
ABSTRACT
In this study, we investigated the effects of planting three types of forage crops in an apple orchard on the soil microbial community structure. The apple orchard was intercropped with native grasses (NG), red clover (RC; Trifolium pratense L.), ryegrass (RE; Trifolium repens L.), and no vegetation (CT control; clean tillage). We obtained soil samples from depths of 0–20, 20–40, and 40–60 cm from the different treatment plots in the orchard and analysed them using a high-throughput DNA sequencing technique. The three forage crops had affected the soil bacterial and fungal community structures. Compared with CT control, intercropping with NG increased the proportion of Acidobacteria and reduced those of Nitrospirae and Verrucomicrobia, whereas intercropping with RC increased the proportions of Nitrospirae and Verrucomicrobia and reduced that of Planctomycetes. Intercropping with RE increased the proportions of Nitrospirae and Chloroflexi, whereas reduced that of Acidobacteria. Furthermore, unlike in the other treatments, intercropping with NG increased the proportion of Zygomycota in the 0–20-cm soil layer. Intercropping with RC increased the proportion of Chytridiomycota in all the three soil layers, whereas intercropping with RE increased the proportion of Basidiomycota in the 20–40-cm soil layer. Collectively, these findings suggest that intercropping with forage crops, especially RC, in an apple orchard, could alter soil microbial community structure. In our previous study, we showed that microbial sole-carbon-source utilisation is changed by intercropping with forage crops; thus, it can be considered as an effective approach to improve the efficiency of soil C cycling in the apple orchard by altering the microbial community structure.
Introduction
Numerous studies on microbial genomics (Duarte et al. Citation1998; Rastogi et al. Citation2010), metabolic features (Degens & Harris Citation1997; Chaerun et al. Citation2011), and biofilm characteristics (Bossio et al. Citation1998; Watzinger et al. Citation2008) have shown that vegetation type affects the soil microbial community structure. Plants can influence the composition of the microbial community in the underlying soil regardless of the soil type (Miethling et al. Citation2000), since essential nutrients for soil microbes often originate from plant litter or root exudates (Grayston et al. Citation1998). Differences in plant exudates, as well as litter quantity and quality, can alter soil carbon sequestration and thereby affect soil microbial community structure (De Deyn et al. Citation2008). Therefore, these effects are closely related to various plant-based ecophysiological traits such as variation in short-term carbon (C) cycling (Ward et al. Citation2009). Additionally, plants can form symbiotic relationships with soil microorganisms (Bever Citation2003), such as in the case of nitrogen-fixing bacteria in legumes, and such relationships alter community structure by favouring certain microbes over others. Furthermore, plant roots physically alter the soil structure, creating varying microhabitats that are suitable for different microbes (Bardgett et al. Citation2005); the presence of mycorrhizae is known to directly and indirectly influence the soil microbial diversity (Purin et al. Citation2006). Finally, the characteristic traits that define different plant functional groups (such as variation in carbon to nitrogen ratios between legumes and graminoids) are likely to influence the effects of plants on soil properties, which could in turn affect the microbial community composition.
There is increasing evidence that changes in soil microbial community structure can influence biogeochemical processes such as C cycling (Balser & Firestone Citation2005; Osler & Sommerkorn Citation2007; Strickland et al. Citation2009). Although we still know relatively little regarding the mechanisms underlying this influence (Condron et al. Citation2010), available evidence (Strickland et al. Citation2009) seems to support the ‘functional dissimilarity’ hypothesis, rather than the earlier ‘functional equivalence’ hypothesis. The former suggests that microbial community structure and function are altered by the environment, but species diversity across different communities does not affect the ecosystem process rates (Strickland et al. Citation2009). In contrast, the ‘functional equivalence’ hypothesis suggests that, along with the environment, the species composition of microbial communities affects their function (Strickland et al. Citation2009). In other words, microbial communities that vary in species diversity will also differ in their ability to alter the rates of ecosystem processes. Thus, assessing the composition of soil microflora reveals the response of soil ecosystems to environmental changes: healthy ecosystems are characterised by microbial communities with high structural diversity, whereas communities with low structural diversity have difficulty in responding to environmental fluctuations (Böhme et al. Citation2005).
In our previous study, we showed that planting forage crops, particularly red clover (RC), improved the carbon use efficiency of the soil organisms in an apple orchard (Jiao et al. Citation2013). However, we could not determine whether the intercropping with forage crops could significantly affect the microbial community structure. Thus, in this study, we assessed the effects of changes in soil microbial community structure of an apple orchard by intercropping with forage crops.
