
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
In recent years, the incidence of skin cancer has increased worldwide, presenting a significant burden on healthcare services. Chemotherapy intervention is often not appropriate for all patients due to localized adverse effects on skin physiology. The aim of this study was, therefore, to consider the development of a novel phytochemical-based deformable liposomal formulation suspended in an aqueous gel for the controlled-release of naringenin. Naringenin is an antioxidant, free radical scavenger, anti-inflammatory agent, and immune system modulator thus may be potentially useful as a pharmacological anti-cancer agent. Formulated liposomes incorporating an increasing loading of Tween 20 (from 0% w/w to 10% w/w) demonstrated a significant decrease in deformability index (DI) (80.71 ± 2.02–59.17 ± 4.42 %), indicating an increase in elasticity. The release of naringenin over 24 h was directly affected by Tween-20 concentration, decreasing from 100.72%±4.98% to 79.53%±3.68% for 0% and 2% w/w Tween 20, respectively. Further, the incorporation of deformable liposomes into hydroxyethylcellulose (HEC) and hydroxypropyl methylcellulose (HPMC) gels resulting in a further retardation of naringenin release, 23.21%±1.17% and 19.83%±1.50%, respectively, over 24 h. Incubation of 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate-loaded liposomes with human dermal fibroblast (HDF) and keratinocyte cells demonstrated intracellular accumulation within 2 h, confirming deformable liposomes may be beneficial in improving drug penetration across dermal cells and would be valuable in emerging controlled-release formulations.
1. Introduction
Presently, up to 3 million non-melanoma skin cancers and 132 000 melanoma skin cancers occur worldwide each year (World Health Organisation Citation2017). With an increase in the prevalence of skin cancer, the burden on public healthcare systems, particularly in developed countries is increasing (Diepgen and Mahler Citation2002, Donaldson and Coldiron Citation2011, World Health Organisation Citation2017). Whilst the majority of skin cancers are treatable; poor treatment, in addition to malignant tumors, results in the death of over 9000 people annually (American Cancer Society Citation2017). Existing treatments include local surgery to remove tumors which may not be suitable for all patients, as well as topical creams linked with poor patient compliance stemming from a high dose frequency requirement and unpleasant side effects resulting in treatment failure (Kaplan and Moy Citation2000, Ali et al. Citation2007, Neville et al. Citation2007, Felicio et al. Citation2009). Thus, an ideal formulation would offer a modified release profile for the chemotherapeutic compound to minimize product application with marginal side effects to normal, non-cancerous skin cells.
Many chemotherapeutic agents have poor safety profiles resulting in unpleasant side effects, therefore, effective novel non-toxic agents are urgently required to overcome these issues and improve treatment outcomes (Carey and Burish Citation1988, Kanadaswami et al. Citation2005, Bansal et al. Citation2009). Emerging trends in chemoprevention and chemoprotection have focused on naturally occurring non-toxic anti-oxidant agents (Kanadaswami et al. Citation2005, Hwang et al. Citation2007, Siddiqui et al. Citation2009, Tsai et al. Citation2015). Reactive oxygen species (ROS) play a major role in many pathological conditions including photo-carcinogenesis and immune suppression. The use of anti-oxidants to prevent oxidative skin damage appears to be a promising approach (Albini and Sporn Citation2007, Huang et al. Citation2011, Casey et al. Citation2015). Naringenin, a member of the flavanone group of polyphenols, is an antioxidant, free radical scavenger, anti-inflammatory agent, and immune system modulator and thus may be potentially useful as a pharmacological anti-cancer agent (Chen et al. Citation2003, Huang et al. Citation2011, Casey et al. Citation2015, Kaur and Badhan Citation2015, Citation2017). Furthermore, naringenin has been found to regulate fibrosis. Fibroblasts and myofibroblasts play a critical role in the formation of the extra-cellular matrix and inducing fibrosis within growing tumors (Casey et al. Citation2015). Tissue fibrosis is frequently observed in the tumor microenvironment associated with rapid proliferation of fibroblast cells (Kerkar and Restifo Citation2012).
The use of such naturally occurring compounds for skin cancer management has met with limited success due to their limited oral bioavailability and poor delivery systems (Basnet et al. Citation2012, Zhao et al. Citation2013, Wang et al. Citation2017). Consequently, to achieve maximum clinical efficacy, novel approaches are required to enhance bioavailability for topical drug delivery. Nanoparticles formulation systems have been used in cancer management and therapeutics, primarily to aid in the solubility of the compound (Nishiyama Citation2007, Siddiqui et al. Citation2009). Naringenin is a poorly water soluble compound (41.76 μg/mL ± 0.51 μg/mL (Tsai et al. Citation2015)), and, therefore, a solubility enhancing nanoparticle mediated delivery system would be ideal for dermal delivery (Hsiu et al. Citation2002, Semalty et al. Citation2010, Tsai et al. Citation2015).
The skin with its multilayer anatomy is an efficient physical barrier to the external environment including xenobiotics (Lopez et al. Citation2011). Accordingly, there is a need to develop carrier systems to enhance penetrability. The use of liposomal formulations as drug-delivery vehicles provide a novel approach to the delivery and targeting of the dermal layer with benefits for the controlled delivery of poorly permeable molecules (du Plessis et al. Citation1994, Park et al. Citation2013). Deformable liposomes have successfully been employed in the transdermal delivery of lipophilic and hydrophilic drugs including anti-inflammatory agents, plasmid DNA, anti-tumor agents, and hormones with deeper penetration compared with conventional liposomes (El Maghraby et al. Citation1999, Cevc and Blume Citation2001, Oh et al. Citation2006, Romero et al. Citation2013).
Due to the nature of liposomes, preparations intended for application to the skin will need to be transported in a carrier. Liposomes are compatible with viscosity-increasing agents such as cellulose-based gels including hydroxyethylcellulose (HEC) and hydroxypropyl methylcellulose (HPMC) (Foldvari Citation1996). Aqueous semi-solid polymeric gels, such as those based on HEC and HPMC are often used in transdermal drug delivery and are useful in designing controlled delivery formulations (Valenta and Auner Citation2004, Ghosal and Nanda Citation2013).
This study developed a deformable liposomal gel formulation able to pass through the stratum corneum for targeting toward intracellular uptake into dermal cells. With this intent, HEC and HPMC gels as well as different liposomal formulations with varying loadings of Tween 20 were prepared. The vesicle size, surface charge, encapsulation efficiency, and deformability index (DI) were determined. The release of naringenin from gel, liposomes, and liposomal gel formulations into release media was studied. Deformable liposomes loaded with naringenin were applied to human dermal fibroblast (HDF) and keratinocyte cells to observe liposomal uptake. The stability of these formulations was also evaluated for assessing the clinical value of deformable liposomes.
2. Materials and methods
2.1. Materials
Soy phosphatidylcholine (PC) was obtained from Avanti Polar Lipids (grade ≥99%). HEC, HPMC, cholesterol (grade ≥ 99%), Tween 20, and naringenin (grade ≥ 95%, N5893) were obtained from Sigma-Aldrich (Dorset, England). All other reagents including methanol and chloroform were obtained from Fisher Scientific (Waltham, MA). Ultrapure water was obtained from a Milli-Q purification system (Millipore, Billerica, MA). Polycarbonate filters (pore size 50 and 100 nm) were obtained from Sigma-Aldrich (WHA800309). HDFa isolated from adult skin along with all cell culture reagents (Medium 106 and low serum growth supplement (LSGS) kit consisting of fetal bovine serum, 2% v/v, hydrocortisone 1 µg/mL, human epidermal growth factor, 10 ng/mL, basic fibroblast growth factor, 3 ng/mL, heparin, 10 µg/mL; DMEM media supplemented with 1% L-glutamine, 10% FBS, 1% Penicillin Streptomycin and 0.25% amphotericin) were obtained from Life technologies (Carlsbad, CA). Immortalized human keratinocytes (HaCat) cells were gifted by Dr Andrew Sanders (Cardiff China Medical Research Collaborative, Cardiff University, Henry Wellcome Building, Heath Park, Cardiff, CF14 4XN). Finally, 4′,6-diamidino-2-phenylindole (DAPI) and 1,1′-Dioctadecyl-3,3,3′,3′-Tetramethylindocarbocyanine Perchlorate (DilC) was obtained from ThermoFisher Scientific (D1306 and D282, respectively).
2.2. Methods
2.2.1. Deformable liposome preparation
Liposomes were prepared by adapting the film hydration method developed by Bangham et al. (Citation1965). Soy PC, cholesterol (16:8 µM) and 1 mg/mL naringenin was dispersed in a mixture of chloroform and methanol in a 9:1 v/v ratio in a round bottomed flask (Bangham et al. Citation1965, Hiruta et al. Citation2006, Oh et al. Citation2006, Ita et al. Citation2007, Tsai et al. Citation2015). Up to 10 w/w % of Tween 20 was added to the formulation into the lipid mixing stage to prepare deformable liposomes. The organic solvent was then removed by rotary evaporation for 5 min at 35 °C, and then purged with nitrogen gas. The resultant dry film residue was hydrated by the addition of 4 mL water at room temperature (Pagano and Weinstein Citation1978) and vortexed for 5 min to form multilamellar vesicles (MLVs). The resulting MLV’s were extruded 21 times through polycarbonate membranes with 100-nm diameter pore size, using an Avanti Mini Extruder to produce unilamellar vesicles. The resultant liposomes were equilibrated for 30 min at room temperature prior to characterization studies (Pagano and Weinstein Citation1978, Lasic and Barenholz Citation1996, Ali et al. Citation2013).
2.2.2. Liposome characterization: particle size, polydispersity, and zeta potential
The average liposome diameter and the polydispersity index (measurement of the level of homogeneity of particle sizes) of liposomes were measured by dynamic light scattering (DLS) using a Zetaplus (Brookhaven Instruments, Holtsville, NY). All formulations were diluted with distilled water (1:4 ratio) to ensure intensity adjustment. A polydispersity value < 0.2 indicates a homogenous particle population, whilst values > 0.3 indicates heterogeneity (Song and Kim Citation2006). The liposome surface charge was quantified as zeta potential (ζ) and was determined by photon correlation spectroscopy using a Zetaplus (Brookhaven Instruments). Formulation were diluted three-fold preceding analysis.
