
Abstract
Endothelial cell (EC) dysfunction contributes to COVID-19–associated vascular inflammation and coagulopathy, and the angiotensin-converting enzyme 2 (ACE2) receptor plays a role in EC dysfunction in COVID-19. To expand the understanding of the role of the ACE2 receptor relative to EC dysfunction, this review addresses (1) tissue distribution of the ACE2 protein and its mRNA expression in humans, (2) susceptibility of the capillary ECs to SARS-CoV-2 infection, and (3) the role of EC dysfunction relevant to ACE2 and nuclear factor-κB in COVID-19.
Target audience: All physicians
Learning objectives: After completing the article, the learner should be able to:
1. Recognize how the susceptibility of capillary endothelial cells contributes to widespread pulmonary and end-organ injury in the setting of SARS-CoV-2 infection
2. Discuss how new and preexisting endothelial cell dysfunction and the crosstalk of pathways between the angiotensin-converting enzyme 2 receptor and other endothelial cell receptors account for the intensity and extent of systemic manifestations in COVID-19
Faculty credentials/disclosure: Dr. Jun Zhang received his MD and MS in human pathology and has worked as a research pharmacologist for approximately 30 years. Dr. Kristen M. Tecson received her PhD in statistical science and has worked in the field of cardiovascular research for the past 5 years. Dr. Peter A. McCullough (MD, MPH) is a practicing cardiologist and an internationally recognized authority on the role of chronic kidney disease as a cardiovascular risk state. The authors and planner report no conflicts of interest. This work was partially funded by the Baylor Health Care System Foundation.
Accreditation: The A. Webb Roberts Center for Continuing Medical Education of Baylor Scott & White Health is accredited by the Accreditation Council for Continuing Medical Education to provide continuing medical education for physicians.
Designation: The A. Webb Roberts Center for Continuing Medical Education of Baylor Scott & White Health designates this journal CME activity for a maximum of 1.0 AMA PRA Category 1 CreditTM. Physicians should claim only the credit commensurate with the extent of their participation in the activity.
ABIM MOC: The A. Webb Roberts Center for Continuing Medical Education of Baylor Scott & White Health will submit participant completion information to ACCME, which will be transferred to the American Board of Internal Medicine (ABIM) for Maintenance of Certification (MOC) credit. By entering your ABIM Diplomate number into your profile and completing the required post-test for this activity, you are giving permission for this transfer to take place.
Successful completion of this CME activity, which includes participation in the evaluation component, enables the participant to earn up to 1.0 MOC points in the program. It is the CME activity provider’s responsibility to submit participant completion information to ACCME for the purpose of granting ABIM MOC credit.
Process: To complete this CME activity, read the entire article and then go to https://ce.bswhealth.com/Proceedings2020. You will register for the course, pay any relevant fee, take the quiz, complete the evaluation, and claim your CME credit. For more information about CME credit, email [email protected].
Expiration date: March 1, 2023.
A variety of receptors are expressed on the endothelial cell (EC) surface, performing vital functions to maintain vascular homeostasis. Furthermore, ECs serve as antigen-presenting cells and play an important role in the immune response. Conversely, EC dysfunction resulting from angiotensin-converting enzyme 2 (ACE2) and vitamin D receptors contributes to coronavirus disease 2019 (COVID-19)–associated vascular inflammation and coagulopathy.Citation1,Citation2 Studying severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) in a primate model, Koo et al showed that the development of COVID-19 caused acute interstitial pneumonia with endotheliitis in the lungs.Citation3 These findings strengthen the implications of EC dysfunction in COVID-19. Herein, we provide a review of endothelial receptors that induce or modulate EC dysfunction in COVID-19.