Materials and methods
Study site and soil sampling
The study was conducted at the experimental orchard of the Shenyang Agricultural University (41°88′N, 123°85′E, 76.2 m ASL). The climatic conditions of the site were as follows: an average yearly sunshine duration of 2587.6 h, an annual frost-free period of 146–163 days, and an average annual rainfall of 721 mm. Orchard soil consisted of clay loam (pH, 6.3; soil organic matter, 8.6 g kg−1; available N, 50.6 mg kg−1; available P, 35.2 mg kg−1; available K, 77.0 mg kg−1).
In 2006, an apple cultivar called HanFu (‘Hanfu’/GM256/Malus baccata Borkh) was planted at a 2.0 m (in-row) to 4.0 m (between-row) density. During April 2009, we planted and cultivated 18 different forage crops between the tree rows. Three forage crops that showed the most desirable growth were selected in this study – NG (native grasses), RC (red clover; Trifolium pratense L.), and RE (ryegrass; Trifolium repens L.) – further, one CT (clean tillage) control condition was established, which involved manual removing of weeds every two weeks to ensure the absence of vegetative covering.
On 15 June 2013, we selected four adjacent and flat experimental plots (each 3 m × 165 m) for the four treatments for soil sampling. For each treatment, we randomly selected five sampling sites and collected soil from each treatment condition by using a 5-cm diameter soil corer. Soil sample depths were set at 0–20 cm, 20–40 cm, and 40–60 cm. Five soil samples from the same layer were mixed for further analysis. The soil samples were placed in individually labelled plastic bags and transported on the same day to the laboratory. At the laboratory, each soil sample was sieved using a <2-mm sieve and then stored at −70°C.
DNA extraction, amplification, and sequencing
Genomic DNA was extracted from 250 mg of composite soil samples by using the Soil DNA Kit (D5625–01; Omega Bio-Tek, USA), following the manufacturer's protocols. DNA concentrations were then measured using a protein and nucleic acid analyser (Nano-2000; NanoDrop, USA), and the samples were stored at −20°C.
We determined the diversity and composition of the bacterial communities in our soil samples by using the protocol described by Caporaso et al. (Citation2010). We performed polymerase chain reactions (PCRs) to amplify the V4 region of the 16S rDNA gene. We selected the 515f/806r primer set (BGI Company, China) for the PCRs, enabling accurate phylogenetic and taxonomic information after DNA analysis. The reverse primer contains a 6-bp error-correcting barcode, which is unique to each soil sample. DNA was amplified following a previously described protocol (Magoč & Salzberg Citation2011).
The libraries of partial bacterial 16S rRNA gene fragments were analysed using the Illumina Genome Analyzer System GAII (BGI Company, China) by using sequencing-by-synthesis and 151-index paired-end sequencing. Sequences containing uncalled bases, incorrect primer sequences, or runs of ≥10 identical nucleotides were removed. Furthermore, sequences shorter than 55 bp after quality trimming were discarded.
Next, we determined the diversity and composition of soil fungal communities by using the methods previously described by Schmidt et al. (Citation2013). Paired-end sequencing (2 × 150 bp) was performed on an Illumina MiSeq sequencer.
Data analysis
Pairs of reads were merged using fast length adjustment of short reads, which is used specifically in the cases when the original DNA fragments are shorter than twice the length of the reads (Magoč & Salzberg Citation2011). Sequencing reads were assigned to each sample according to the latter's unique barcodes (Edgar Citation2013).
Sequences were analysed using the Quantitative Insights into Microbial Ecology (QIIME) software package (Wang et al. Citation2007) and the UPARSE pipeline (Edgar Citation2013). We also wrote custom Perl scripts to analyse alpha (within sample) and beta (between sample) diversities.
First, the reads were run through QIIME quality filters. Default settings for Illumina processing in QIIME were used. The quality filters were as follows:
min_per_read_length: minimum number of consecutive high-quality base calls to retain read (as percentage of total read length);
max_bad_run_length: maximum number of consecutive low-quality base calls allowed before truncating a read;
sequence_max_n: maximum number of ambiguous (N) characters allowed in a sequence;
phred_quality_score: last quality score considered low quality.