2.2.3. Liposomal deformability assessment
To assess the deformability of liposome formulations, a 6 mL liposome suspension of a 16 µM PC and 8 µM cholesterol formulated with up to 10% w/w of Tween 20 solution (diluted 1:4 with distilled water) was passed through a polycarbonate filter of (50 nm pore size) using a syringe driver (Cole Parmer, Saint Neots, UK) set at 0.6 mL/min for 10 min. The mean liposome size and the polydispersity index of liposomes were quantified by DLS. The deformability was calculated as the DI (EquationEquation (1))(1)
(1) (Goindi et al. Citation2013, Marwah et al. Citation2020):
(1)
(1)
where D is deformability, Le is size of extruded liposomes (nm), and L is size of liposomes (nm) prior to extrusion.
2.2.4. HPLC methodology
Detection of naringenin was assessed using a reverse phase HPLC method. A Waters Alliance separation module HPLC with UV detection was utilized at an operating wavelength of 287 nm (Wen et al. Citation2010). A Waters Xselect column (5 µm C18 4.6 × 150 mm column) was used with a 10 μL sample injected at room temperature. The mobile phase comprised of a 50:50 ratio of 0.1% TFA in water to acetonitrile at a flow rate of 1 mL/min.
Stock solutions and standard solutions of naringenin were prepared with both water and ethanol ranging from 0.05 to 250 µg/mL. The method was fully validated and resulted in a final calibration curve with an R2 of 0.9393 and a polynomial equation of y = (–3 × 107 x2) + (9 × 106 x) was obtained.
2.2.5. Determination of entrapment efficiency
The entrapment efficiency of naringenin-loaded deformable liposomes was determined following centrifugation of samples and quantifying drug in the supernatant. To separate the encapsulated drug from the free drug, liposomes were centrifuged at 18 000 rpm for 30 min at 4 °C in an Optimatm MAX-XP ultracentrifuge. The supernatant was then analyzed using HPLC-UV analysis to determine the encapsulation efficiency of naringenin in liposomal formulations (EquationEquation (2)(2)
(2) :
(2)
(2)
where E is the encapsulation efficiency (%), Dt is the total drug content (mg), and Ds is drug content in supernatant (mg).
2.2.6. Differential scanning calorimetry investigations of naringenin and naringenin-lipid blends
To evaluate thermal properties materials including melting temperatures, phase transitions and heat capacity changes of liposomes, naringenin and lipid ratios, surfactant, and drug mixtures corresponding to that of the liposome formulation, were investigated in the solid state using a TA Instruments Q200 Thermal Analysis Differential scanning calorimetry (DSC). An amount of 3 mg of naringenin was weighed into T-Zero aluminum pans and hermetically sealed. All experimental runs began with an initial temperature of 0 °C, purged under nitrogen gas, with a scan rate of 10 °C/min up to 300 °C.
2.2.7. Naringenin loaded aqueous gel formulation
Aqueous gels in distilled water were prepared using HEC (3% w/v) and HPMC (3% w/v) and mixed overnight using a mechanical mixer (Polytron PT 3100D) at a speed of 3000 rpm. Gels with a final naringenin loading of 1% w/w were produced.
2.2.8. In-vitro release of naringenin from formulations
Naringenin release from gels, liposomes and liposomal gels was quantified over 24 h using a variety of methods.
2.2.8.1 Side-by-side diffusion study
To evaluate the effect of inclusion of Tween 20 on the release of naringenin from liposomal formulations, a side-by-side diffusion cell (PermeGear diffusion cell, Hellertown, PA) was maintained at 35 °C. Release was observed over a 24 h period from a naringenin aqueous solution (0.1 mg/mL) as well as naringenin-loaded gels and liposomes (final loading for liposomes formulated with 0, 2, 6, and 10% w/w Tween 20: 0.91, 0.85, 0.75, and 0.64 mg/mL respectively). The donor side of the diffusion cell was loaded with 10 mL of each formulation and release across a membrane with 50 nm pore size (Whatman®) into the receiver side containing 100 mL of PBS with a stirrer was quantified at defined time points over 24 h with volume replacement (0.5 mL) using HPLC-UV quantification.
2.2.8.2 Liposomal gel release study
To investigate naringenin released from naringenin-loaded liposomes loaded into polymer gels, a permeable insert models system was employed (Marwah et al. Citation2020). This system was used to evaluate if naringenin/liposomes loaded with naringenin were able to diffuse out from the gel rather than to compare release rates. A 4 cm2 cylindrical cell culture Thincert™ insert (400 µm pore size) was filled with 1 mL of formulation and placed into a 6-well Thincert™ plate. Release into 4 mL of PBS from solution, and liposomal gels was quantified. HEC and HPMC (3% w/v of polymer) were loaded with non-surfactant loaded liposomes formulated or liposomes formulated with 2% w/w Tween 20. Thincert™ plates were maintained at 35 °C on a shaking plate and release media was sampled with volume replacement (0.5 mL) over 24 h and analyzed using HPLC quantification with UV analysis.
2.2.9. In-vitro drug release kinetics
Numerous kinetic drug release mathematical models were used in the evaluation of drug release from the formulations (EquationEquations (3–6)). The best-fit (R2) to the mathematical models described below established the appropriate release kinetics:
(3)
(3)
where Mt/M∞ is the drug fraction released at time t and k0 is the zero-order release constant.
(4)
(4)
where Mt/M∞ is the drug fraction released at time t and k1 is the first-order release constant.
(5)
(5)
where Mt/M∞ is the drug fraction released at time t and kH is the Higuchi constant.
(6)
(6)
where Ct/C is fraction of drug released at time t, k is the release rate constant. The value of n is important in understanding drug release mechanisms. When n≤ 0.45, drug release is diffusion controlled and termed as ‘Fickian’ diffusion, conversely when n> 0.89, the diffusion is indicating erosion-controlled drug release (class-II kinetics). When 0.45<n≤ 0.89, the diffusion is a multifaceted mixture of both processes termed anomalous transport. Regardless, in all cases this is based on the assumption of release from a cylinder and applied to cumulative release rates < 60% (Korsmeyer et al. Citation1983).
Mathematical models to assess release kinetics were fit using Microsoft Excel®. Zero order, first order, Higuchi and Korsmeyer–Peppas release profiles were applied to release from naringenin solution and naringenin loaded liposome solution after which regression analysis techniques were used to determine the likely drug-release. The release kinetic model displaying the highest r2 metric (≥0.95) was determined to be the release mechanism.
2.2.10. Development of an in-vitro dermal model
To develop an in-vitro system to represent human dermal tissue two dermal cell lines were used to assess cellular toxicity and cellular uptake of deformable liposomes. HDFa were cultured in Medium 106 supplemented with LSGS. Human epidermal keratinocytes (HaCat) cells sustained in high glucose supplemented DMEM media (as outlined in materials and methods). Media was replaced every 3 d unless at 70–80% confluency in which case media was discarded and Trypsin/EDTA added to detach cells. Cells were incubated for 5 min, following which 3 mL growth media was added to neutralize the trypsin. Cells were then centrifuged at 1200 rpm for 10 min and resuspended in media prior to use in the following experimental studies.
2.2.11. Cellular toxicity of naringenin toward HDFa and HaCat cells
The cytotoxicity profile of naringenin toward HDFa and HaCat cells was determined with a (2,3-bis-(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide (XTT) assay (Scudiero et al. Citation1988). Cell viability after exposure to increasing concentrations from 0.1 to 100 µM of naringenin for 24 h was observed. Cells were seeded at a density of 50 × 103 cells per well into a 96-well plate and grown for 3 d. Media was removed, and cells were exposed to 100 µL of 0.1–100 µM naringenin and incubated at 37 °C. After 24 h, 25 µL of a 12.5:1 (XTT: menadione) was added to each well and incubated for a further 3 h at 37 °C prior to the absorbance being read at 450 nm (Tecan, Männedorf, Switzerland). Assessment of naringenin toxicity toward cells was conducted after analysis of changes in XTT absorbance with changing drug concentration.
2.2.12. Intracellular deformable liposomal uptake assay on HDFa and HaCat cells
Conventional and deformable liposomes were formulated with the addition of 25 µg DilC during the lipid mixing stage. Liposomes were centrifuged at 18 000 g for 30 min to remove unentrapped DilC. Coverslips were prepared by coating with poly-l-lysine (0.01% w/v) and allowing to dry for 30 min before the seeding of cells at a density of 100 × 103 cells per coverslip. Cells were treated after 24 h, with DilC loaded liposomes diluted with 1 part of supplemented media (as detailed in materials) and incubated for 2 h at 37 °C. Subsequently, coverslips were washed and fixed with 4% w/v paraformaldehyde for 5 min at room temperature. Coverslips were then mounted onto glass slides with the addition of a DAPI-containing mounting media. Cover slips were analyzed with an upright confocal microscope (Leica SP5 TCS II MP) and visualized with a 40× oil immersion objective. Images were obtained using a helium-neon laser at 633 or 461 nm to visualize DilC and DAPI, respectively.
2.2.13. Liposome stability
The stability of liposomes was determined following evaluation of vesicle size over a 28-d period, stored in glass vials in a stability cabinet maintained at 25 ± 2 °C (Froilabo, Meyzieu, France) at a humidity of 60%±5%. Vesicle sizes were determined on day 1, 2, 7, 14, 21, and 28 by DLS. Additionally, the encapsulation efficiency of naringenin loaded liposomes were assessed over 4 weeks as outlined in Section 2.2.4.
2.2.14. Statistical analysis
All results are presented as mean ± standard deviation (SD). Replicates of at least three independent studies were used for all studies. For multiwell plate cells assays, replicates of six were used for each experimental condition, repeated 3 times. A one-way ANOVA was used to determine any statistically significant difference between means tested (p ≤ 0.05) with a post-hoc Tukey’s multiple comparisons test applied to evaluate differences between groups. All calculations were completed on Graphpad version 7 (GraphPad Inc., La Jolla, CA).