ENDOTHELIAL DYSFUNCTION RELATED TO THE ACE2 RECEPTOR IN COVID-19
Since ACE2 was identified as the functional receptor for SARS-CoV,Citation4 the outbreak of SARS-CoV-2 in Wuhan, China, ignited a new investigation of ACE2. Important commonalities between SARS-CoV and SARS-CoV-2 exist, including the use of the ACE2 receptor for entry into host cells.Citation5–8 However, unlike SARS-CoV, the SARS-CoV-2 receptor-binding domain has a 10- to 20-fold higher ACE2 binding affinity.Citation7,Citation9 Upon infection by a virus, cells of the host can present surface receptors as pattern recognition receptors (PRRs) to detect molecules that are shared by pathogens but distinguishable from host molecules as pathogen-associated molecular patterns (PAMPs). With COVID-19, the ACE2 receptor is a PRR and can be used by alveolar cells of the lung and ECs of blood vessels to recognize a PAMP. As a result, ACE2 leads to diffuse alveolar damage through COVID-19 pneumonia, as well as multiorgan involvement, all of which originated from EC dysfunction in the initial phase of infection.Citation1
TISSUE DISTRIBUTION OF ACE2 PROTEIN AND ITS MRNA EXPRESSION IN HUMANS
ACE2 protein and its mRNA expression display ACE2 on human tissues. Hamming et al detected ACE2 protein in ECs from arteries and veins, as well as on lung alveolar epithelial cells and enterocytes of the small intestine.Citation4 Therefore, the localization of ACE2 protein in the lung, immune organs, and systemic small vessels makes it a primary target of SARS-CoV-2. Additionally, Lely et al found ACE2 in tubular and glomerular epithelium as well as in vascular smooth muscle cells and ECs of interlobular arteries in healthy controls. They also found neoexpression of ACE2 in glomerular and peritubular capillary endothelium in samples from patients with renal disease/transplants.Citation10 Li et al reported that the lowest ACE2 expression was in the blood vessels, blood, spleen, bone marrow, and brain, whereas the highest was in the heart, kidneys, small intestine, testis, thyroid, and adipose tissue. The lungs, colon, liver, bladder, and adrenal gland had mid-level expression.Citation6 Chen et al found that Asian women had significantly higher ACE2 expression than men and other racial/ethnic groups.Citation5 Additionally, they determined that ACE2 expression had an inverse relationship with age and that it was significantly lower in patients with type 2 diabetes mellitus. Their results suggest a negative correlation between ACE2 expression and viral infection, an argument that is supported by women’s lower observed rate of COVID-19–associated death.Citation5,Citation11 Regardless of sex, higher levels of ACE2 may improve the prognosis of COVID-19 by reducing inflammation and thrombosis.Citation12
SUSCEPTIBILITY OF THE BLOOD VESSELS TO SARS-COV-2INFECTION
Like the epithelial cells of the respiratory tract, vascular ECs also use ACE2 as a cellular receptor to bind the viral spike protein of SARS-CoV-2. The respiratory tract is susceptible to viral infection, and thus the lungs are believed to be the main target organ of SARS-CoV-2. However, the blood vessels are susceptible to SARS-CoV-2 infection as well. The density of ACE2 receptors is inversely related to COVID-19 mortality. While the virus uses ACE2 for entry, the ACE2 receptor itself is necessary for the pulmonary vasculature to respond to and survive acute respiratory distress syndrome (ARDS). This pathophysiology explains why the infection is fulminant in those with a reduced density of ACE2 receptors, including the elderly and those with comorbidities. Conversely, those on antecedent angiotensin-converting enzyme inhibitors with chronically upregulated ACE2 have relative protection from COVID-19 mortality.Citation13
Using human endothelial cells in vitro, Zhang et al determined that ACE2 protects endothelial function and inhibits the inflammatory response. Specifically, they found that ACE2 inhibited monocyte adhesion to human umbilical vein ECs and was associated with reduced monocyte chemoattractant protein-1 (MCP-1), vascular cell adhesion molecule-1 (VCAM-1), and E-selectin protein.Citation12 Fraga-Silva et al evaluated the role of ACE2 in thrombus formation in a spontaneously hypertensive rat and showed that ACE2 is protective against thrombosis.Citation14 They highlighted the function of Ang II as a prothrombotic agent that induces inflammatory/profibrotic pathways and Ang-(1-7) as an antithrombotic peptide.Citation14 ACE2 cleaves Ang II to Ang-(1-7), which exerts vasodilating, antiinflammatory, and antifibrotic effects.Citation9 Additionally, Ang-(1-7) can increase the antiplatelet agent nitric oxide and prostacyclin synthesis.Citation14
The susceptibility of the blood vessels is also relevant to the renin-angiotensin system (RAS), a regulator of vascular function. Within the RAS pathway, Ang I-converting enzyme (ACE1) and ACE2 balance local vasoconstriction and vasodilation through the ACE1/Ang-II/Ang II type 1 axis and ACE2/Ang1-7/MAS axis, respectively, with a high ACE2/ACE1 ratio being protective against EC dysfunction.Citation11
It is of interest to know whether the abundant expression of ACE2 in the vascular system may serve as a route of spread and replication, and whether SARS-CoV-2 can spread throughout the body.Citation4 In this context, Bourgonje et al stated that type II pneumocytes together with the capillary endothelium may be a primary site of SARS-CoV-2 entrance, resulting in damage to those cells.Citation9 Recently, Lin et al hypothesized that the clinical manifestation of SARS-CoV-2 may be divided into three phases: the viremia phase, the acute pneumonia phase, and the recovery phase. In the initial phase, SARS-CoV-2 may enter the peripheral blood from the lungs and attack organs expressing ACE2.Citation15 Taken together, it is reasonable to assume that the blood vessels are susceptible to SARS-CoV-2 infection.
