
ABSTRACT
An open question with regard to the community ecology of arbuscular mycorrhizal fungi (AMF) concerns how to best amplify AMF in the soil, which contains a large proportion of DNA from AM extra-radical mycelium and spores. However, to date, a direct comparison of AMF primers for soil samples, which would systematically assess their amplification efficiency, is still missing. In our present study, we compared and characterized four widely used primer sets targeting AMF 18S rDNA or SSU-ITS-LSU rDNA from three soil samples as follows: (1) SSUmAf/LSUmAr + SSUmCf/LSUmBr, (2) GeoA2/Geo11 + NS31/AM1, (3) AML1/AML2 + NS31/AM1 and (4) AMV4.5NF/AMDGR. These primer sets were compared in terms of the proportion of Glomeromycota detected, AMF diversity and community composition. Our data revealed that the newly combined primer set 3 was the most suitable one for amplifying AMF from soil samples. It yielded the highest AMF alpha diversity, and was very specific to Glomeromycota. Primer set 2 was unable to amplify Claroideoglomus from soil 1, which was the dominant AMF clade as proved by other three primer sets. Primer set 4 demonstrated its instability among different soil samples, since the proportion of AMF in total sequences varied from 5% to 83%. Although primer set 1 showed the highest proportion of AMF (95–100%) in the soil samples, it captured the lowest AMF diversity, and the operational taxonomic units obtained by this primer set were only 36.4% of that by primer set 4. Taken together, our data suggested that AMF diversity in soil samples could be underestimated by primer set 1, 2 and 4. Our result confirmed the important role of the choice of AMF primers for analyzing AMF communities in soil and explored the most suitable one for amplifying AMF from soil samples.
Introduction
Arbuscular mycorrhizal fungi (AMF) form a symbiosis with approximately 80% of terrestrial vascular plants in the world (Brundrett Citation2009). These associations can affect global carbon (C), phosphorus (P) and nitrogen (N) cycling (Smith & Read Citation2008). Moreover, they can also affect plant community structure by their key role at the soil–plant interface (van der Heijden et al. Citation1998; Stein et al. Citation2009) and improve host plant pathogen defense (Zhang et al. Citation2008) and drought stress tolerance (Borowicz Citation2010).
Despite the important role of AMF in terrestrial ecosystems, little is known about their biodiversity and distribution patterns across different environments. Most studies on AMF assemblages, in which either morphological or molecular methods have been used, have exclusively focused on intraradical fungi or fungal spores (Hempel et al. Citation2007; Li et al. Citation2009; Dumbrell et al. Citation2011; Öpik et al. Citation2013). Kohout et al. (Citation2014) compared some commonly used primer sets for AMF from root samples. However, the specific study targeted root material and did not consider soil samples where the extra-radical mycelium may represent a feature responsible for many of the above-referenced functions. Moreover, root-associated AMF communities could underestimate alpha diversity because of host preferences, such that different plant species choose different AMF communities (Vandenkoornhuyse et al. Citation2003; Alguacil et al. Citation2009; Öpik et al. Citation2009). It is striking that there are no AMF sequence types detected in the roots of different plants from the same place that have not been found in the soil – but not necessarily vice versa (Hempel et al. Citation2007; Alguacil et al. Citation2011). Therefore, it may be preferable to assess AMF biodiversity in an ecosystem when DNA is directly extracted from soil, which contains a large proportion of DNA from AM extra-radical mycelium and spores. Therefore, molecular methods targeting AMF in the soil are important and need to be subjected to rigorous testing. However, to date, only limited number of studies detected AMF biodiversity from soil samples with these primers, and found that some of the primers have a high proportion of non-AMF through polymerase chain reaction (PCR) amplifying (Lumini et al. Citation2010). A direct comparison of these primers, which would systematically assess their amplification efficiency, is still missing. The aim of our study was therefore to identify the most suitable primers for AMF detecting on soil samples. Moreover, we also aimed to characterize and assess the specificity and selectivity of commonly used primer sets.