3. Results and discussion
As the incidence of skin cancer is rising, novel formulations able to successfully deliver therapeutic agents, whilst reducing side effects, are essential to decrease the burden on already stretched healthcare services. This study proposed the development a topical deformable liposome gel formulation for the controlled release of the natural compound naringenin, in view of its possible ‘anti-cancer’ properties (Chen et al. Citation2003, Huang et al. Citation2011, Casey et al. Citation2015).
The therapeutic potential of flavonoids such as naringenin is restricted due to their limited bioavailability, poor solubility, and inefficient delivery systems. Liposomes may prove useful as delivery agents as they can improve such compounds’ solubility profile and enhance their bioavailability (Nishiyama Citation2007, Siddiqui et al. Citation2009). Further, deformable liposomes incorporating surfactant have been found to be valuable in dermal drug delivery as they can improve compound solubility whilst reducing the rate of drug degradation and can be formulated for targeted, controlled drug release (Cevc Citation1996). The skin is an effective physical barrier to the external environment, however, deformable liposomes have been reported to overcome this barrier and penetrate the skin. Liposomes have already been used in the transdermal delivery of both lipophilic and hydrophilic drugs including anti-inflammatory agents, plasmid DNA, antioxidants , anti-tumor agents, and hormones (El Maghraby et al. Citation1999, Cevc and Blume Citation2001, Oh et al. Citation2006, Romero et al. Citation2013). Liposomes can also be formulated to improve drug deposition within the skin when the skin itself is the target and systemic absorption is not favorable (Cevc Citation1996).
Dermal application of liposomes requires an additional carrier due to the liquid nature of the formulation. Liposomes compatibility has previously been established with viscosity-increasing agents such as cellulose-based gels including HEC and HPMC (Foldvari Citation1996). Such polymers are recognized as safe in topical, dermal, and transdermal delivery (Patton et al. Citation2007, Hascicek et al. Citation2009, Forbes et al. Citation2011).
This study aimed to develop naringenin-loaded deformable liposomes with a controlled release profile to be loaded into an aqueous gel. Liposome characteristics, naringenin release profile, liposome stability as well as intracellular uptake was assessed. Naringenin liposomes were formulated with PC and cholesterol with up to 10% w/w loading of Tween 20 and the effect of Tween 20 on the characteristics of liposomes was observed.
3.1. Liposome characterization: particle size, polydispersity, zeta potential, entrapment efficiency, and DI
Liposome formulation was characterized by observing liposome size, polydispersity, zeta potential, entrapment efficiency, and DI. These are important determinants of liposome stability and compound delivery. As the surfactant loading in the bilayer of naringenin-free liposomes increased, liposome diameter decreased from 366.16 nm ± 34.32 nm for liposomes formulated with no surfactant to 188.68 nm ± 11.32 nm for liposomes formulated with 10% w/w Tween 20 (). An increase in surfactant loading in the bilayer of naringenin-loaded liposomes observed a decrease in liposome diameter, from 392.45 nm ± 32.44 nm for liposomes formulated without surfactant compared with 235.60 ± 9.31 nm for liposomes formulated with 10% w/w Tween 20 (). The decrease in size was statistically significant for naringenin-free liposomes and naringenin-loaded liposomes formulated with no surfactant compared with liposomes loaded with 2%, 6%, and 10% w/w Tween 20 (p ≤ 0.0001). Additionally, naringenin-loaded liposomes had a larger diameter than naringenin-free liposomes. Naringenin is hydrophobic, therefore, was added in at the lipid mixing stage. The inclusion of drug in the bilayer may have caused an increase in liposome size by increasing bilayer hydrophobicity as it had caused the bilayer to have less interaction with the aqueous phase.
Figure 1. Liposome size distribution and polydispersity of naringenin-free liposomes and naringenin-loaded liposomes. Liposome size distribution and polydispersity, determined by DLS, comparing (A) naringenin-free liposomes and (B) naringenin-loaded formulations with Tween 20 (0–10 % w/w). Liposomes were prepared by the dry film hydration method and naringenin added during the lipid mixing stage. Data represent mean ± SD. n = 3 independent batches. ****indicates statistical comparison between the size of liposome formulated with 0 % w/w Tween 20 and all other loadings of Tween 20 with a p ≤ 0.0001.
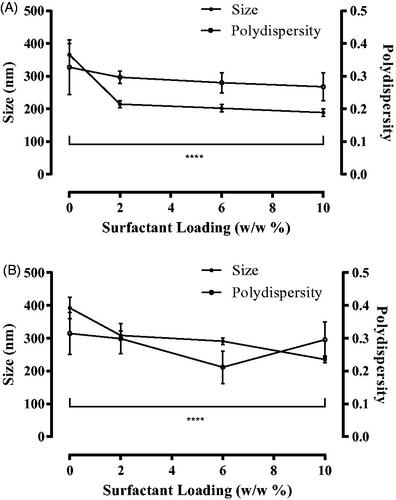
Surfactants decrease liposome size in comparison to ‘conventional’ liposomes (Goindi et al. 2013, Tsai et al. Citation2015, Marwah et al. Citation2020) due to the surfactant having a destabilizing effect within the bilayer. Furthermore, the amphiphilic nature of surfactant allows for an increased interaction of the phospholipid bilayer with the aqueous phase allowing for the formation of more liposomes of a smaller diameter (El Zaafarany et al. Citation2010). The inclusion of surfactant within liposomes has been reported to decrease liposome size in comparison to conventional liposomes previously; a study including either Phospholipon® 90G and both Tween 80 and Span 80 within the formulation observed a size reduction from 207 to 139 nm after addition of the surfactants was described (Goindi et al. Citation2013).
A homogenous liposomal size distribution is essential as final liposome size will influence the tissue distribution in-vivo as well as influence drug release kinetics. A polydispersity up to 0.3 is indicative of a homogenously sized liposome formulation (Chen et al. Citation2012, Goindi et al. Citation2013, Kang et al. Citation2013). Liposomes formulated without surfactant were beyond this range (0.33 ± 0.08 and 0.31 ± 0.03 for naringenin-free and naringenin-loaded liposomes, respectively), however, liposomes formulated with surfactant had a polydispersity below 0.3, and, therefore, can be considered homogenous (). Further, the polydispersity for naringenin-loaded liposomes was similar to respective naringenin-free liposomes, and not statistically different (p > 0.05), suggesting that the inclusion of Tween 20 within the liposome formulation appeared to improve homogeneity.
The zeta potential (ζ) indicates the magnitude of electrostatic repulsion between nearby, similarly charged particles within a dispersion. Therefore, it is a central parameter affecting stability of liposomal formulations. The formulated liposomes in this study demonstrated a near neutral charge (). Whilst a neutral liposomal surface charge is vital to prevent skin irritation (Prausnitz and Langer Citation2008), this may allow for particle flocculation due the lack of repulsion between vesicles (Weiner et al. Citation1992). Naringenin liposomal formulations formulated between 0 and 10% w/w Tween 20 were found to have general decrease in zeta potential values from 4.12 to −0.22 mV, respectively. A study using Tween 80 in the liposomal formulation also found that in increase in surfactant loading did not affect liposomal charge. Although, contrasting with this study observing more positive zeta potential values, negative zeta potential values were observed (between −2.2 and −16 mV) (Tsai et al. Citation2015). This may be due to differences in formulation parameters.
Table 1. Zeta potential of liposome formulations with up to 10% w/w loading of Tween 20.
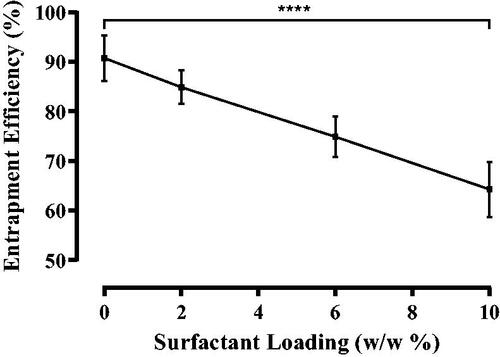
The impact of surfactant addition on the amount of naringenin entrapped was studied. As surfactant loading increased from 0% w/w to 10% w/w, naringenin entrapment decreased from an efficiency of 90.8%±4.6% to 64.3%±5.6% (). Inclusion of surfactant in the bilayer of the liposome may have prevented drug inclusion within the bilayer implying the surfactant has a higher affinity to the lipids (Levy et al. Citation1991, Casas and Baszkin Citation1992). Tween 20, with larger molecular weight of 1227.54 g/mol compared to that of naringenin (272.26 g/mol), may be assumed to be better poised to displace naringenin from the bilayer. Furthermore, studies using Tween 80 instead of Tween 20 were able to achieve a 99% entrapment efficiency (Tsai et al. Citation2015). This may be due to the difference in the surfactant chains. Tween 80 has one more unsaturated bond causing a kink in the chain. This may then allow for the accommodation of greater naringenin.
Figure 2. Entrapment efficiency of naringenin in liposomes formulated with up to 10% w/w Tween 20. Entrapment efficiency (%) of naringenin in liposomes formulated with varying amounts of Tween 20 (0–10% w/w) Data represent mean ± SD. n = 3 independent batches. ****indicates statistical comparison between the entrapment efficiency of liposome formulated with 0 % w/w Tween 20 and all other loadings of Tween 20 with a p ≤ 0.0001.