Mollica and colleagues explained that SARS-CoV-2 uses the ACE2 receptor for cell entry, in combination with the host’s transmembrane serine protease 2 (TMPRSS2).Citation16 TMPRSS2 is a protein expressed by epithelial cells and cleaves the viral S glycoprotein, which facilitates viral activation.Citation8 Hence, TMPRSS2 is crucial for SARS-CoV-2’s cell entry and pathogenesis. Further, expression of TMPRSS2 is androgen regulated and may explain the differential symptom severity of COVID-19 by gender.Citation17
ROLE OF ENDOTHELIAL DYSFUNCTION RELEVANT TO ACE2 AND NUCLEAR FACTOR-κB IN COVID-19
Nuclear factor-κB (NF-κB) can activate proinflammatory genes, contributing to endothelial apoptosis and activation.Citation18 Additionally, NF-κB regulates the transcription of proinflammatory cytokines (tumor necrosis factor [TNF]-α, interleukin [IL]-1Β, IL-2, IL-6, and IL-12) and leukocyte adhesion molecules (E-selectin, VCAM-1, and ICAM-1).Citation19
Li et al demonstrated that lipopolysaccharide (LPS) administration decreased ACE2 expression in the lungs, caused severe lung injury (inflammation, edema, and hemorrhage), and resulted in elevated TNF-α and IL-1ß levels in bronchoalveolar lavage fluid in an animal study.Citation20 Conversely, ACE2 overexpression suppressed the inflammatory response and mitigated the LPS-induced lung injury. Further, they found that while LPS administration increased phosphorylation of NF-κB p50 and p65 and decreased Iκ B-α expression, overexpression of ACE2 suppressed the phosphorylation and enhanced IκB-α expression.Citation20
COEXISTENCE OF ACE2 NEW ENDOTHELIAL DYSFUNCTION AND PREEXISTING ENDOTHELIAL DYSFUNCTION
Escher et al first demonstrated that severe COVID-19 infection is associated with EC activation. The EC activation in this case is based on the increased von Willebrand factor (vWF), which indicates massive endothelial stimulation and damage with release of vWF from Weibel-Palade bodies.Citation21 Varga et al first provided pathology of EC dysfunction in COVID-19, as exemplified by findings of viral particles in ECs and endothelial apoptosis.Citation22 It is thus of interest to compare the clinical features of patients reported by the two groups. For the former, the single male case was previously healthy and the treatment resulted in clinical improvement. The latter studied samples from three patients; all had hypertension, one was also a renal transplant recipient with coronary artery disease, and another had diabetes and obesity. The two patients with the greatest comorbidity loads died from multisystem organ failure and circulatory failure, respectively. Because EC dysfunction is frequently present in patients with hypertension, atherosclerosis, diabetes, and obesity, these case studies’ differences raise fundamental questions regarding ACE2-induced EC dysfunction in the former vs preexisting EC dysfunction in the latter.Citation19 The coexistence of two categories of EC dysfunction could increase severe capillary permeability and cause a procoagulant state and EC apoptosis, leading to endotheliitis in many organs (systemic vasculitis), as well as venous thromboembolism. Therefore, the coexistence of new EC dysfunction combined with preexisting EC dysfunction could partially account for differences in morbidity and mortality in COVID-19.