In our present study, we compared and characterized three widely used PCR primer sets targeting the AMF 18S rDNA (GeoA2/Geo11, AML1/AML2, NS31/AM1 and AMV4.5NF/AMDGR) and one primer set targeting the SSU-ITS-LSU rDNA (SSUmAf-LSUmAr/SSUmCf-LSUmBr) from three soil samples which have obvious difference in their characters (). In order to verify the primer stability in different soil samples, we used clone libraries of AMF from three soil samples, which were apparently different in terms of soil properties, vegetation and climatic conditions (Table S1). In order to comprehensively analyze the AMF community structure and detect as much AMF diversity as possible, we particularly compared the specificity and selectivity of these primer sets in the following features: the proportion of Glomeromycota detected, AMF diversity and community composition.
Table 1. Primer sets for 18S rDNA of AMF.
Materials and methods
Soil samples and DNA extraction
Soil samples were collected from three different grasslands (0–20 cm) with a gradient of degradation. Soil 1, the most degraded sample, was obtained from Silver City, Gansu Province (36.991650N, 104.867567E); soil 2 was from Ping An City, Qinghai Province (36.450333N, 102.070750E); the sample from the least degraded site was soil 3 obtained from Guanghe County, Gansu Province (35.448850N, 103.468683E) (Table S1). The field study did not involve any privately owned land or protected area of land (such as national park), and the sampling did not involve any endangered or protected species. Therefore, no specific permits were required for the described field studies. For each soil sample, three replicates of genomic DNA were extracted from the same amount of fresh soil (500 mg) by using a FastDNA Spin kit for soil (Bio 101, Vista, CA, USA) according to the manufacturer's recommendations.
PCR primer sets and amplification
In order to select suitable primer sets for detecting AMF diversity from soil samples, soil clone library analyses were used in our study. lists the primers targeting AMF ribosomal DNA, and shows the position of these primers on rDNA of AMF. Four primer sets were constructed by these primers as follows: (1) primer set 1 consisted of four primers used for nested PCR: SSUmAf/LSUmAr + SSUmCf/LSUmBr; (2) primer set 2 consisted of GeoA2/Geo11 and NS31/AM1; (3) primer set 3 consisted of AML1/AML2 and NS31/AM1; and (4) primer set 4 was made up of AMV4.5NF/AMDGR.
Figure 1. Positions of the primers on rDNA of AMF.
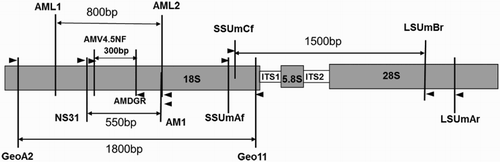
Nested PCR was performed with primer sets 1, 2 and 3. The first PCR was performed within a 25-µL reaction system containing 2.5 µL 10× PCR buffer, 1.5 mM MgCl2, 0.2 mM dNTPs (deoxy-ribonucleoside triphosphate), 0.5 mM of each primer, 1.5 U Ex Taq polymerase (Takara, Japan), 1 µL DNA template and 7.5 μg bovine serum albumin. Briefly, following a denaturation step at 94°C for 3 min, the amplification was carried out with 35 cycles at a melting temperature of 94°C for 30 s, an annealing temperature of 58°C for 1 min and an extension temperature of 72°C for 1.5 min. Finally, an extension step at 72°C for 10 min was followed. For primer set 4, the PCR condition was as same as above except for the extension time (45 s). Subsequently, single-banded amplicon obtained from the first PCR amplification was diluted at a ratio of 1:10, and the diluted PCR product was then used as the template in second PCR with the same condition except for the extension time (45 s). All the PCR products from the same soil sample were mixed into one composite PCR products, afterward, PCR products were separated by gel electrophoresis (1.5% agarose in 0.5× TAE [Tris/Acetate/EDTA]), bands were excised, and purified using the QIAEX II Gel Extraction Kit (QIAGEN Sciences, Germantown, MD, USA).