The deformability of each liposome formulation was determined following extrusion through a polycarbonate filter (pore size of 50 nm). The DI is defined as the degree the liposomes deformed; the more the liposomes deformed, the less elastic the liposomes are due to their inability to regain their larger size pre-extrusion. Liposomal DI following extrusion statistically significantly decreased (p ≤ 0.0001) as surfactant loading increased in the naringenin-free liposomes, from 72.74%±6.46% for liposomes formulated with no Tween 20 to 29.60%±2.59% for liposomes formulated with 10% w/w Tween 20. Naringenin loaded liposomes formulated with Tween 20 observed a statistically significant decrease in DI (p ≤ 0.0001) from 80.71%±2.02% for liposomes formulated with no Tween 20 compared with 59.17%±4.42% for liposomes formulated with 10% w/w Tween 20 (). These observations indicate liposomal bilayers had elastic properties as they could deform to pass through an opening lesser than its own diameter although, to a certain degree regaining its size (Marwah et al. Citation2020). It is essential to address the original size of liposome. Those formulated with Tween 20 were of a smaller diameter in the first instance thus would require less deformation to fit through the membrane pores. However, the membrane pore size was always smaller than that of liposome diameter and the DI equation requires the ratio of pre to post extrusion size. Furthermore, naringenin incorporation in the liposome formulation did not affect the DI compared with liposomes formulated without. A previous study formulating liposomes with Phospholipon® 90G and both Tween 80 and Span 80 observed a size reduction with inclusion of surfactant to decrease the DI from 51.4 ± 3.6 to 17.3 ± 5.2 (Goindi et al. Citation2013).
Figure 3. Deformability index for naringenin-free liposomes and naringenin-loaded liposomes. Deformability index following extrusion through 50 nm membranes for naringenin-free liposomes and naringenin loaded liposomes with increasing surfactant loading up to a maximum of 10% w/w. Liposomes were prepared to adapt the dry film method adding the surfactant and adding naringenin during the lipid mixing stage. The preparation was vortexed and then extruded through the membranes. Data represent mean ± SD. n = 3 independent batches. **** indicates statistical comparison between the DI of liposome formulations with a p ≤ 0.0001.
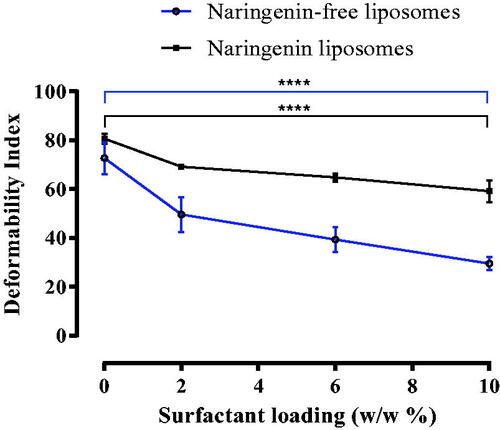
Surfactant has a propensity for highly curved structures (e.g. micelles and liposomes), thus diminishing the energy required for particle deformation allowing for vesicle deformation. The surfactant reduces the energy required for particle deformation and accommodate vesicle shape changes upon physical stress (Trotta et al. Citation2004, Marwah et al. Citation2020). These surfactants may have interacted with the PC with strong affinity but in reversible mode allowing for deformability when under stress (Oh et al. Citation2006).
The energy required for liposomes to deform (Gompper and Kroll Citation1995, Fresta and Puglisi Citation1996, Trotta et al. Citation2002) reversibly was supplied in the form of increasing pressure due to the movement of the syringe driver. Surfactant is able to retain energy (Trotta et al. Citation2002), thus increases liposome capacity to deform reversibly, thus as surfactant loading increased, an increased reversible deformation was observed. Liposomes did not completely regain their original size as some energy would be lost due to the friction of particles being forced through the pores (Vajjha et al. Citation2010, Marwah et al. Citation2020). Additionally, in-vivo, liposomes would likely diffuse through the skin following the transepidermal gradient (Gompper and Kroll Citation1995, Cevc Citation1996, Trotta et al. Citation2002, Goindi et al. Citation2013). Moreover, skin is warmer than room temp (35 °C compared to 20 °C) and since temperature dictates the energy term of enthalpy, liposomes would have additional energy to be even more flexible and cross through the SC.
3.2. Differential scanning calorimetry investigations of naringenin and naringenin lipid blends
DSC is extensively used in its application in understanding the thermal characteristics of materials where an insight into a range of thermal properties including melting temperatures, phase transitions, and heat capacity changes can be obtained. Naringenin showed a sharp endothermic peak (Tm) at 253 °C (). To substantiate the association of naringenin with the lipid/surfactant complex, DSC analysis was performed on, the lipid blend, and the naringenin–lipid/surfactant blend. In the lipid/surfactant mix a very small peak at 40 °C is noticeable, a larger peak at 172 °C and a medium peak at 212 °C. On the other hand, naringenin–lipid/surfactant complex showed a small peak at 40 °C and a large peak at 152 °C, differing from the peaks of the individual components of the complex (Supplementary Figure 1). It is evident that the original peaks of naringenin and phospholipids disappear from the thermogram of complex and the phase transition temperature is lower than that of naringenin as there is no sharp peak around 253 °C.
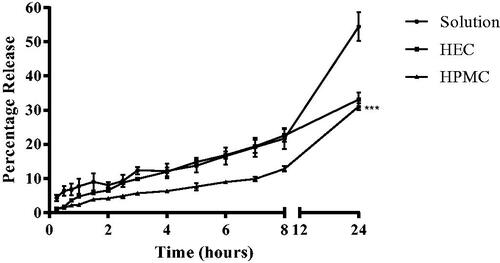
Figure 4. In-vitro percentage naringenin release profiles from aqueous gels. Naringenin release profiles from HEC and HPMC (3% w/v) aqueous gels with 1% w/v naringenin and naringenin solution over 24 hours. Data represent mean ± SD. n = 3 independent batches. ***indicates statistical comparison between the naringenin release of liposome formulations with a p ≤ 0.001.
The Tm of Naringenin corresponds with similar studies (Khan et al. Citation2015, Semalty et al. Citation2010). Differences between reported values and those observed here may be due to differences in naringenin sample purity. Furthermore, Semalty et al. observed the association of naringenin with soy PC. PC showed a smaller peak at 64.45 °C and major peaks at 83.21 and 107.90 °C. They suggested the first peak was probably due to the hot movement of the phospholipids polar head group. The second peak was assumed to be due to phase transition from gel to liquid crystalline state. The non-polar hydrocarbon tail of phospholipids may be melted during this phase, yielding a sharp peak. This melting might have occurred in two phases which subsequently gave the next peak. The naringenin–PC complex showed two peaks at 51.23 and 62.21 °C, which is different from the peaks of the individual components of the complex. Therefore Semalty et al. also found that the original peaks of naringenin and PC disappear from the thermogram of the blend and the phase transition temperature is lower than that of naringenin alone.
This interaction may be a result of hydrophobic interaction and/or hydrogen bonding (Semalty et al. Citation2010). The hydroxyl groups of the phenol rings of naringenin may be involved in hydrogen bonding and the aromatic rings may be involved in any hydrophobic interaction. Consequently, the major sharp peaks of phospholipids disappear and decrease the phase transition temperature. These DSC data are supported with the results of DSC thermograms of the phospholipid complexes of some phytoconstituents including silybin, puerarin, and curcumin in which the thermogram of the complex also exhibited a single peak which was different from the peak of phytoconstituents and the phospholipids (Yanyu et al. Citation2006, Maiti et al. Citation2007, Kumar et al. Citation2008, Li et al. Citation2008).
3.3. Naringenin release studies
Liposomes intended for topical application require a carrier. Liposomes have previously been established to be compatible with safe viscosity-increasing agents such as cellulose-based gels (Foldvari Citation1996, Patton et al. Citation2007, Hascicek et al. Citation2009, Forbes et al. Citation2011). Naringenin-loaded HEC and HPMC gels were formulated as well as liposome preparations and liposomal gel and release over 24 h observed.
3.3.1. Naringenin release studies from gel formulations
Gels appeared to retard the release of naringenin with HPMC having a greater effect than HEC in this phenomenon (). Over 24 h, the solution gave 54.5%±4.2% release whilst HEC and HPMC saw a 33.1%±2.0% and 31.1%±1.0% release, respectively. The release profile was significantly different between HPMC and both solution and HEC (p ≤ 0.001). There was no significant difference in the release profile of naringenin from solution and the HEC gel implying HPMC is better at compound retardation in this case. Comparison between the gels found that HEC at its respective loading of polymer in the HPMC gels, consistently resulted in a faster release of drug (at the time point of 3 h, 9.8%±0.5% was released in comparison to 5.6%±0.2%). Therefore, drug release was faster from the HEC gel compared to the HPMC gel.
The addition of water-insoluble drugs can increase the water uptake by the dosage form thus weakening network integrity thus drug loading will influence network integrity (Panomsuk et al. Citation1996, Nafee et al. Citation2003). In this case, it appears the HEC matrix eroded/swelled quicker than HPMC giving a faster rate of release. This is in contrast with a study comparing the release of miconazole from a 1.5% w/v HEC formulation with a 3% HPMC w/v formulation where a faster erosion was observed from the HPMC matrix (even at double the polymer loading) (Nafee et al. Citation2003). This highlights how the physiochemical properties of the drug, the polymer and the interaction between the two, influences drug release from the formulation.
An increased rate of naringenin release was observed from the HEC gel compared with the HPMC gel formulation over the 24 h observed (0.49 ± 0.13 compared with 0.06 ± 0.02 based on the Korsmeyer–Peppas model) (Supplementary Table 1). Naringenin is released from gel by the creation of pores dues to swelling, as viscosity increases polymer chains become more resistant to movement as they are physically restricted consequently taking longer to dissipate into the media thus slowing drug release. The Korsmeyer–Peppas model (highest R2 and lowest AIC) best described naringenin release from both gel formulations. For both HEC and HPMC gels, the diffusion release exponent value suggested naringenin release was non-Fickian thus a complex mixture of diffusion and erosion controlled drug release or class-II kinetics (diffusion not based on concentration gradient) often described as anomalous transport (Peppas and Sahlin Citation1989) (Korsmeyer et al. Citation1983). A study by Ritger and Peppas found both Fickian and anomalous release from swellable devices (Ritger and Peppas Citation1987). Additionally, a study using the polymer HPC observed both Non-Fickian and super case 11 transport (Alfrey et al. Citation1966, Ranga Rao et al. Citation1988).
At temperatures below the Tg, the polymer chains are not sufficiently able to move to permit immediate penetration of the solvent in the polymer core (Masaro and Zhu Citation1999). This implies that, in our results, when Non-Fickian transport was observed, the polymer chains were unable to move sufficiently and that at those particular loadings of polymer, the gel was in a glassy state.