SUSCEPTIBILITY OF CAPILLARY ENDOTHELIAL CELLS TO COVID-19 INFECTION
Throughout vascular beds, ECs’ ability to express adhesion molecules and modulate coagulation varies.Citation23 For example, capillary ECs strongly express major histocompatibility complex classes I and II and ICAM-1, whereas large vessel ECs express little or none.Citation23 Further, differences in biomarkers associated with EC activation may be partially explained by differences in the endothelial functional phenotype of capillaries and small and large blood vessels. For example, E-selectin expression has been observed in small- to medium-sized veins, but not in the aorta. Further, it has been shown to be preferentially expressed by cytokine-activated capillary ECs, yet large vessels constitutively express it. This indicates that capillary ECs may be more susceptible to immune attack.Citation23
Buja et al summarized 23 autopsy reports from individuals who died from COVID-19 across five centers in the United States. The most prominent morphological feature of COVID-19, other than distinctive acute interstitial pneumonia with diffuse alveolar damage, was microvascular damage across several body systems, resulting in multiorgan failure. Pulmonary pathology revealed fibrin-rich thrombi in capillaries and small blood vessels.Citation24 Although some small thrombi were observed in a few small pulmonary artery branches, the pulmonary arteries at the hilum in the lungs had none. Endothelial dysfunction manifested as increased vascular permeability, resulting in thickened alveolar capillaries, vascular leakage leading to edema and hemorrhage, and capillary leakage leading to fibrinous exudates, fibrin precipitate, fibrin deposition in and outside capillaries, and fibrin thrombi within capillaries and small vessels, such as microthrombi in pulmonary arterioles and fibrin-platelet thrombus in renal glomerular capillaries.Citation24 In addition, EC dysfunction may also result in entrapped neutrophils within alveolar capillaries.Citation24 Hence, the special functional phenotype of capillary ECs may partially account for the susceptibility of capillary ECs to COVID-19 infection. This observed pathology may be explained by the hemagglutination that occurs as SARS-CoV-2’s spike protein attaches to red blood cells via adhesion molecule CD147.Citation25
INDUCTION OF EC DYSFUNCTION IN COVID-19 THROUGH THE NF-kB PATHWAY
The precise pathways by which SARS-CoV-2 binding with the ACE2 receptor induces EC dysfunction are still not clear, but likely involve the NF-kB pathway. In view of diffuse pulmonary EC injury leading to impairment of the alveolar-capillary barrier and increased microvascular permeability in the pathogenesis of ARDS, Li and colleagues used Sprague-Dawley rats and pulmonary microvascular ECs to investigate whether up-regulation of ACE2 expression prevents LPS-induced pulmonary inflammation and cytotoxicity via the mitogen-activated protein kinase (MAPK)/NF-kB signal pathway. Their results showed that the ACE2/Ang-(1-7)/Mas axis prevents LPS-induced apoptosis of pulmonary microvascular ECs by inhibiting phospho-c-jun N-terminal kinases (JNK)/NF-kB pathways.Citation26 As previously discussed, ACE1 and ACE2 balance vasoconstrictive (ACE1/Ang-II/AT1-axis) and vasodilator (ACE2/Ang1-7/MAS-axis) actions in the RAS.Citation11,Citation26 ACE2 is expressed in the endothelium, lung, heart, intestine, and kidney as well as the epithelial cells of oral mucosa and the tongue.Citation11 Following SARS-CoV-2 infection, ACE2 is down-regulated, resulting in ACE1/ACE2 imbalance, RAS activation, cytokine storm, immune response, increased permeability, edema, fibrosis, and thrombosis.Citation11 Further, the ACE1/ACE2 pathway is suggested in ARDS because inflammation, blood coagulation, and fibrinolysis occur from an activated RAS.Citation26 Libby and Lüscher reiterated that COVID-19 is an endothelial disease, highlighting cellular events (thrombosis, fibrinolysis, balance between vasodilation and vasoconstriction, balance between proinflammation and antiinflammation, balance between prooxidant and antioxidant, barrier function, and cytokine storm) in COVID-19.Citation27