Cloning and sequencing analysis
The PCR products were cloned into pMD18-T vector (Promega, Madison, WI, USA) and then transformed into competent cells of Escherichia coli DH-5a (Takara, Japan) strain following the manufacturer's protocols. Approximately 60 positive clones in each library were screened by restriction fragment length polymorphism (RFLP) with restriction endonucleases of HinfI and MboI (Fermentas, Hanover, MD, USA) according to the manufacturer's protocols. The clone representative of each RFLP type detected in each library was selected and sequenced using vector primers on an ABI 3730 automatic sequencer (SinoGenoMax, China).
Statistical analysis
In order to determine operational taxonomic units (OTUs), all the sequences were analyzed using the computer program DOTOUR (Schloss & Handelsman Citation2005) with an evolutionary distance of 0.03. The OTUs defined at 97% sequence similarity (Dumbrell et al. Citation2011) were used to perform rarefaction analysis and calculate the diversity (Shannon) indices. Rarefaction curves were constructed with the software EstimateS 8.00 (Buée et al. Citation2009). The Shannon indices of diversity were calculated from the equation: H = −∑pi(ln pi), where pi is the proportion of sequences belonging to each OTU relative to the total number of sequences (Pivato et al. Citation2007; Alguacil et al. Citation2011). Sequences were aligned by Clustal W, and phylogenetic trees were constructed using MEGA version 4.0 (Tamura et al. Citation2007) and subjected to phylogenetic inference using the neighbor-joining algorithm followed by 1000 cycles of bootstrap sampling. The V4 region of 18S rDNA sequences was used to construct the phylogenetic tree of primer sets 1–3. For primer set 4 (SSUmAf/LSUmAr + SSUmCf/LSUmBr), we only selected the D1 and D2 regions of LSU rDNA (800 bp) to build phylogenetic tree with a similarity value of 95% (Li et al. Citation2009), due to the relatively limited long AMF sequences in the Genbank. Sequences generated in this study were registered in Genbank under the accession numbers from JQ218147 to JQ218225.
Results
AMF community richness
In our present study, the 18S rDNA region was amplified by primer sets 2, 3 and 4, while the 28S rDNA region was obtained by primer set 1. The rarefaction curve of each library tended to approach the saturation plateau (Figure S1). It revealed that the clone number for primer set 1 (50 clones) and that for primer sets 2, 3 and 4 (60 clones) provided a reasonable coverage of AMF diversity.
Comparison of AMF proportion of available 18S rDNA sequences in clone libraries
Using primer sets 2, 3 and 4, we obtained 58, 60 and 60 sequences of 18S rDNA from soil sample 1; 60, 56 and 57 sequences for soil sample 2; 60, 59 and 58 sequences for soil sample 3, respectively. Meanwhile, 47, 50 and 48 sequences of SSU-ITS-LSU rDNA were obtained from soil samples 1, 2 and 3 using primer set 1, respectively.
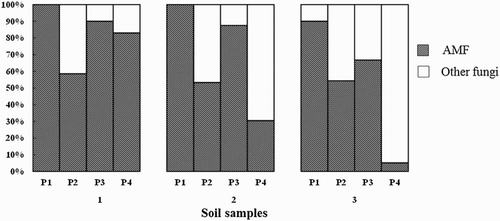
In spite of the AMF primer specificity, other sequences outside the Glomeromycota were detected. shows the proportion of AMF sequences obtained with each primer set in the different soil samples. Among these primer sets, primer set 1 demonstrated the highest AMF sequence proportion (95–100%) in all soil samples. The AMF sequence proportion of primer set 3 (66.7–90%) was higher than that of primer set 2 (54.38–87.50%). In soil 1, the AMF sequence proportion of primer set 3 (90%) was much higher than that of primer set 2 (58.62%). The same trend was observed in soil 2, where the AMF sequence proportion of primer sets 3 and 2 was 87.5% and 53.3%, respectively. For these three primer sets, there was no obvious difference in terms of the AMF sequence proportion among soil samples. On the contrary, the proportion of AMF sequences was significantly different from sample to sample using primer set 4. For example, the AMF proportion of soil 1 was 83% using primer set 4, whereas it was only 30.5% and 5% from soils 2 and 3, respectively.