Diffusion of solution out of the polymer is dependent on temperature, pressure, solute size, and viscosity. Diffusion in polymers is complex with the rate of diffusion between that in liquids and in solids. It is dependent on the concentration and degree of swelling of polymers. Solvent diffusion is associated with the physical properties of the polymer network and the interactions between the polymer and solvent (Masaro and Zhu Citation1999). Drug release from aqueous gels has been suggested to be governed by a swelling-controlled mechanism in which the drug releases into the media due to the simultaneous absorption of water by the gel causing the gel to dissipate into the media thus releasing drug and due to desorption of drug from the gel (Ranga Rao and Padmalatha Devi Citation1988, Bouwstra and Junginger Citation1993, Sinha Roy and Rohera Citation2002, Nafee et al. Citation2003). The rate-controlling factor mediating drug release is the resistance of the polymer to a change in shape owing to an increase in volume (Ranga Rao and Padmalatha Devi Citation1988). The membrane prevented the gel from swelling completely, whilst water could move across it, the polymer did not have space to swell as the donor compartment was filled to near capacity.
3.3.2. Naringenin release from liposomes
Release of naringenin from 100 µg/10 mL solution, liposomes, and liposomes formulated with either 2%, 6%, or 10% w/w of Tween 20 across a membrane was studied over a 24 h period ().
Figure 5. In-vitro percentage naringenin cumulative release profiles from solution and liposomal formulations. Naringenin release profiles from solution and liposomes formulated with 0%, 2%, 6%, or 10 % w/w Tween 20 over 24 h. Liposomes were prepared adapting the dry film method adding the surfactant and naringenin during the lipid mixing stage. A diffusion cell dialysis system was used to evaluate in-vitro drug release. Data represent mean ± SD. n = 3 independent batches. **** indicates statistical comparison between the naringenin release of liposome formulations with a p ≤ 0.0001, **indicates statistical comparison between the naringenin release of liposome formulations with a p ≤ 0.01.
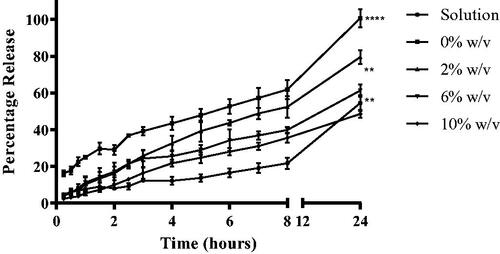
A higher percentage of naringenin was released from liposomes compared with drug solutions. Over the course of 24 h, the aqueous solution resulted in a release of 54.5% ± 4.2% whilst liposomes formulated with 0%, 2%, 6%, and 10% w/w of Tween 20 demonstrated a release of 100.7%±5.0%, 79.5%±3.7%, 61.3%±3.4%, and 48.5%±2.1%, respectively. The cumulative percentage released after 24 h was significant between the solution and liposomes loaded with 0% and 2% w/w of Tween 20 as well as between all liposomal formulations (p ≤ 0.0001 and p ≤ 0.01 between liposomes loaded with 6% and 10% Tween 20). As the loading of Tween 20 increased, the release from the liposomes slowed. An increased rate of release was observed from the liposomes loaded with naringenin compared with the naringenin solution over the 24 h observed (Supplementary Table 2). Thus, as the loading of surfactant increased, the rate of naringenin release decreased (from 2.35 ± 0.50% to 0.91 ± 0.27% for liposomes loaded with 0% and 10% of Tween 20, respectively, based on the Korsmeyer–Peppas model). Thus, surfactant appears to decrease drug release. This may be attributed to the decrease in drug entrapment; therefore, drug release would be expected to be slower as a consequence of a reduced concentration gradient. Furthermore, the drug is hydrophobic therefore may be less likely to diffuse out of the hydrophobic liposome bilayer into the surrounding media. Furthermore, reversible binding of the drug released from the liposome reduces the driving force for drug transport across the dialysis membrane leading to a slower overall apparent release rate (Modi and Anderson Citation2013). In this case, this reversible binding was greater as surfactant loading increased resulting in a slower rate of release for liposome formulated with 10% w/w Tween 20 in comparison to liposomes formulated with no Tween 20. A study comparing the deposition of naringenin in the skin from deformable liposomes and saturated solution found elastic liposomes (zero-order release) formulated with Tween 80 increased the deposition of naringenin in the skin significantly (by about 7.3 ∼ 11.8-fold) compared with the saturated aqueous solution. Another study also found deformable liposomes were able to increase drug permeation into the skin in comparison to a conventional cream (2-fold) (Goindi et al. Citation2013). It has been suggested that the mechanism of the in-vitro release seems to be the formation of transient pores in the lipid bilayer, through which drugs are released from the inner aqueous core of the liposomes to the extra-liposomal medium (Wang et al. Citation2017).
The Korsmeyer–Peppas model best described release kinetics from all liposomal formulations (highest R2 and lowest AIC). The diffusion release exponent values suggested release was a complex mixture of diffusion and erosion controlled drug release or class-II kinetics often termed anomalous transport (Peppas and Sahlin Citation1989).
3.3.3. Liposomal gel naringenin release studies
Naringenin release from the liposomal gels was observed over 24 h ().
Figure 6. In-vitro percentage naringenin release profiles from liposomal gels. Naringenin release profiles using a permeable insert system with permeable inserts of a 400 nm pore size from gels loaded with either Tween 20-free or deformable liposomes formulated with 2% w/w Tween 20 over 24 h. F1: HEC and Tween 20-free liposomes, F2: HEC and elastic liposomes, F3: HPMC and Tween 20-free liposomes, F4: HMPC and elastic liposomes. Gels were prepared using 3% w/v loading of either HEC or HPMC with a 1% w/w of naringenin loading. Liposomes were prepared adapting the dry film method. Data represent mean ± SD. n = 3 independent batches. ** indicates statistical comparison between the naringenin release from HEC and HPMC gel loaded with either 0% or 2% w/w Tween 20 with a p ≤ 0.01.
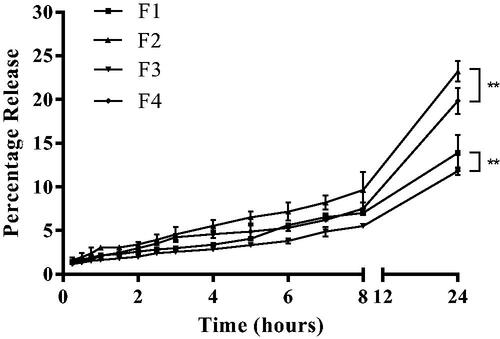
Liposomal gels formulated with HEC were observed to give a slightly higher release of naringenin over 24 h (up to 23%). Furthermore, a higher percentage of drug was released from the deformable liposome (up to 23%). Over the course of 24 h formulation F1, F2, F3, and F4 gave a release of 13.9% ± 2.0%, 23.2% ± 4.1%, 11.8% ± 0.4%, and 19.8% ± 1.5%, respectively. The cumulative percentage released after 24 h was significant between the solution and all liposomal gels (p ≤ 0.0001). The difference was also significant between F1 and F2 as well as between F2 and F3. F1 and F3 contained no surfactant in the liposome and F2 and F4 contained 2% w/w of Tween 20. Therefore, release in the liposomal gels appeared to be affected by presence of surfactant in liposomal gels formulated with HEC but not HPMC.
This indicated that either deformable liposomes were better able to move through the HEC gel compared with naringenin-free liposomes or that the presence of surfactant increased the solubility of drug thus encouraging release from the liposome bilayer. A study observing lidocaine HCL release from liposome-loaded gels had similar results with hydrogel formulations having a faster release rate of lidocaine HCl compared to liposomal gel formulations (Glavas-Dodov et al. Citation2002). Drug properties (solubility, log P) as well as liposome stability when formulated as part of a gel system determines the entire system behavior and thus drug release (Mourtas et al. Citation2007).
3.4. Cellular toxicity of naringenin toward HDFa and HaCat cells
Topical formulations are applied directly on to the skin and the numerous connective layers making up the skin determine drug delivery. The skin primarily consists of the epidermis, dermis, and subcutaneous layers with each layer having a unique combination of cells, connective tissue, components, and functions. Skin cancers progress in the upper layers of the skin including both the dermal and epidermal layer, thus the impact of formulation systems on these tissue layers should be considered.
The toxicity and penetrability of naringenin formulations on skin layers were evaluated by adopting two in-vitro cell culture systems, namely HaCat and human fibroblast cells (HDFa) (Marwah et al. Citation2020). An XTT assay was performed to measure cell viability on both cell lines after exposure of cells to different concentrations of naringenin for 24 h (Supplementary Figure 2).
The concentration of naringenin was increased from 0.1 to 100 µM causing a decrease in HDFa cell viability with statistical significance (p ≤ 0.05). This would be due to toxicity and apoptosis (Bae et al. Citation2008, Tanigawa et al. Citation2014). There is limited data on the cytotoxicity of naringenin toward dermal tissues however a previous study observed the protective effects of naringenin to HDFa cells exposed to external stressors including UVA and UVB radiation (Fernández-García Citation2014). Conversely, HaCat cell viability was upheld across the concentration range of 0.1–100 µM with no statistically significant difference was observed (p ≥ 0.05). Thus, at these concentrations, naringenin was not toxic to this line of keratinocytes. Moreover, previous studies reported naringenin to impart shielding effects in HaCat cells exposed to external stressors including UVA and UVB radiation (El‐Mahdy et al. Citation2008).
3.5. Cellular liposomal uptake assay into HDFa and HaCat cells
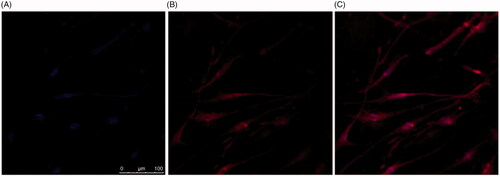
An important aim of our studies was to demonstrate uptake of naringenin-loaded deformable liposomes into a cell culture skin model. These liposomes, formulated with DilC, were incubated with HaCat () and HDFa () for 2 h with both HaCat and HDFa cells seeded onto collagen-coated coverslips cells to evaluate cellular uptake. The cellular localization of these liposomes was determined using confocal microscopy. Following the 2-h incubation with cells, intracellular localization of labelled liposomes was evident, confirming the successful liposomal uptake into HaCat and HDFa cells.