Recently, Hariharan et al indicated that an activated NF-κB pathway is involved in patients with severe COVID-19 symptoms, particularly those who are elderly and/or have metabolic syndrome (obesity, type 2 diabetes, and atherosclerosis).Citation28 Various cells, including the cardiovascular system and other organs in COVID-19, could undergo activation of NF-κB.Citation28 Hirano and Murakami suggested that SARS-CoV-2 itself activates NF-kB via PPRs, and thus hyperactivation of the NF-kB pathway is involved in the phenotype of COVID-19.Citation29 Consistent with this, in 2016, de Wit et al pointed out that the NF-kB pathway is involved in the pathogenesis of SARS-CoV and Middle East respiratory syndrome coronavirus.Citation30 They elucidated that the innate immune response is activated by the detection of PAMPs, and this occurs via a host’s PRRs. Following PRR-mediated detection of a PAMP, the resulting interaction of PRRs with mitochondrial antiviral-signaling protein activates NF-κB through a signaling cascade involving several kinases. Activated NF-κB translocates to the nucleus, where it induces the transcription of proinflammatory cytokines.Citation30 One of the major pathways for NF-kB activation after coronavirus infection is the myeloid differentiation primary-response protein 88–dependent pathway in the early phase of NF-κB activation, which leads to the production of inflammatory cytokines.Citation30 It also regulates innate and adaptive immune functions, involving CD4+ T-helper cells.Citation31 NF-kB triggers EC activation and makes the endothelium more susceptible to apoptosis, but protective, antiinflammatory genes help regulate activation and apoptosis.Citation23
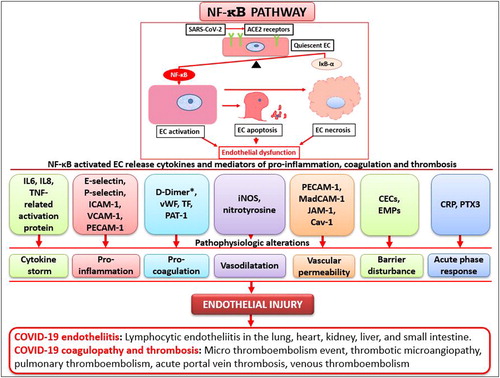
As illustrated in , imbalance between NF-kB and its protective gene, NF-kB inhibitor-a (IkB-a), induces EC activation, followed by EC apoptosis and EC necrosis. Therefore, a possible sequence in the pathogenesis of EC dysfunction is activation of NF-kB, followed by the release of proteins by activated EC and morphologic derangement. The interactions among the alterations of pathophysiology (e.g., cytokine storm, proinflammation, procoagulation, vasodilatation, increased vascular permeability, barrier disturbance, and acute phase response) eventually lead to irreversible endothelial injury and result in clinically relevant complications, such as COVID-19–associated endotheliitis, coagulation, and thrombus.
Figure 1. Proposed nuclear factor-κB (NF-κB) pathway by which angiotensin-converting enzyme 2 (ACE2) receptors induce endothelial dysfunction in COVID-19. The balance of NF-kB and its protective gene (IkB-a) maintain endothelial quiescence under normal conditions, but after SARS-Cov-2 binds with ACE2 receptors on the surface of endothelial cells (ECs) and NF-kB activation, the IkB-a genes are insufficient to counteract the action of NF-kB, and ECs become activated. If uncontrolled, ECs progress to apoptosis, necrosis, and endothelial dysfunction. The EC phenotypic changes via the NF-κB pathway result in the release of a variety of molecules/proteins (e.g., IL6, IL8, TNF-related activation protein; E-selectin, P-selectin, ICAM-1, VCAM-1, PECAM-1; D-Dimer, vWF, TF, PAT-1; iNOS, nitrotyrosine; PECAM-1, MadCAM-1, JAM-1, Cav-1; CECs, EMPs; CRP, PTX3). These molecules can directly act on ECs to induce a cytokine storm, inflammation, coagulation, vasodilatation, increased vascular permeability, barrier disturbance, and acute phase response. This endothelial injury can clinically manifest as COVID-19–associated endotheliitis, coagulopathy, and thrombosis. Note that D-dimers are not released from activated ECs and they are one of the fragments produced when plasma cleaves fibrin to break down clots.
SIMILAR PATHWAYS OF THE ACE2 RECEPTOR AND OTHER ENDOTHELIAL RECEPTORS RESPONSIBLE FOR INDUCTION OF EC DYSFUNCTION
Endothelial receptors share similar characteristics in the induction of EC dysfunction. In view of previous EC dysfunction in preexisting comorbidities, we hypothesize that a cross-talk of pathways between ACE2 and other receptors may transpire and that unexpected signal transfers in those pathways may occur. It is reported that coronaviruses have been shown to involve three MAPK pathways for viral pathogenesis: the JNK, the P38 MAPK, and the extracellular signal-regulated kinase (ERK1/2) pathway.Citation28 The P38 MAPK pathway regulates the translation of TNF-α and IL-1β, which can activate NF-κB noncanonically.Citation28 Interestingly, it was reported that P38 MAPKs mediate cross-talk activation of NF-κB signaling.Citation32 Of particular importance in this context is that inhibition of p38 could be used as a therapeutic approach for COVID-19 to attenuate proinflammatory cytokines (e.g., IL-6, TNF-α, and IL-1β).Citation33 This principle is based on SARS-CoV-2 inducing inflammation by directly activating p38, while downregulating inflammation by inhibiting p38. Two mechanisms may be involved: (1) Ang II signals proinflammatory, provasoconstrictive, prothrombotic activity through p38 MAPK activation, which is countered by Ang 1-7 down-regulation of p38 activity; and (2) SARS-CoV-2 may directly up-regulate p38 activity via a viral protein.Citation33 Furthermore, elevated p38 MAPK activity in the endothelium has been implicated in platelet aggregation, arterial thrombosis, and apoptosis of endothelial cells.Citation33
