2017.01.012.
- Katsnelson MA, Rucker LG, Russo HM, Dubyak GR. 2015. K + efflux agonists induce NLRP3 inflammasome activation independently of Ca2+ signaling. J Immunol. 194(8):3937–3952. doi: 10.4049/jimmunol.1402658.
- Kendall RL, Holian A. 2021. The role of lysosomal ion channels in lysosome dysfunction. Inhal Toxicol. 33(2):41–54. doi: 10.1080/08958378.2021.1876188.
- Kendall RL, Holian A. 2023. Cholesterol-dependent molecular mechanisms contribute to cationic amphiphilic drugs’ prevention of silica-induced inflammation. Eur J Cell Biol. 102(2):151310. doi: 10.1016/j.ejcb.2023.151310.
- Kendall RL, Ray JL, Hamilton RF, Jr., Holian A. 2023. Self-replicating murine ex vivo cultured alveolar macrophages as a model for toxicological studies of particle-induced inflammation. Toxicol Appl Pharmacol. 461:116400. doi: 10.1016/j.taap.2023.116400.
- Khan N, Lakpa KL, Halcrow PW, Afghah Z, Miller NM, Geiger JD, Chen X. 2019. BK channels regulate extracellular Tat-mediated HIV-1 LTR transactivation. Sci Rep. 9(1):12285. doi: 10.1038/s41598-019-48777-y.
- Leso V, Fontana L, Romano R, Gervetti P, Iavicoli I. 2019. Artificial stone associated silicosis: a systematic review. Int J Environ Res Public Health. 16(4):568. doi: 10.3390/ijerph16040568.
- Leung CC, Yu IT, Chen W. 2012. Silicosis. Lancet. 379(9830):2008–2018. doi: 10.1016/S0140-6736(12)60235-9.
- Liscum L, Faust JR. 1989. The intracellular transport of low density lipoprotein-derived cholesterol is inhibited in Chinese hamster ovary cells cultured with 3-β-[2-(diethylamino)ethoxy]androst-5-en-17-one. J Biol Chem. 264(20):11796–11806. doi: 10.1016/s0021-9258(18)80136-3.
- Lu F, Liang Q, Abi-Mosleh L, Das A, De Brabander JK, Goldstein JL, Brown MS. 2015. Identification of NPC1 as the target of U18666A, an inhibitor of lysosomal cholesterol export and Ebola infection. Elife. 4: e12177. doi: 10.7554/eLife.12177.
- Marty A. 1981. Ca-dependent K channels with large unitary conductance in chromaffin cell membranes. Nature. 291(5815):497–500. doi: 10.1038/291497a0.
- McCauliff LA, Langan A, Li R, Ilnytska O, Bose D, Waghalter M, Lai K, Kahn PC, Storch J. 2019. Intracellular cholesterol trafficking is dependent upon NPC2 interaction with lysobisphosphatidic acid. Elife. 8: e50832. doi: 10.7554/eLife.50832.
- Migliaccio CT, Buford MC, Jessop F, Holian A. 2008. The IL-4Ralpha pathway in macrophages and its potential role in silica-induced pulmonary fibrosis. J Leukoc Biol. 83(3):630–639. doi: 10.1189/jlb.0807533.
- Mindell JA. 2012. Lysosomal acidification mechanisms. Annu Rev Physiol. 74(1):69–86. doi: 10.1146/annurev-physiol-012110-142317.
- Muñoz-Planillo R, Kuffa P, Martínez-Colón G, Smith BL, Rajendiran TM, Núñez G. 2013. K(+) efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity. 38(6):1142–1153. doi: 10.1016/j.immuni.2013.05.016.
- Oh S, Paknejad N, Hite RK. 2020. Gating and selectivity mechanisms for the lysosomal K(+) channel TMEM175. Elife. 9: e53430. doi: 10.7554/eLife.53430.
- Oh S, Stix R, Zhou W, Faraldo-Gómez JD, Hite RK. 2022. Mechanism of 4-aminopyridine inhibition of the lysosomal channel TMEM175. Proc Natl Acad Sci U S A. 119(44):e2208882119. doi: 10.1073/pnas.2208882119.
- Olesen SP, Munch E, Moldt P, Drejer J. 1994. Selective activation of Ca(2+)-dependent K + channels by novel benzimidazolone. Eur J Pharmacol. 251(1):53–59. doi: 10.1016/0014-2999(94)90442-1.
- Pallotta BS, Magleby KL, Barrett JN. 1981. Single channel recordings of Ca2+-activated K + currents in rat muscle cell culture. Nature. 293(5832):471–474. doi: 10.1038/293471a0.
- Pavan C, Delle Piane M, Gullo M, Filippi F, Fubini B, Hoet P, Horwell CJ, Huaux F, Lison D, Lo Giudice C, et al. 2019. The puzzling issue of silica toxicity: are silanols bridging the gaps between surface states and pathogenicity? Part Fibre Toxicol. 16(1):32. doi: 10.1186/s12989-019-0315-3.
- Pavan C, Santalucia R, Leinardi R, Fabbiani M, Yakoub Y, Uwambayinema F, Ugliengo P, Tomatis M, Martra G, Turci F, et al. 2020. Nearly free surface silanols are the critical molecular moieties that initiate the toxicity of silica particles. Proc Natl Acad Sci U S A. 117(45):27836–27846. doi: 10.1073/pnas.2008006117.