Figure 2. The proportion of AMF sequences obtained from primer set 1, 2, 3 and 4 (P1, P2, P3 and P4) in three soil samples.
Comparison of AMF diversity
shows a comparison of OTUs0.03 and Shannon's index (H) among three soil samples using the four primer sets. The obtained AMF sequences were grouped into OTUs at the similarity level of 97%.
Figure 3. The OTUs and Shannon's index (H) of the Glomeromycota in three soil samples using the four primer sets (P1, P2, P3 and P4).
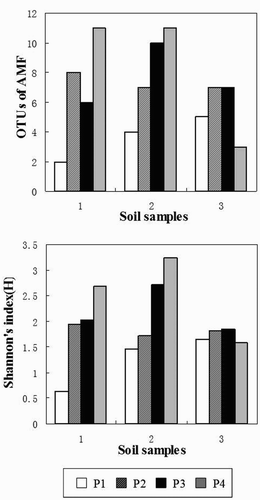
The number of AMF OTUs0.03 was highly variable among the four primer sets within the same soil sample. For example, the number of OTUs0.03 of soils 1 and 2 was highest using primer set 4; was intermediate using primer sets 2 and 3; was lowest using primer set 1. Interestingly, the number of OTUs0.03 of soil 3 was highest using primer sets 2 and 3; was intermediate using primer set 1; was lowest using primer set 4. Additionally, there was no difference between the number of OTUs0.03 for primer sets 2 and 3 in the soil samples.
There was a slight difference in terms of OTUs0.03 among the three soil samples using the same primer set except for primer set 4, for which OTUs0.03 of soil 3 was lower than that of soils 1 and 2.
The Shannon's index (H) displayed a similar trend as the OTUs0.03 except that primer set 3 yielded a higher Shannon's index (H) than primer set 2 in all soil samples.
Comparison of AMF community structure
Cluster analyses based on the phylogenetic tree with respect to the similarity of AMF communities were divided into nine distinct clusters in soil 1 using primer sets 2, 3 and 4 (Figure S2). In , set 2 shows the percentage of 18S rDNA sequences in each cluster. It reflects the divergence in the specificity of primer sets on AMF. For example, sequences of clusters 3 and 5 could be only amplified with primer set 4, while cluster 8 was the specific sequence for primer set 2. In addition, primer sets 3 and 4 have the same dominant clusters while primer set 2 was the different one. More specifically, the dominant types in primer sets 3 and 4 were clusters 7, 9 and 1, but the most abundant types for primer set 2 were clusters 8 and 4. Moreover, the primer set 2 was cluster 8-specific, whereas this cluster was not successfully amplified by primer sets 3 and 4. On the other hand, primer sets 3 and 4 could specifically amplify cluster 9, which was identified as the group B (Claroideoglomus) of AMF, while this cluster was not amplified by primer set 2. It is important to note that the group B could be considered as the most abundant type in this soil sample because 84% of the total sequences amplified by primer set 1 belonged to group B (Figure S3).
Figure 4. Proportion of 18S rDNA sequences in each cluster using primer sets 2, 3 and 4.
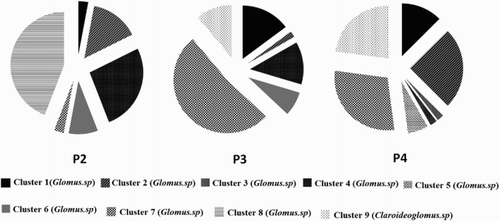
Taken together, although each primer set had different specificity for AMF, primer sets 3 and 4 were in well agreement with the community structure of AMF, whereas the results from using primer set 2 diverged from that of other primer sets.