Next, we used the UPARSE pipeline to select operational taxonomic units (OTUs) by creating an OTU table. Sequences with 97% similarity were assigned to the OTUs. We selected a representative sequence for each OTU by conducting sequence comparison across multiple databases (International Nucleotide Sequence Databases: NCBI, EMBL, DDBJ). We then used the Ribosomal Database Project (RDP) classifier to assign taxonomic data to those sequences (Caporaso et al. Citation2011) that were added to estimate the phyla constitution of each soil sample. We plotted the most abundant bacterial phyla in a histogram.
Based on the bacterial and fungal abundances of each soil sample, we performed transverse clustering in accordance with species and longitudinal clustering between the 12 soil samples. After clustering, we could identify the bacterial and fungal species that were more abundant in our soil samples.
To compare the effects of the four treatment conditions on the soil microbial communities in the apple orchard, we analysed beta diversity, a measure of the phenotypic relatedness between two communities (Caporaso et al. Citation2011). This metric allowed us to record the changes in community structure due to the treatment conditions across our soil samples. To obtain the beta diversity, we first calculated the Bray–Curtis distance and performed a principle coordinate analysis (PCoA) on the statistic. PCoA is a technique that extracts and visualises the most informative components of variation from multidimensional data (Wang et al. Citation2007); this analysis was necessary for our complex raw data. Beta diversity was assessed by analysing the graphed results of the PCoA, wherein the samples that were further apart on the graph were considered to have higher beta diversity.
Results
Effects of forage crops on OTU abundances
Compared with the control CT condition ((a)), the NG treatment increased soil bacterial diversity in the 0–20-cm soil layer, but decreased it in the 20–40-cm and 40–60-cm soil layers. However, compared to CT, the RC treatment did not affect the soil bacterial diversity in the 0–20, 20–40, and 40–60-cm soil layers. The RE treatment improved soil bacterial diversity only in the 20–40-cm layer.
Figure 1. The number of bacterial (a) and fungal (b) operational taxonomic unit (OTU) number in the apple orchard soils intercropped with different forage species, as well as in that with no vegetation, that is, clean tillage. (Apple trees intercropped with NG, RC, and RE; CT, clean tillage in the apple orchard).
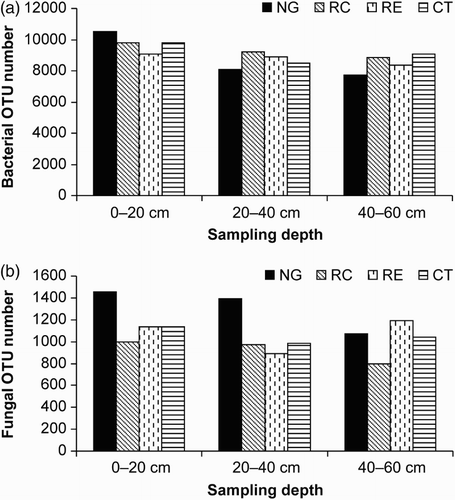
The NG treatment increased the fungal diversity compared with that in the CT control across all the three soil depths ((b)). The RC treatment had little effect on fungal diversity in the 20–40-cm soil depth but reduced soil fungal diversity at the 0–20-cm and 40–60-cm soil layers compared with that in the CT. Furthermore, the RE treatment had no effect on fungal diversity at the 0–20-, 20–40-, and 40–60-cm soil layers, respectively, compared with that in the CT control.
Microbial genera abundance clustering
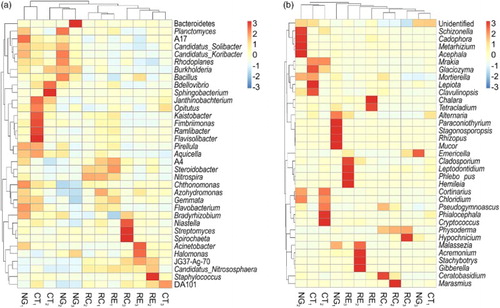
The results of our clustering analyses revealed marked differences in the relative abundances of different bacterial and fungal genera among the four soil samples ((a)). NG and CT treatments led to an increase in the bacterial genera compared with the RC and RE treatments at the 0–20-cm and 20–40-cm soil layers. In contrast, at the 40–60-cm soil layer, the planting of the three forage crops resulted in greater bacterial diversity than that in the CT control.