- Pavan C, Sydor MJ, Bellomo C, Leinardi R, Cananà S, Kendall RL, Rebba E, Corno M, Ugliengo P, Mino L, et al. 2022. Molecular recognition between membrane epitopes and nearly free surface silanols explains silica membranolytic activity. Colloids Surf B Biointerfaces. 217:112625. doi: 10.1016/j.colsurfb.2022.112625.
- Pfau JC, Schneider JC, Archer AJ, Sentissi J, Leyva FJ, Cramton J. 2004. Environmental oxygen tension affects phenotype in cultured bone marrow-derived macrophages. Am J Physiol Lung Cell Mol Physiol. 286(2):L354–362. doi: 10.1152/ajplung.00380.2002.
- Platt N, Shepherd D, Weng Y, Churchill GC, Galione A, Platt FM. 2022. Release of acidic store calcium is required for effective priming of the NLRP3 inflammasome. Bio Rxiv. 2022.01.06.475262. doi: 10.1101/2022.01.06.475262.
- Qian H, Wu X, Du X, Yao X, Zhao X, Lee J, Yang H, Yan N. 2020. Structural basis of low-pH-dependent lysosomal cholesterol egress by NPC1 and NPC2. Cell. 182(1):98–111 e118. doi: 10.1016/j.cell.2020.05.020.
- Reiners JJ, Jr., Kleinman M, Kessel D, Mathieu PA, Caruso JA. 2011. Nonesterified cholesterol content of lysosomes modulates susceptibility to oxidant-induced permeabilization. Free Radic Biol Med. 50(2):281–294. doi: 10.1016/j.freeradbiomed.2010.11.006.
- Rockman ME, Vouga AG, Rothberg BS. 2020. Molecular mechanism of BK channel activation by the smooth muscle relaxant NS11021. J Gen Physiol. 152(6). e201912506. doi: 10.1085/jgp.201912506.
- Saikia S, Parker EJ, Koulman A, Scott B. 2007. Defining paxilline biosynthesis in Penicillium axillin: functional characterization of two cytochrome P450 monooxygenases. J Biol Chem. 282(23):16829–16837. doi: 10.1074/jbc.M701626200.
- Sydor MJ, Kendall RL, Holian A. 2023. Cholesterol content regulates silica-induced lysosomal membrane permeability. Front Toxicol. 5:1112822. doi: 10.3389/ftox.2023.1112822.
- Wang W, Zhang X, Gao Q, Lawas M, Yu L, Cheng X, Gu M, Sahoo N, Li X, Li P, et al. 2017. A voltage-dependent K(+) channel in the lysosome is required for refilling lysosomal Ca(2+) stores. J Cell Biol. 216(6):1715–1730. doi: 10.1083/jcb.201612123.
- Weber K, Schilling JD. 2014. Lysosomes integrate metabolic-inflammatory cross-talk in primary macrophage inflammasome activation. J Biol Chem. 289(13):9158–9171. doi: 10.1074/jbc.M113.531202.
- Wie J, Liu Z, Song H, Tropea TF, Yang L, Wang H, Liang Y, Cang C, Aranda K, Lohmann J, et al. 2021. A growth-factor-activated lysosomal K(+) channel regulates Parkinson’s pathology. Nature. 591(7850):431–437. doi: 10.1038/s41586-021-03185-z.
- Wierenga KA, Wee J, Gilley KN, Rajasinghe LD, Bates MA, Gavrilin MA, Holian A, Pestka JJ. 2019. Docosahexaenoic Acid Suppresses Silica-Induced Inflammasome Activation and IL-1 Cytokine Release by Interfering With Priming Signal. Front Immunol. 10:2130. doi: 10.3389/fimmu.2019.02130.
- Xu H, Ren D. 2015. Lysosomal physiology. Annu Rev Physiol. 77(1):57–80. doi: 10.1146/annurev-physiol-021014-071649.
- Yamamura H, Ohi Y, Muraki K, Watanabe M, Imaizumi Y. 2001. BK channel activation by NS-1619 is partially mediated by intracellular Ca2+ release in smooth muscle cells of porcine coronary artery. Br J Pharmacol. 132(4):828–834. doi: 10.1038/sj.bjp.0703885.
- Yang X, Wang G, Cao T, Zhang L, Ma Y, Jiang S, Teng X, Sun X. 2019. Large-conductance calcium-activated potassium channels mediate lipopolysaccharide-induced activation of murine microglia. J Biol Chem. 294(35):12921–12932. doi: 10.1074/jbc.RA118.006425.
- Zhong XZ, Sun X, Cao Q, Dong G, Schiffmann R, Dong XP. 2016. BK channel agonist represents a potential therapeutic approach for lysosomal storage diseases. Sci Rep. 6(1):33684. doi: 10.1038/srep33684.
- Zhou Y, Lingle CJ. 2014. Paxilline inhibits BK channels by an almost exclusively closed-channel block mechanism. J Gen Physiol. 144(5):415–440. doi: 10.1085/jgp.201411259.
- Zhou Y, Xia XM, Lingle CJ. 2020. The functionally relevant site for paxilline inhibition of BK channels. Proc Natl Acad Sci U S A. 117(2):1021–1026. doi: 10.1073/pnas.1912623117.
- Ziglari T, Wang Z, Holian A. 2021. Contribution of particle-induced lysosomal membrane hyperpolarization to lysosomal membrane permeabilization. Int J Mol Sci. 22(5):2277. doi: 10.3390/ijms22052277.