Discussion
The primer set 3 consisted of AML1/AML2 and NS31/AM1, and was found to work best with regard to AMF amplification from soils in terms of proportion of Glomeromycota, AMF diversity and community composition. More specifically, the proportion of AMF taxa amplified (66.7–90%) did not change much across soil samples and was much higher than that of other primer sets (). Moreover, the community structure of the samples obtained from this primer set were in correspondence with those of primer sets except for the primer set 2, which has more mismatches compared with other primer sets and could not amplify Claroideoglomus in the soil. To our knowledge, we, for the first time, used these two primer pairs together to characterize the soil-associated AMF communities.
In previous studies, the primer pair AML1/AML2 has been found to be more specific to the Glomeromycota, and it provides better coverage across the Glomeromycota, including Glomus group B (Claroideoglomus), group C (Diversisporaceae), Archaeosporaceae and Paraglomeraceae (Lee et al. Citation2008; Alguacil et al. Citation2011). On the contrary, the main limitation of the primer pair NS31/AM1 is that it can exclude sequences from these types (Helgason et al. Citation1999; Öpik et al. Citation2009). Little is known about the diversity and distribution of these sequence types when amplified by the combined primer set (AML1/AML2 + NS31/AM1). In the present study, we found that the specific combined primer sets were able to obtain sequences from both Glomus group B (Claroideoglomus) and group C (Diversisporaceae) (Figure S4). However, the sequence belongs to Claroideoglomus could not be found by primer set 4, and sequence of Diversisporaceae could not be detected by primer set 2 (Figure S4). Furthermore, previous studies have also shown the rarity of Archaeosporaceae and Paraglomeraceae groups in natural area (Gai et al. Citation2006; Santos et al. Citation2006; Lee et al. Citation2008). Therefore, AMF diversity could not be significantly underestimated by using the NS31/AM1 region. Instead, the primer set 3 can provide more data of Glomeromycota natural community in the 18S rDNA region than other primers.
In the present study, we showed that the primer set 2, including GeoA2/Geo11 and NS31/AM1, was not suitable for amplification of AMF from soils due to the following reasons. Firstly, the AMF proportion was not high enough (53.30–58.60%) (). Therefore, many non-specific sequences could be amplified by this primer set, weakening the quality of sequencing. On the other hand, primer set 2 showed more bias in the specificity of AMF compared with other primer sets. In our present study, primer set 2 was unable to amplify group B in soil 1, which was the dominant type of AMF proved together by other primer sets. On the contrary, we found that the primer set 2 was cluster 8-specific, and nearly 44.12% of total sequences belonged to this type, whereas this cluster was not successfully amplified by primer sets 3 and 4 (). It indicated that primer set 2 could give a completely different community structure of AMF.
We also used the primer pair NS31/AM1 in a single treatment. However, we only obtained faint bands irrespective of PCR conditions. It indicated that the efficiency of this primer pair was much lower than that of the other primers in the soil sample, which was consistent with previous findings (Kowalchuk et al. Citation2002). In addition, the recent 454 pyrosequencing has proved that only 37.59% of total sequences amplified by this primer pair belong to Glomeromycota (Lumini et al. Citation2010). Therefore, we abandoned further pursuing this primer set.
In this study, we used a set of PCR primers (SSUmAf/SSUmCf-LSUmAr/LSUmBr) as the ‘standard’ means of assessing AMF diversity. Because this primer set has been shown to be suitable for amplifying DNA from the whole Glomeromycota and had the highest specificity compared to other primers in plants and soils, leading to its role as the basis for AMF DNA barcoding (Wang et al. Citation2011; Kohout et al. Citation2014). However, little is known about the specificity, selectivity or stability of this primer set for DNA from soil. We asked if primer set 1 was also suitable for AMF diversity analysis from different soil samples. There were two types of AMF using the primer set 1 in soil 1, whereas there were more than six types using other primer sets. In addition, there were many more OTUs and higher diversity by other primer sets than primer set 1. Therefore, in our present study, the primer set 1 was not suitable for AMF amplification from soil sample.