Figure 2. Relative abundances of different bacterial (a) and fungal (b) genera in the apple orchard soils intercropped with different forage species, as well as in that with no vegetation (clean tillage). (Apple trees intercropped with NG, RC, and RE; CT, clean tillage in the apple orchard; 1, 2, and 3 represent soils at depths of 0–20 cm, 20–40 cm, and 40–60 cm, respectively).
Effect of forage crops on microbial community composition
We found that the forage species affected the soil bacterial community structures per phylum compared with those in the CT control ((a)). NG treatment showed greater proportions of Acidobacteria (19.6–22.6%), but smaller proportions of Nitrospirae (0.9–2.3%) and Verrucomicrobia (4.3–8.1%). RC treatment increased the proportions of Nitrospirae (3.0–4.3%) and Verrucomicrobia (9.3–11.4%), but reduced those of Planctomycetes (3.2–5.1%). However, RE treatment showed greater proportions of Nitrospirae (3.2–3.9%) and Chloroflexi (4.1–9.5%), but smaller proportions of Acidobacteria (14.5–17.5%). Furthermore, sampling depth influenced the bacterial community composition for all the four treatment groups. The abundances of Actinobacteria and Chloroflexi increased, whereas those of Bacteroidetes, Planctomycetes, and Proteobacteria decreased with increasing soil depth.
Figure 3. Relative abundances of different bacterial (a) and fungal (b) classes in the apple orchard soils intercropped with different forage species, as well as in that with no vegetation (clean tillage). (Apple trees intercropped with NG, RC, and RE; CT, clean tillage in the apple orchard; 1, 2, and 3 represent soil depths of 0–20 cm, 20–40 cm, and 40–60 cm, respectively).
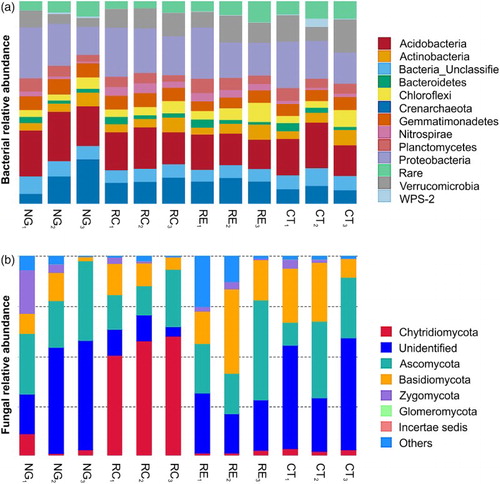
The three forage crops also markedly affected the soil fungal community structures per phylum ((b)). NG treatment showed greater proportions of Zygomycota (21.8%) in the 0–20-cm soil layer, RC treatment showed greater proportions of Chytridiomycota (50.8–60.2%) in all the three soil layers, and RE treatment showed greater proportions of Basidiomycota (42.2%) in the 20–40-cm soil layer. Thus, RC treatment had a greater influence on the fungal community structures than the other treatments, particularly in the deeper soil layers (20–40 and 40–60 cm).
Effects of forage crops on beta diversity
The PCA values of the first and second PCs were 11.09% and 10.20% for bacteria. Their eigenvalues were larger than the pooled average of all eigenvalues (Ramette Citation2007), indicating their significance. Our analysis of beta diversity yielded two significant findings with regard to soil bacterial community ((a)). First, the three forage crops influenced soil bacterial communities compared with those in the CT control, with the effects of NG and RC treatments being greater than those of RE treatment. Second, we found very similar bacterial communities in the 0–20 and 20–40-cm soil layers in the RC treatment; the roots are distributed at these depths.
Figure 4. Principal coordinate analysis of bacterial (a) and fungal (b) communities in the apple orchard soils intercropped with different forage species, as well as in that with no vegetation (clean tillage). (Apple trees intercropped with NG, RC, and RE; CT, clean tillage in the apple orchard; 1, 2, and 3 represent soil depths of 0–20 cm, 20–40 cm, and 40–60 cm, respectively.
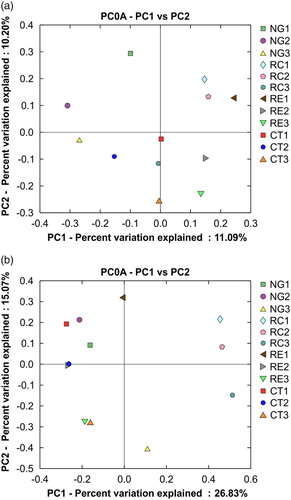
The PCA values of the first and second PCs were 26.83% and 15.07% for fungi, and their eigenvalues were also significant. Although all the three forage crops influenced soil fungal communities, the influence of RC was the greatest across all the three soil layers ((b)).