The purinergic P2Y1 receptor (P2Y1R) on ECs induces EC activation and mediates leukocyte adhesion and TNF-α production.Citation34 The other purinergic receptors, P2X7 and P2X4, are also involved in high glucose and palmitate-mediated EC activation and EC dysfunction.Citation35 Exposure of human umbilical vein ECs to high glucose and palmitate causes activation of P2X7 and P2X4, which results in increased intracellular reactive oxygen species and reduced endothelial nitric oxide synthase, which contribute to EC dysfunction.Citation35 The activation of these two receptors leads to EC activation via increased expression of IL-6, ICAM-1, VCAM-1, IL-8, and cyclooxygenase-2.Citation35 A proposed mechanism for the EC activation and dysfunction involves activation of p38-MAPK.Citation35
Heparan sulfate-containing proteoglycan receptors (dengue virus receptors) are present on ECs. The interactions of the receptors with dengue virus induce EC dysfunction (endothelial barrier disturbance, vascular leakage, increased endothelial cell permeability, EC apoptosis, and death).Citation36 NF-κB is known to be activated by infection with dengue virus because it participates in the regulation of proinflammatory mediators, including inducible nitric oxide synthase (iNOS) and TNF-α, which play a role in induction of EC dysfunction. Therefore, the distinct pathway for NF-κB activation is involved.Citation37 Clausen and colleagues found that heparan sulfate interacts with a component of the spike protein called the receptor binding domain and that heparan sulfate promotes the interaction between the spike protein and ACE2, indicating that SARS-CoV-2 infection depends on both heparan sulfate and ACE2.Citation38
Protease-activated receptor-1 (PAR1) is expressed on ECs and induces endothelial barrier disturbance.Citation39 A postulated mechanism for the disturbance is attributed to thrombin, since PAR1 is the major effector of thrombin signaling in ECs. Thrombin binds to and cleaves the N-terminus of PAR1, which unmasks a new N-terminal domain to serve as a tethered ligand. Therefore, thrombin activation of PAR1 is responsible for EC dysfunction.Citation39 In addition, endothelial barrier permeability is affected by thrombin promoting p38 MAP kinase and NF-activation, which increases ICAM-1, IL-6, IL-8, and MCP1 cytokines in ECs.Citation39
Safia and colleagues showed that, in vitro, beta-adrenergic receptor stimulation reduced EC apoptosis and decreased the level of reactive oxygen species generation via the NF-κB/IκBa pathway.Citation40 Their study demonstrated that hyperglycemia could be an apoptotic stimulus to trigger NF-κB release and activation.Citation40
CONCLUSION
This essay highlights fundamental issues regarding the endothelial ACE2 receptor as it pertains to EC dysfunction in COVID-19. Specifically, it reviewed the susceptibility of capillary ECs to SARS-CoV-2 infection, the coexistence of new and preexisting EC dysfunction, EC dysfunction in COVID-19 through the NF-kB pathway, and the cross-talk of pathways between the ACE2 receptor and other endothelial receptors (e.g., purinergic receptor, heparin sulfate–containing proteoglycan receptor, protease-activated receptor, and beta-adrenergic receptor). It is of special interest that both heparan sulfate and ACE2 are necessary for SARS-CoV-2 infection. Thus, SARS-CoV-2 infection is tightly linked to endothelial injury and dysfunction, which in turn promote thrombosis with hemagglutination.
- Zhang J, Tecson KM, McCullough PA. Endothelial dysfunction contributes to COVID-19-associated vascular inflammation and coagulopathy. Rev Cardiovasc Med. 2020;21(3):315–319. doi:10.31083/j.rcm.2020.03.126.
- Zhang J, McCullough PA, Tecson KM. Vitamin D deficiency in association with endothelial dysfunction: implications for patients with COVID-19. Rev Cardiovasc Med. 2020;21(3):339–344. doi:10.31083/j.rcm.2020.03.131.
- Koo B-S, Oh H, Kim G, et al. Transient lymphopenia and interstitial pneumonia with endotheliitis in SARS-CoV-2-infected macaques. J Infect Dis. 2020;222(10):1596–1600. doi:10.1093/infdis/jiaa486.
- Hamming I, Timens W, Bulthuis ML, Lely AT, Navis G, van Goor H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J Pathol. 2004;203(2):631–637. doi:10.1002/path.1570.
- Chen J, Jiang Q, Xia X, et al. Individual variation of the SARS-CoV-2 receptor ACE2 gene expression and regulation. Aging Cell. 2020;19(7):1–12. doi:10.1111/acel.13168.