Figure 7. Localization of DilC labelled liposomes loaded with naringenin and 2% w/w Tween 20 suspended in HPMC gels in HaCat cells. Cells were grown on the coverslips for 2 d. Cell nuclei were visualized using (A) DAPI. Liposomes were formulated with DilC for visualization (B). Liposome localization within the cell is shown in the merged image (C).
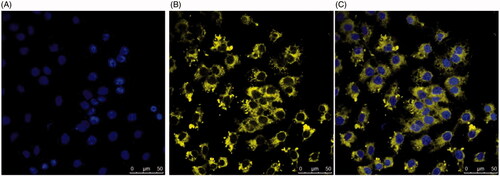
Figure 8. Localization of DilC labelled liposomes loaded with naringenin and 2% w/w Tween 20 suspended in HPMC gel in HDFa cells. Cells were grown on the coverslips for 2 d. Cell nuclei were visualized using (A) DAPI. Liposomes were formulated with DilC for visualization (B). Liposome localization within the cell is shown in the merged image (C).
Liposome uptake into cells is governed by particle size and charge (Patil et al. Citation2007, Kyung et al. Citation2009, Chen et al. Citation2010). Suggested approaches of liposome uptake include endocytosis, adsorption, fusion of the lipid bilayer with the cell plasma membrane, and lipid transfer (Martin and MacDonald Citation1976, Pagano and Weinstein Citation1978). These uptake methods are not mutually exclusive thus a combination of them may have occurred in this study (Pagano and Weinstein Citation1978). The cell membrane surface is associated with anionic glycosaminoglycan side chains, accordingly, interaction between these side chains and positively charged liposomes are ionic (Panyam and Labhasetwar Citation2003). Liposomes applied to the cells had a ζ of 3.30 ± 1.09 suggesting an ionic interaction may have transpired. A previous study formulating chemotherapy in the treatment of malignant melanoma using Normal HDFs detected an increased uptake of cationic liposomes compared with neutral liposomes owing to the electrostatic interaction with the anionic phospholipid cell membrane (Ito et al. Citation2007).
The confocal microscopy studies showed the potential of delivery of deformable liposomes to relevant dermal tissues using in-vitro cell culture techniques. Nonetheless, application of such formulations could also be assessed using ex-vivo human or animal dermal tissues. The definitive aim of this delivery system was to improve dermal cell uptake and with a controlled release of active agent, thus from a regulatory perspective, pharmacokinetic data are not required as drug is not intended to enter the blood stream (European Agency for the Evaluation of Medicinal Products Citation2000).
This study did not report skin penetration. The liposomal gel formulation recommended to take into further studies is that loaded with 2% w/w Tween 20. To ascertain the extent of carrier and drug permeation a skin strip test could be appropriate (Schäfer-Korting et al. Citation2007). This encompasses the use of adhesive tape to strip the skin layer by layer to quantify lipid and drug on each layer (Weigmann et al. Citation1999). Additionally, the most appropriate animal model for human skin is considered to be porcine skin tissue, however, sample-to-sample variability as well as variances in the lipid dermal matrices results in an altered permeability profile restricting the wider human translational objective (Dick and Scott Citation1992, Schmook et al. Citation2001, Godin and Touitou Citation2007). Finally, use of skin tissue samples would help conclude on whether complete liposomes deliver naringenin to dermal cells or if delivery is a result of liposomes mixing with lipids in the stratum corneum.
3.6. Stability of deformable liposomes
The medium-term storage of naringenin-loaded liposomes formulated with 2% w/w Tween 20 was evaluated after storage in stability cabinets maintained at 25 ± 2 °C (Froilabo, France) at a humidity of 60% ± 5. The effect of this size () and naringenin encapsulation efficiency () was evaluated. No statistically significant difference in the size of all naringenin-loaded liposomes during the storage period was observed (). However, previous reports observed aggregation is common upon liposomal formulation storage resulting in vesicle size growth (Lentz et al. Citation1987) particularly evident with neutral liposomes (Weiner et al. Citation1992). Inclusion of surfactant appears to prevent this phenomenon correlating with similar studies (Seras et al. Citation1992, Marwah et al. Citation2020). This may be due to the destabilizing effect of surfactant on the lipid bilayer reducing the energy required to expand the interface, thus allowing conservation of smaller vesicles.
Figure 9. Stability of naringenin-loaded liposomes as determined by size. Size of naringenin-loaded liposomes formulated with 0-10% w/w Tween 20, using DLS, formulated with up to 10% w/w Tween 20 measured on various days (1, 7, 14, 21, and 28). Data represent mean ± SD. n = 6 independent batches.
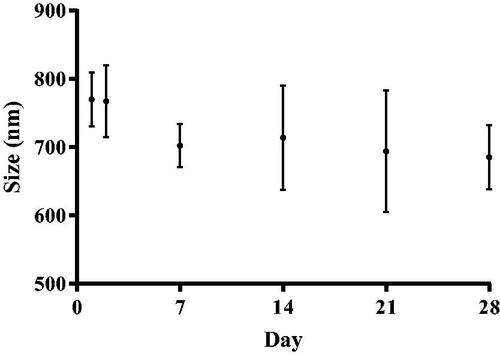
Figure 10. Liposome encapsulation efficiency for naringenin. Liposome encapsulation efficiency for naringenin in liposomes formulated with 2 % w/w Tween 20 liposomes over 28 d. Liposomes were prepared adapting the dry film method adding the surfactant and drug during the lipid mixing stage. The preparation was then washed via centrifugation. The quantity of naringenin in supernatant over 28 d was then analyzed by HPLC coupled with UV detection to assess liposome stability. Data represent mean ± SD. n = 6 independent batches.
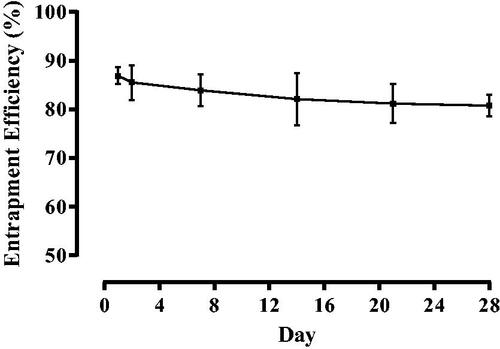
Furthermore, encapsulation efficiency appears to decrease non-significantly from 86.89% ± 1.72 to 85.51% ± 3.59 to 83.92% ± 3.23 to 82.09% ± 5.39 to 81.19% ± 3.97 to 80.76% ± 2.21, for liposomes formulated with 2% w/w Tween 20 (). This suggests drug leaching is independent of surfactant loading. However, Tween 20 is able to increase compound solubility, therefore, as not all would be entrapped within the bilayer, this may allow naringenin to solubilize within the liposomal media (Almog et al. Citation1986). Therefore, as the loading of Tween 20 increased, this would increase the amount of free Tween 20 resulting in more naringenin being able to solubilize in the liposome media.
4. Conclusion
The occurrence of skin cancer is increasing thus novel treatments to overcome limited therapeutic options is essential. Deformable liposomes in an aqueous gel carrier applied topically for the direct dermal delivery of compounds would be useful in the delivery of compounds across the stratum corneum at a controlled rate whilst minimizing side effects. The use of anti-oxidants to prevent oxidative skin damage is gaining evidence as a favorable strategy in the treatment of a range of skin concerns (Albini and Sporn Citation2007, Huang et al. Citation2011, Casey et al. Citation2015). Naringenin is a major flavanone in extracted from grapefruit with roles as an antioxidant, free radical scavenger, anti-inflammatory agent, and immune system modulator thus offers potential as a pharmacological anti-cancer agent (Chen et al. Citation2003, Huang et al. Citation2011, Casey et al. Citation2015). The formulation of such compounds has had limited success owing to a poor solubility profile, limited bioavailability and ineffective delivery systems. We developed an original deformable liposome formulation loaded with naringenin suspended in an aqueous gel and systemically investigated the encapsulation efficiency, uptake and in-vitro release of naringenin from the formulated liposomes. This study observed deformable liposomes are valuable in enhancing the bioavailability of naringenin as well as offering controlled release (Nishiyama Citation2007; Siddiqui et al. Citation2009). We established that aqueous gels hindered the release of naringenin compared to naringenin solution. Furthermore, increasing the loading of Tween 20 in the liposomal bilayer, liposome size decreased and elasticity increased however, naringenin encapsulation decreased. This reducing in naringenin encapsualtion may be a result of Tween 20 competing for space within the bilayer or Tween 20 increasing the solubilization capacity of naringenin. Naringenin release from liposomes observed liposomes retarded the release of drug with complete release not observed within 24 h. Furthermore, we demonstrated liposomes were uptaken into epidermal keratinocytes and dermal fibroblasts within 2 h. This study demonstrates liposomes formulated with Tween 20 suspended in either HEC or HPMC carriers are valuable in the development of a controlled release formulation with potential for dermal drug delivery necessary to overcome patient compliance concerns thus improving skin cancer treatment results.
Supplemental Material
Download MS Word (139.2 KB)Acknowledgments
The authors thank the MRC for providing funds for this project. We would also like to acknowledge Aston Research Centre for Healthy Ageing and for assisting with the collection of confocal imaging data.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Albini, A. and Sporn, M.B., 2007. The tumour microenvironment as a target for chemoprevention. Nature reviews cancer, 7 (2), 139–147.
- Alfrey, T., Jr, Gurnee, E., and Lloyd, W., 1966. Diffusion in glassy polymers. Journal of polymer science part C: polymer symposia, 12 (1), 249–261.
- Ali, M.H., et al., 2013. The role of lipid geometry in designing liposomes for the solubilisation of poorly water soluble drugs. International journal of pharmaceutics, 453 (1), 225–232.