Discussion
Our study results suggest that different forage crops (native mixed grasses, RC [Leguminosae], and RE [Poaceae]) could alter the soil bacterial and fungal diversities () and community structures ( and ). Specifically, soils planted with NG showed increased Acidobacteria abundance; this species has a rich plant species diversity (Foesel et al. Citation2014) and polysaccharide-rich stems and leaves, which could release carbohydrates into the soil (Rawat et al. Citation2012). Soils treated with RC showed increased Nitrospirae abundance, which is likely related to the nitrogen substrates exuded by the symbiotic nitrogen-fixing bacteria in the RC roots (Sawada et al. Citation2003). These bacteria are able to convert nitrogen (in the air) into ammonium nitrogen and provide nitrogen nutrition for plants and other soil microorganisms (Høgh-Jensen & Schjoerring Citation2001). Furthermore, in the deep soil layers (40–60 cm), a clear difference in the abundance of bacterial species was apparent between the control soil and the soils from the forage crop treatment ((a)), particularly in the case of RC. These results suggest that the forage crops exert their effect on soil microbial community composition via their roots, corroborating the findings of previous studies (Breulmann et al. Citation2012). RC might have been especially effective owing to its deeper root distribution (Jiao et al. Citation2013).
Numerous studies have focused on the relationship between variation in microbial community structure and the changes in the microbial ecosystem functions (Hooper et al. Citation2005; Wardle Citation2006; Gessner et al. Citation2010). In our study, we found that planting RC clearly altered the bacterial ((a)) and fungal ((b)) community structures. When combined with our previous study findings indicating that RC increases carbon use efficiency in soil microbes (Jiao et al. Citation2013), our data support the view that changes to essential microbial functions (i.e. carbon use) are correlated with soil microbial abundance (Hättenschwiler et al. Citation2005).
Furthermore, we found that RC changed soil fungal communities more prominently than either NG or RE across all soil depths ((b)). Given that fungi play an essential role in the conversion of soil nutrients and the decomposition of organic matter (Brodie et al. Citation2003), changes to fungal community structure might influence many key ecosystem processes. Thus, our results suggest that RC might also increase the carbon use efficiency and other nutrient transformation processes by altering the fungal community structure, particularly in deeper soil layers where its roots are distributed.
Although we have shown that forage crops increase soil bacterial/fungal abundance and diversity, as well as carbon use efficiency (Jiao et al. Citation2013), we were unable to elucidate the relationship between the variation in microbial community structure and carbon use. Further research involving the use of new technologies is warranted to elucidate the complex interactions occurring within diverse soil microbial communities and to understand how such interactions might affect the ecosystem function (McGuire & Treseder Citation2010). For example, metagenomics can be used to analyse the gene functions of the total DNA isolated from the soil; however, such studies will not be able to elucidate the ecological functions of more specific microbial groups (Daniel Citation2005). Stable isotope probing can couple the analyses of complex microbial species composition with physiological mechanisms. Such an integrative approach might provide further direct evidence and can link microbial community shifts with changes in the major ecological processes (Radajewski et al. Citation2000).
In summary, our results showed that forage crops influenced the abundance and diversity of soil bacterial and fungal communities. Considering the results obtained in our previous study, we propose that the cultivation of forage crops, especially RC, in an intercropping system could alter the microbial community structure; this could be associated with the changes in the microbial sole-carbon-source utilisation. Thus, planting forage crops is an effective approach to improve the microbial community structure and functions of orchard soil.