- Li MY, Li L, Zhang Y, Wang XS. Expression of the SARS-CoV-2 cell receptor gene ACE2 in a wide variety of human tissues. Infect Dis Poverty. 2020;9(1):45. doi:10.1186/s40249-020-00662-x.
- Shang J, Wan Y, Luo C, et al. Cell entry mechanisms of SARS-CoV-2. Proc Natl Acad Sci USA. 2020;117(21):11727–11734. doi:10.1073/pnas.2003138117.
- Hoffmann M, Kleine-Weber H, Schroeder S, et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181(2):271–280.e8. doi:10.1016/j.cell.2020.02.052.
- Bourgonje AR, Abdulle AE, Timens W, et al. Angiotensin-converting enzyme 2 (ACE2), SARS-CoV-2 and the pathophysiology of coronavirus disease 2019 (COVID-19). J Pathol. 2020;251(3):228–248. doi:10.1002/path.5471.
- Lely AT, Hamming I, van Goor H, Navis GJ. Renal ACE2 expression in human kidney disease. J Pathol. 2004;204(5):587–593. doi:10.1002/path.1670.
- Gemmati D, Bramanti B, Serino ML, et al. COVID-19 and individual genetic susceptibility/receptivity: role of ACE1/ACE2 genes, immunity, inflammation and coagulation. Might the double X-Chromosome in females be protective against SARS-CoV-2 compared to the single X-Chromosome in males? Int J Mol Sci. 2020;21(10):3474. doi:10.3390/ijms21103474.
- Zhang YH, Zhang YH, Dong XF, et al. ACE2 and Ang-(1-7) protect endothelial cell function and prevent early atherosclerosis by inhibiting inflammatory response. Inflamm Res. 2015;64(3-4):253–260. doi:10.1007/s00011-015-0805-1.
- Palazzuoli A, Mancone M, De Ferrari GM, et al. Antecedent administration of angiotensin-converting enzyme inhibitors or angiotensin II receptor antagonists and survival after hospitalization for COVID-19 syndrome. J Am Heart Assoc. 2020;9(22):e017364. doi:10.1161/JAHA.120.017364.
- Fraga-Silva RA, Sorg BS, Wankhede M, et al. ACE2 activation promotes antithrombotic activity. Mol Med. 2010;16(5-6):210–215. doi:10.2119/molmed.2009.00160.
- Lin L, Lu L, Cao W, Li T. Hypothesis for potential pathogenesis of SARS-CoV-2 infection-a review of immune changes in patients with viral pneumonia. Emerg Microbes Infect. 2020;9(1):727–732. doi:10.1080/22221751.2020.1746199.
- Mollica V, Rizzo A, Massari F. The pivotal role of TMPRSS2 in coronavirus disease 2019 and prostate cancer. Future Oncol. 2020;16(27):2029–2033. doi:10.2217/fon-2020-0571.
- Stopsack KH, Mucci LA, Antonarakis ES, et al. TMPRSS2 and COVID-19: serendipity or opportunity for intervention? Cancer Discov. 2020;10(6):779–782. doi:10.1158/2159-8290.CD-20-0451.
- Zhang J, Defelice AF, Hanig JP, Colatsky T. Biomarkers of endothelial cell activation serve as potential surrogate markers for drug-induced vascular injury. Toxicol Pathol. 2010;38(6):856–871. doi:10.1177/0192623310378866.
- Teixeira TM, da Costa DC, Resende AC, Soulage CO, Bezerra FF, Daleprane JB. Activation of Nrf2-antioxidant signaling by 1,25-dihydroxycholecalciferol prevents leptin-induced oxidative stress and inflammation in human endothelial cells. J Nutr. 2017;147(4):506–513. doi:10.3945/jn.116.239475.
- Li Y, Zeng Z, Cao Y, et al. Angiotensin-converting enzyme 2 prevents lipopolysaccharide-induced rat acute lung injury via suppressing the ERK1/2 and NF-κB signaling pathways. Sci Rep. 2016;6(1):27911. doi:10.1038/srep27911.
- Escher R, Breakey N, Lämmle B. Severe COVID-19 infection associated with endothelial activation. Thromb Res. 2020;190:62. doi:10.1016/j.thromres.2020.04.014.
- Varga Z, Flammer AJ, Steiger P, et al. Endothelial cell infection and endotheliitis in COVID-19. Lancet. 2020;395(10234):1417–1418. doi:10.1016/S0140-6736(20)30937-5.