- Ali, S.M., et al., 2007. Poor adherence to treatments: a fundamental principle of dermatology. Archives of dermatology, 143 (7), 912–915.
- Almog, S., et al., 1986. Kinetic and structural aspects of reconstitution of phosphatidylcholine vesicles by dilution of phosphatidylcholine-sodium cholate mixed micelles. Biochemistry, 25 (9), 2597–2605.
- American Cancer Society. 2017. Cancer facts and figures. Atlanta, GA: American Cancer Society.
- Bae, J.Y., et al., 2008. Epigallocatechin gallate hampers collagen destruction and collagenase activation in ultraviolet-B-irradiated human dermal fibroblasts: involvement of mitogen-activated protein kinase. Food and chemical toxicology: toxicology, 46 (4), 1298–1307.
- Bangham, A.D., Standish, M.M., and Watkins, J.C., 1965. Diffusion of univalent ions across the lamellae of swollen phospholipids. Journal of molecular biology, 13 (1), 238–252.
- Bansal, T., et al., 2009. Emerging significance of flavonoids as P-glycoprotein inhibitors in cancer chemotherapy. Journal of pharmacy & pharmaceutical sciences, 12 (1), 46–78.
- Basnet, P., et al., 2012. Liposomal delivery system enhances anti-inflammatory properties of curcumin. Journal of pharmaceutical sciences, 101 (2), 598–609.
- Bouwstra, J. and Junginger, H., 1993. Hydrogels. Encyclopaedia of pharmaceutical technology, 441–465.
- Carey, M.P. and Burish, T.G., 1988. Etiology and treatment of the psychological side effects associated with cancer chemotherapy: a critical review and discussion. Psychological bulletin, 104 (3), 307–325.
- Casas, M. and Baszkin, A., 1992. Interactions of a non-ionic surfactant with mixed phospholipid—oleic acid monolayers. Surface potential and surface pressure studies at constant area. Colloids and surfaces, 63 (3–4), 301–309.
- Casey, S.C., et al., 2015. Cancer prevention and therapy through the modulation of the tumor microenvironment. Seminars in cancer biology, 35, S199–S223.
- Cevc, G., 1996. Transfersomes, liposomes and other lipid suspensions on the skin: permeation enhancement, vesicle penetration, and transdermal drug delivery. Critical reviews™ in therapeutic drug carrier systems, 13 (3–4), 257–388.
- Cevc, G. and Blume, G., 2001. New, highly efficient formulation of diclofenac for the topical, transdermal administration in ultradeformable drug carriers, Transfersomes. Biochimica et biophysica acta (BBA) – biomembranes, 1514 (2), 191–205.
- Chen, C. C., et al., 2010. Effects of lipophilic emulsifiers on the oral administration of lovastatin from nanostructured lipid carriers: physicochemical characterization and pharmacokinetics. European journal of pharmaceutics and biopharmaceutics: official journal of arbeitsgemeinschaft fur pharmazeutische verfahrenstechnik e.V, 74 (3), 474–482.
- Chen, C., et al., 2003. Epigallocatechin-3-gallate-induced stress signals in HT-29 human colon adenocarcinoma cells. Carcinogenesis, 24 (8), 1369–1378.
- Chen, Y., et al., 2012. Preparation of curcumin-loaded liposomes and evaluation of their skin permeation and pharmacodynamics. Molecules, 17 (5), 5972–5987.
- Dick, I.P. and Scott, R.C., 1992. Pig ear skin as an in‐vitro model for human skin permeability. The journal of pharmacy and pharmacology, 44 (8), 640–645.
- Diepgen, T.L. and Mahler, V., 2002. The epidemiology of skin cancer. The British journal of dermatology, 146 (61), 1–6.
- Donaldson, M.R. and Coldiron, B.M., 2011. No end in sight: the skin cancer epidemic continues, Seminars in cutaneous medicine and surgery. Seminars in cutaneous medicine and surgery, 30 (1), 3–5.
- du Plessis, J., Weiner, N., and Müller, D., 1994. The influence of in vivo treatment of skin with liposomes on the topical absorption of a hydrophilic and a hydrophobic drug in vitro. International journal of pharmaceutics, 103 (2), R1–R5.
- El‐Mahdy, M.A., et al., 2008. Naringenin protects HaCaT human keratinocytes against UVB‐induced apoptosis and enhances the removal of cyclobutane pyrimidine dimers from the genome. Photochemistry and photobiology, 84 (2), 307–316.
- El Maghraby, G.M., Williams, A.C., and Barry, B.W., 1999. Skin delivery of oestradiol from deformable and traditional liposomes: mechanistic studies. The journal of pharmacy and pharmacology, 51 (10), 1123–1134.
- European Agency for the Evaluation of Medicinal Products 2000. Note for Guidance on the Investigation of Bioavailability and Bioequivalence, Note for guidance on the investigation of bioavilability and bioequivalence. London: European Agency for the Evaluation of Medicinal Products.
- El Zaafarany, G.M., et al., 2010. Role of edge activators and surface charge in developing ultradeformable vesicles with enhanced skin delivery. International journal of pharmaceutics, 397 (1–2), 164–172.
- Felicio, L., et al., 2009. Long-term follow-up of topical 5-aminolaevulinic acid photodynamic therapy diode laser single session for non-melanoma skin cancer. Photodiagnosis and photodynamic therapy, 6 (3–4), 207–213.
- Fernández-García, E., 2014. Photoprotection of human dermal fibroblasts against ultraviolet light by antioxidant combinations present in tomato. Food & function, 5 (2), 285–290.
- Foldvari, M., 1996. Effect of vehicle on topical liposomal drug delivery: petrolatum bases. Journal of microencapsulation, 13 (5), 589–600.
- Forbes, C.J., et al., 2011. Non-aqueous silicone elastomer gels as a vaginal microbicide delivery system for the HIV-1 entry inhibitor maraviroc. Journal of controlled release: official journal of the controlled release society, 156 (2), 161–169.
- Fresta, M. and Puglisi, G., 1996. Application of liposomes as potential cutaneous drug delivery systems. In vitro and in vivo investigation with radioactively labelled vesicles. Journal of drug targeting, 4 (2), 95–101.
- Ghosal, K. and Nanda, A., 2013. Development of diclofenac potassium gel from hydrophobically modified HPMC. Iranian polymer journal, 22 (6), 457–464.
- Glavas-Dodov, M., et al., 2002. Release profile of lidocaine HCl from topical liposomal gel formulation. International journal of pharmaceutics, 242 (1–2), 381–384.
- Godin, B. and Touitou, E., 2007. Transdermal skin delivery: predictions for humans from in vivo, ex vivo and animal models. Advanced drug delivery reviews, 59 (11), 1152–1161.
- Goindi, S., et al., 2013. Development of novel elastic vesicle-based topical formulation of cetirizine dihydrochloride for treatment of atopic dermatitis. Aaps pharmscitech, 14 (4), 1284–1293.
- Gompper, G. and Kroll, D.M., 1995. Driven transport of fluid vesicles through narrow pores. Physical review E, statistical physics, plasmas, fluids, and related interdisciplinary topics, 52 (4), 4198–4208.
- Hascicek, C., Bediz-Ölçer, A., and Gönül, N., 2009. Preparation and evaluation of different gel formulations for transdermal delivery of meloxicam. Turkish journal of pharmaceutical sciences, 6, 177–186.
- Hiruta, Y., et al., 2006. Novel ultra-deformable vesicles entrapped with bleomycin and enhanced to penetrate rat skin. Journal of controlled release: official journal of the controlled release society, 113 (2), 146–154.
- Hsiu, S.L., et al., 2002. Comparison of metabolic pharmacokinetics of naringin and naringenin in rabbits. Life sciences, 70 (13), 1481–1489.
- Huang, Y.C., Yang, C.H., and Chiou, Y.L., 2011. Citrus flavanone naringenin enhances melanogenesis through the activation of Wnt/β-catenin signalling in mouse melanoma cells. Phytomedicine: international journal of phytotherapy and phytopharmacology, 18 (14), 1244–1249.
- Hwang, J. T., et al., 2007. Apoptotic effect of EGCG in HT-29 colon cancer cells via AMPK signal pathway. Cancer letters, 247 (1), 115–121.
- Ita, K.B., et al., 2007. Dermal delivery of selected hydrophilic drugs from elastic liposomes: effect of phospholipid formulation and surfactants. The journal of pharmacy and pharmacology, 59 (9), 1215–1222.
- Ito, A., et al., 2007. 4-S-Cysteaminylphenol-loaded magnetite cationic liposomes for combination therapy of hyperthermia with chemotherapy against malignant melanoma. Cancer science, 98 (3), 424–430.
- Kanadaswami, C., et al., 2005. The antitumor activities of flavonoids. In vivo, 19, 895–909.
- Kang, S.N., et al., 2013. Enhancement of liposomal stability and cellular drug uptake by incorporating tributyrin into celecoxib-loaded liposomes. Asian journal of pharmaceutical sciences, 8 (2), 128–133.
- Kaplan, B. and Moy, R.L., 2000. Effect of perilesional injections of PEG-interleukin-2 on basal cell carcinoma. Dermatologic surgery, 26 (11), 1037–1040.
- Kaur, M. and Badhan, R.K., 2015. Phytoestrogens modulate breast cancer resistance protein expression and function at the blood-cerebrospinal fluid barrier. Journal of pharmacy & pharmaceutical sciences, 18 (2), 132–154.
- Kaur, M. and Badhan, R.K., 2017. Phytochemical mediated-modulation of the expression and transporter function of breast cancer resistance protein at the blood-brain barrier: an in-vitro study. Brain research, 1654, 9–23.
- Kerkar, S.P. and Restifo, N.P., 2012. Cellular constituents of immune escape within the tumor microenvironment. Cancer research, 72 (13), 3125–3130.
- Khan, A.W., et al., 2015. Enhanced dissolution and bioavailability of grapefruit flavonoid Naringenin by solid dispersion utilizing fourth generation carrier. Drug development and industrial pharmacy, 41 (5), 772–779.