Acknowledgements
We would like to thank Editage for providing editorial assistance.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Balser TC, Firestone MK. 2005. Linking microbial community composition and soil processes in a California annual grassland and mixed-conifer forest. Biogeochemistry. 73:395–415. doi: 10.1007/s10533-004-0372-y
- Bardgett RD, Bowman WD, Kaufmann R, Schmidt SK. 2005. A temporal approach to linking aboveground and belowground ecology. Trends Ecol Evol. 20:634–641. doi: 10.1016/j.tree.2005.08.005
- Bever JD. 2003. Soil community feedback and the coexistence of competitors: conceptual frameworks and empirical tests. New Phytol. 157:465–473. doi: 10.1046/j.1469-8137.2003.00714.x
- Böhme L, Langer U, Böhme F. 2005. Microbial biomass, enzyme activities and microbial community structure in two European long-term field experiments. Agric Ecosyst Environ. 109:141–152. doi: 10.1016/j.agee.2005.01.017
- Bossio DA, Scow KM, Gunapala N, Graham KJ. 1998. Determinants of soil microbial communities: effects of agricultural management, season, and soil type on phospholipid fatty acid profiles. Microb Ecol. 36:1–12. doi: 10.1007/s002489900087
- Breulmann M, Schulz E, Weißhuhn K, Buscot F. 2012. Impact of the plant community composition on labile soil organic carbon, soil microbial activity and community structure in semi-natural grassland ecosystems of different productivity. Plant Soil. 352:253–265. doi: 10.1007/s11104-011-0993-6
- Brodie E, Edwards S, Clipson N. 2003. Soil fungal community structure in a temperate upland grassland soil. FEMS Microbiol Ecol. 45:105–114. doi: 10.1016/S0168-6496(03)00126-0
- Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Peña AG, Goodrich JK, Gordon JI, et al. 2010. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 7:335–336. doi: 10.1038/nmeth.f.303
- Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Lozupone CA, Turnbaugh PJ, Fierer N, Knight R. 2011. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc Natl Acad Sci USA. 108:4516–4522. doi: 10.1073/pnas.1000080107
- Chaerun SK, Pangesti NP, Toyota K, Whitman WB. 2011. Changes in microbial functional diversity and activity in paddy soils irrigated with industrial wastewaters in Bandung, West Java Province, Indonesia. Water, Air, Soil Pollut. 217:491–502. doi: 10.1007/s11270-010-0603-x
- Condron L, Stark C, O'Callaghan M, Clinton P, Huang Z. 2010. The role of microbial communities in the formation and decomposition of soil organic matter. In: Dixon GR, Tilston EL, editors. Soil microbiology and sustainable crop production. Dordrecht: Springer; p. 81–118.
- Daniel R. 2005. The metagenomics of soil. Nat Rev Microbiol. 3:470–478. doi: 10.1038/nrmicro1160
- De Deyn GB, Cornelissen JH, Bardgett RD. 2008. Plant functional traits and soil carbon sequestration in contrasting biomes. Ecol Lett. 11:516–531. doi: 10.1111/j.1461-0248.2008.01164.x
- Degens BP, Harris JA. 1997. Development of a physiological approach to measuring the catabolic diversity of soil microbial communities. Soil Biol Biochem. 29:1309–1320. doi: 10.1016/S0038-0717(97)00076-X
- Duarte GF, Rosado AS, Seldin L, Keijzer-Wolters AC, van Elsas JD. 1998. Extraction of ribosomal RNA and genomic DNA from soil for studying the diversity of the indigenous bacterial community. J Microbiol Methods. 32:21–29. doi: 10.1016/S0167-7012(98)00004-9
- Edgar RC. 2013. UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat Methods. 10:996–998. doi: 10.1038/nmeth.2604
- Foesel BU, Nägele V, Naether A, Wüst PK, Weinert J, Bonkowski M, Lohaus G, Polle A, Alt F, Oelmann Y, et al. 2014. Determinants of Acidobacteria activity inferred from the relative abundances of 16S rRNA transcripts in German grassland and forest soils. Environ Microbiol. 16:658–675. doi: 10.1111/1462-2920.12162
- Gessner MO, Swan CM, Dang CK, McKie BG, Bardgett RD, Wall DH, Hättenschwiler S. 2010. Diversity meets decomposition. Trends Ecol Evol. 25:372–380. doi: 10.1016/j.tree.2010.01.010
- Grayston SJ, Wang S, Campbell CD, Edwards AC. 1998. Selective influence of plant species on microbial diversity in the rhizosphere. Soil Biol Biochem. 30:369–378. doi: 10.1016/S0038-0717(97)00124-7
- Hättenschwiler S, Tiunov AV, Scheu S. 2005. Biodiversity and litter decomposition in terrestrial ecosystems. Annu Rev Ecol Evol Syst. 36:191–218. doi: 10.1146/annurev.ecolsys.36.112904.151932
- Høgh-Jensen H, Schjoerring JK. 2001. Rhizodeposition of nitrogen by red clover, white clover and ryegrass leys. Soil Biol Biochem. 33(4):439–448. doi: 10.1016/S0038-0717(00)00183-8
- Hooper DU, Chapin FS, Ewel JJ, Hector A, Inchausti P, Lavorel S, Lawton JH, Lodge DM, Loreau M, Naeem S, et al. 2005. Effects of biodiversity on ecosystem functioning: a consensus of current knowledge. Ecol Monogr. 75:3–35. doi: 10.1890/04-0922
- Jiao K, Qin S, Lyu D, Liu L, Ma H. 2013. Red clover intercropping of apple orchards improves soil microbial community functional diversity. Acta Agric Scand Sect B. 63:466–472.