- Zhang J, Hanig JP, De Felice AF. Biomarkers of endothelial cell activation: candidate markers for drug-induced vasculitis in patients or drug-induced vascular injury in animals. Vascul Pharmacol. 2012;56(1-2):14–25. doi:10.1016/j.vph.2011.09.002.
- Buja LM, Wolf DA, Zhao B, et al. The emerging spectrum of cardiopulmonary pathology of the coronavirus disease 2019 (COVID-19): report of 3 autopsies from Houston, Texas, and review of autopsy findings from other United States cities. Cardiovasc Pathol. 2020;48:107233. doi:10.1016/j.carpath.2020.107233.
- Scheim D. Ivermectin for COVID-19 treatment: clinical response at quasi-threshold doses via hypothesized alleviation of CD147-mediated vascular occlusion. SSRN. 2020;1–22. doi:10.2139/ssrn.3636557.
- Li Y, Cao Y, Zeng Z, et al. Angiotensin-converting enzyme 2/angiotensin-(1-7)/Mas axis prevents lipopolysaccharide-induced apoptosis of pulmonary microvascular endothelial cells by inhibiting JNK/NF-κB pathways. Sci Rep. 2015;5(1):8209. doi:10.1038/srep08209.
- Libby P, Lüscher T. COVID-19 is, in the end, an endothelial disease. Eur Heart J. 2020;41(32):3038–3044. doi:10.1093/eurheartj/ehaa623.
- Hariharan A, Hakeem AR, Radhakrishnan S, Reddy MS, Rela M. The role and therapeutic potential of NF-kappa-B pathway in severe COVID-19 patients. Inflammopharmacology. 2020:1–10. doi:10.1007/s10787-020-00773-9.
- Hirano T, Murakami M. COVID-19: a new virus, but a familiar receptor and cytokine release syndrome. Immunity. 2020;52(5):731–733. doi:10.1016/j.immuni.2020.04.003.
- de Wit E, van Doremalen N, Falzarano D, Munster VJ. SARS and MERS: recent insights into emerging coronaviruses. Nat Rev Microbiol. 2016;14(8):523–534. doi:10.1038/nrmicro.2016.81.
- Liu T, Zhang L, Joo D, Sun SC. NF-κB signaling in inflammation. Signal Transduct Target Ther. 2017;2(1):e17023. doi:10.1038/sigtrans.2017.23.
- Saccani S, Pantano S, Natoli G. p38-Dependent marking of inflammatory genes for increased NF-kappa B recruitment. Nat Immunol. 2002;3(1):69–75. doi:10.1038/ni748.
- Grimes JM, Grimes KV. p38 MAPK inhibition: a promising therapeutic approach for COVID-19. J Mol Cell Cardiol. 2020;144:63–65. doi:10.1016/j.yjmcc.2020.05.007.
- Cardoso TC, Pompeu TE, Silva CLM. The P2Y1 receptor-mediated leukocyte adhesion to endothelial cells is inhibited by melatonin. Purinergic Signal. 2017;13(3):331–338. doi:10.1007/s11302-017-9565-4.
- Sathanoori R, Swärd K, Olde B, Erlinge D. The ATP receptors P2X7 and P2X4 modulate high glucose and palmitate-induced inflammatory responses in endothelial cells. PLoS One. 2015;10(5):e0125111. doi:10.1371/journal.pone.0125111.
- Dalrymple N, Mackow ER. Productive dengue virus infection of human endothelial cells is directed by heparan sulfate-containing proteoglycan receptors. J Virol. 2011;85(18):9478–9485. doi:10.1128/JVI.05008-11.
- Cheng YL, Lin YS, Chen CL, et al. Dengue virus infection causes the activation of distinct NF-κB pathways for inducible nitric oxide synthase and TNF-α expression in RAW264.7 cells. Mediators Inflamm. 2015;2015:274025. doi:10.1155/2015/274025.
- Clausen TM, Sandoval DR, Spliid CB, et al. SARS-CoV-2 infection depends on cellular heparan sulfate and ACE2. Cell. 2020;183(4):1043–1057. doi:10.1016/j.cell.2020.09.033.
- Grimsey NJ, Trejo J. Integration of endothelial protease-activated receptor-1 inflammatory signaling by ubiquitin. Curr Opin Hematol. 2016;23(3):274–279. doi:10.1097/MOH.0000000000000232.
- Safi SZ, Shah H, Qvist R, et al. Beta adrenergic receptors stimulation attenuates phosphorylation of NF-κB and IκBα in hyperglycemic endothelial cells. Cell Physiol Biochem. 2018;51(3):1429–1436. doi:10.1159/000495591.