- Korsmeyer, R.W., et al., 1983. Mechanisms of solute release from porous hydrophilic polymers. International journal of pharmaceutics, 15 (1), 25–35.
- Kumar, M., Ahuja, M., and Sharma, S.K., 2008. Hepatoprotective study of curcumin-soya lecithin complex. Scientia pharmaceutica, 76 (4), 761–774.
- Kyung, O.Y., et al., 2009. Toxicity of amorphous silica nanoparticles in mouse keratinocytes. Journal of nanoparticle research, 11 (1), 15–24.
- Lasic, D. D. and Barenholz, Y., 1996. Handbook of nonmedical applications of liposomes: theory and basic sciences. Boca Raton, FL: CRC Press.
- Lentz, B.S., Carpenter, T.J., Aiford, D.R., 1987. Spontaneius fusion of phosphatidycholine small unilamellar vesicles in the fluid phase. Biochemistry, 26 (17), 5389–5397.
- Levy, M.Y., Benita, S., and Baszkin, A., 1991. Interactions of a non-ionic surfactant with mixed phospholipid—oleic acid monolayers. Studies under dynamic conditions. Colloids and surfaces, 59, 225–241.
- Li, Y., et al., 2008. Comparative physicochemical characterization of phospholipids complex of puerarin formulated by conventional and supercritical methods. Pharmaceutical research, 25 (3), 563–577.
- Lopez, R.F., et al., 2011. Enhancing the transdermal delivery of rigid nanoparticles using the simultaneous application of ultrasound and sodium lauryl sulfate. Biomaterials, 32 (3), 933–941.
- Maiti, K., et al., 2007. Curcumin–phospholipid complex: preparation, therapeutic evaluation and pharmacokinetic study in rats. International journal of pharmaceutics, 330 (1–2), 155–163.
- Martin, F.J. and MacDonald, R.C., 1976. Lipid vesicle-cell interactions. I. Hemagglutination and hemolysis. The journal of cell biology, 70 (3), 494–505.
- Marwah, M., et al., 2020. Intracellular uptake of EGCG-loaded deformable controlled release liposomes for skin cancer. Journal of liposome research, 30 (2), 136–149.
- Masaro, L. and Zhu, X.X., 1999. Physical models of diffusion for polymer solutions, gels and solids. Progress in polymer science, 24 (5), 731–775.
- Modi, S. and Anderson, B.D., 2013. Determination of drug release kinetics from nanoparticles: overcoming pitfalls of the dynamic dialysis method. Molecular pharmaceutics, 10 (8), 3076–3089.
- Mourtas, S., et al., 2007. Liposomal drugs dispersed in hydrogels. Effect of liposome, drug and gel properties on drug release kinetics. Colloids and surfaces B, biointerfaces, 55 (2), 212–221.
- Nafee, N.A., et al., 2003. Mucoadhesive buccal patches of miconazole nitrate: in vitro/in vivo performance and effect of ageing. International journal of pharmaceutics, 264 (1–2), 1–14.
- Neville, J.A., Welch, E., and Leffell, D.J., 2007. Management of nonmelanoma skin cancer in 2007. Nature clinical practice oncology, 4 (8), 462–469.
- Nishiyama, N., 2007. Nanomedicine: nanocarriers shape up for long life. Nature nanotechnology, 2 (4), 203–204.
- Oh, Y.K., et al., 2006. Skin permeation of retinol in Tween 20-based deformable liposomes: in-vitro evaluation in human skin and keratinocyte models. The journal of pharmacy and pharmacology, 58 (2), 161–166.
- Pagano, R.E. and Weinstein, J.N., 1978. Interactions of liposomes with mammalian cells. Annual review of biophysics and bioengineering, 7, 435–468.
- Panomsuk, S.P., et al., 1996. A study of the hydrophilic cellulose matrix: effect of drugs on swelling properties. Chemical and pharmaceutical bulletin, 44 (5), 1039–1042.
- Panyam, J. and Labhasetwar, V., 2003. Biodegradable nanoparticles for drug and gene delivery to cells and tissue. Advanced drug delivery reviews, 55 (3), 329–347.
- Park, S. I., et al., 2013. Polymer-hybridized liposomes of poly (amino acid) derivatives as transepidermal carriers. Colloids and surfaces B, biointerfaces, 110, 333–338.
- Patil, S., et al., 2007. Protein adsorption and cellular uptake of cerium oxide nanoparticles as a function of zeta potential. Biomaterials, 28 (31), 4600–4607.
- Patton, D., et al., 2007. Preclinical safety assessments of UC781 anti-human immunodeficiency virus topical microbicide formulations. Antimicrobial agents and chemotherapy, 51 (5), 1608–1615.
- Peppas, N.A. and Sahlin, J.J., 1989. A simple equation for the description of solute release. III. Coupling of diffusion and relaxation. International journal of pharmaceutics, 57 (2), 169–172.
- Prausnitz, M.R. and Langer, R., 2008. Transdermal drug delivery. Nature biotechnology, 26 (11), 1261–1268.
- Ranga Rao, K.V., Devi, K.P., and Buri, P., 1988. Cellulose matrices for zero-order release of soluble drugs. Drug development and industrial pharmacy, 14 (15–17), 2299–2320.
- Ranga Rao, K.V. and Padmalatha Devi, K., 1988. Swelling controlled-release systems: recent developments and applications. International journal of pharmaceutics, 48, 1–13.
- Ritger, P.L. and Peppas, N.A., 1987. A simple equation for description of solute release II. Fickian and anomalous release from swellable devices. Journal of controlled release, 5 (1), 37–42.
- Romero, H.L., et al., 2013. Admission and capacity planning for the implementation of one-stop-shop in skin cancer treatment using simulation-based optimization. Health care management science, 16 (1), 75–86.
- Schäfer-Korting, M., Mehnert, W., and Korting, H. C., 2007. Lipid nanoparticles for improved topical application of drugs for skin diseases. Advanced drug delivery reviews, 59 (6), 427–443.
- Schmook, F.P., Meingassner, J.G., and Billich, A., 2001. Comparison of human skin or epidermis models with human and animal skin in in-vitro percutaneous absorption. International journal of pharmaceutics, 215 (1–2), 51–56.
- Scudiero, D.A., et al., 1988. Evaluation of a soluble tetrazolium/formazan assay for cell growth and drug sensitivity in culture using human and other tumor cell lines. Cancer research, 48 (17), 4827–4833.
- Semalty, A., et al., 2010. Preparation and characterization of phospholipid complexes of naringenin for effective drug delivery. Journal of inclusion phenomena and macrocyclic chemistry, 67 (3–4), 253–260.
- Seras, M., et al., 1992. Kinetic aspects of the solubilization of non-ionic monoalkyl amphiphile-cholesterol vesicles by octylglucoside. Chemistry and physics of lipids, 63 (1–2), 1–14.
- Siddiqui, I.A., et al., 2009. Introducing nanochemoprevention as a novel approach for cancer control: proof of principle with green tea polyphenol epigallocatechin-3-gallate. Cancer research, 69 (5), 1712–1716.
- Sinha Roy, D. and Rohera, B.D., 2002. Comparative evaluation of rate of hydration and matrix erosion of HEC and HPC and study of drug release from their matrices. European journal of pharmaceutical sciences, 16 (3), 193–199.
- Song, Y. K. and Kim, C. K., 2006. Topical delivery of low-molecular-weight heparin with surface-charged flexible liposomes. Biomaterials, 27 (2), 271–280.
- Tanigawa, T., et al., 2014. (+)-Catechin protects dermal fibroblasts against oxidative stress-induced apoptosis. BMC complementary and alternative medicine, 14, 133.
- Trotta, M., et al., 2002. Elastic liposomes for skin delivery of dipotassium glycyrrhizinate. International journal of pharmaceutics, 241 (2), 319–327.
- Trotta, M., et al., 2004. Deformable liposomes for dermal administration of methotrexate. International journal of pharmaceutics, 270 (1–2), 119–125.
- Tsai, M.J., et al., 2015. Preparation and characterization of naringenin-loaded elastic liposomes for topical application. PLoS One, 10 (7), e0131026.
- Vajjha, R.S., Das, D.K., and Kulkarni, D.P., 2010. Development of new correlations for convective heat transfer and friction factor in turbulent regime for nanofluids. International journal of heat and mass transfer, 53 (21–22), 4607–4618.
- Valenta, C. and Auner, B.G., 2004. The use of polymers for dermal and transdermal delivery. European journal of pharmaceutics and biopharmaceutics: official journal of arbeitsgemeinschaft fur pharmazeutische verfahrenstechnik e.V, 58 (2), 279–289.
- Wang, Y., et al., 2017. Enhanced solubility and bioavailability of naringenin via liposomal nanoformulation: preparation and in vitro and in vivo evaluations. Aaps pharmscitech, 18 (3), 586–594.
- Weigmann, H., et al., 1999. Determination of the horny layer profile by tape stripping in combination with optical spectroscopy in the visible range as a prerequisite to quantify percutaneous absorption. Skin pharmacology and applied skin physiology, 12 (1–2), 34–45.
- Weiner, N., Egbaria, K., and Ramachandran, C., 1992. Topical delivery of liposomally encapsulated interferon evaluated by in vitro diffusion studies and in a cutaneous herpes guinea pig model. In: O. Braun-Falco, H.C. Korting, and H.I. Maibach, eds. Liposome dermatics: griesbach conference. Berlin, Heidelberg: Springer Berlin Heidelberg, 242–250.
- Wen, J., et al., 2010. Preparation and physicochemical properties of the complex of naringenin with hydroxypropyl-beta-cyclodextrin. Molecules, 15 (6), 4401–4407.
- World Health Organisation. 2017. Ultraviolet radiation (UV): Skin cancers. Geneva, Switzerland: World Health Organisation.
- Yanyu, X., et al., 2006. The preparation of silybin–phospholipid complex and the study on its pharmacokinetics in rats. International journal of pharmaceutics, 307 (1), 77–82.
- Zhao, Y. Z., et al., 2013. Selection of high efficient transdermal lipid vesicle for curcumin skin delivery. International journal of pharmaceutics, 454 (1), 302–309.