- Magoč T, Salzberg SL. 2011. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics. 27:2957–2963. doi: 10.1093/bioinformatics/btr507
- McGuire KL, Treseder KK. 2010. Microbial communities and their relevance for ecosystem models: decomposition as a case study. Soil Biol Biochem. 42:529–535. doi: 10.1016/j.soilbio.2009.11.016
- Miethling R, Wieland G, Backhaus H, Tebbe CC. 2000. Variation of microbial rhizosphere communities in response to crop species, soil origin, and inoculation with Sinorhizobium meliloti L33. Microb Ecol. 40(1):43–56. doi: 10.1007/s002480000021
- Osler GH, Sommerkorn M. 2007. Toward a complete soil C and N cycle: incorporating the soil fauna. Ecology. 88:1611–1621. doi: 10.1890/06-1357.1
- Purin S, Filho OK, Stürmer SL. 2006. Mycorrhizae activity and diversity in conventional and organic apple orchards from Brazil. Soil Biol Biochem. 38:1831–1839. doi: 10.1016/j.soilbio.2005.12.008
- Radajewski S, Ineson P, Parekh NR, Murrell JC. 2000. Stable-isotope probing as a tool in microbial ecology. Nature. 403:646–649. doi: 10.1038/35001054
- Ramette A. 2007. Multivariate analyses in microbial ecology. FEMS Microbiol Ecol. 62:142–160. doi: 10.1111/j.1574-6941.2007.00375.x
- Rastogi G, Osman S, Vaishampayan PA, Andersen GL, Stetler LD, Sani RK. 2010. Microbial diversity in uranium mining-impacted soils as revealed by high-density 16S microarray and clone library. Microb Ecol. 59:94–108. doi: 10.1007/s00248-009-9598-5
- Rawat SR, Männistö MK, Bromberg Y, Häggblom MM. 2012. Comparative genomic and physiological analysis provides insights into the role of Acidobacteria in organic carbon utilization in Arctic tundra soils. FEMS Microbiol Ecol. 82:341–355. doi: 10.1111/j.1574-6941.2012.01381.x
- Sawada H, Kuykendall LD, Young JM. 2003. Changing concepts in the systematics of bacterial nitrogen-fixing legume symbionts. J Gen Appl Microbiol. 49:155–179. doi: 10.2323/jgam.49.155
- Schmidt PA, Bálint M, Greshake B, Bandow C, Römbke J, Schmitt I. 2013. Illumina metabarcoding of a soil fungal community. Soil Biol Biochem. 65:128–132. doi: 10.1016/j.soilbio.2013.05.014
- Strickland MS, Lauber C, Fierer N, Bradford MA. 2009. Testing the functional significance of microbial community composition. Ecology. 90:441–451. doi: 10.1890/08-0296.1
- Wang Q, Garrity GM, Tiedje JM, Cole JR. 2007. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol. 73:5261–5267. doi: 10.1128/AEM.00062-07
- Ward SE, Bardgett RD, McNamara NP, Ostle NJ. 2009. Plant functional group identity influences short-term peat land ecosystem carbon flux: evidence from a plant removal experiment. Funct Ecol. 23:454–462. doi: 10.1111/j.1365-2435.2008.01521.x
- Wardle DA. 2006. The influence of biotic interactions on soil biodiversity. Ecol Lett. 9:870–886. doi: 10.1111/j.1461-0248.2006.00931.x
- Watzinger A, Stemmer M, Pfeffer M, Rasche F, Reichenauer TG. 2008. Methanotrophic communities in a landfill cover soil as revealed by [13C] PLFAs and respiratory quinones: impact of high methane addition and landfill leachate irrigation. Soil Biol Biochem. 40:751–762. doi: 10.1016/j.soilbio.2007.10.010