Primer set 4 has lately been used in soil samples because it has high proportion of AMF in soil samples (76.4%) and can amplify sequences from a broad spectrum of Glomeromycota (Glomerales, Diversisporales, Archaeosporales and paraglomerales) (Lumini et al. Citation2010). Unfortunately, this primer set showed a huge difference among three soil samples in our present study. The proportion of AMF varied from 5% to 83%, leading to an unstable value of OTUs varying from 4 to 12 (Figures and ). Therefore, in spite of this primer set having amplified a similar selection of AMF as primer sets 3 and 1, it was still not suitable for soil sample mainly because of it has great instability among different soil samples. However, this primer set showed higher diversity than the other primers. Therefore, it could be used when no huge difference existed among the soil samples.
In conclusion, we compared four primer sets in three soil samples, and our data suggested that the primer set 3 was the most suitable one due to its high specificity, high proportion of target sequences, and less amplification bias, probably reflecting more closely the real community structure. It is indisputable that each primer set has a different amplification bias because of different affinities for specific fungal taxa. We also discussed the advantages and disadvantages of these primers. Therefore, our results provided useful information on primer selection for the study of AMF diversity at the ecosystem level.
Supplemental file
Download MS Word (382.5 KB)Acknowledgements
We are grateful to Dr Stavros D. Veresoglou for valuable comments on an earlier version of this manuscript and grateful to Zhou Wen-Ping for help in the laboratory.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Alguacil MM, Roldán A, Torres MP. 2009. Assessing the diversity of AM fungi in arid gypsophilous plant communities. Environ Microbiol. 11:2649–2659. doi: 10.1111/j.1462-2920.2009.01990.x
- Alguacil MM, Torres MP, Torrecillas E, Díaz G, Roldán A. 2011. Plant type differently promote the arbuscular mycorrhizal fungi biodiversity in the rhizosphere after revegetation of a degraded, semiarid land. Soil Biol Biochem. 43:167–173. doi: 10.1016/j.soilbio.2010.09.029
- Borowicz VA. 2010. The impact of arbuscular mycorrhizal fungi on strawberry tolerance to root damage and drought stress. Pedobiologia. 53:265–270. doi: 10.1016/j.pedobi.2010.01.001
- Brundrett MC. 2009. Mycorrhizal associations and other means of nutrition of vascular plants: understanding the global diversity of host plants by resolving conflicting information and developing reliable means of diagnosis. Plant Soil. 320:37–77. doi: 10.1007/s11104-008-9877-9
- Buée M, Reich M, Murat C, Morin E, Nilsson RH, Uroz S, Martin F. 2009. 454 pyrosequencing analyses of forest soil reveal an unexpected high fungal diversity. New Phytol. 184:449–456. doi: 10.1111/j.1469-8137.2009.03003.x
- Dumbrell AJ, Ashton1 PD, Aziz1 N, Feng G, Nelson1 M, Dytham C, Fitter AH, Helgason T. 2011. Distinct seasonal assemblages of arbuscular mycorrhizal fungi revealed by massively parallel pyrosequencing. New Phytol. 3:794–804. doi: 10.1111/j.1469-8137.2010.03636.x
- Gai JP, Christie P, Feng G, Li XL. 2006. Twenty years of research on community composition and species distribution of arbuscular mycorrhizal fungi in China: a review. Mycorrhiza. 16:229–239. doi: 10.1007/s00572-005-0023-8
- van der Heijden MGA, Klironomos JN, Ursic M, Moutoglis P, Streitwolf-Engel R, Boller T, Wiemken A, Sanders IR. 1998. Mycorrhizal fungal diversity determines plant biodiversity, ecosystem variability and productivity. Nature. 396:69–72. doi: 10.1038/23932
- Helgason T, Daniell TJ, Husband R, Fitter AH, Young JPW. 1998. Ploughing up the wood-wide web? Nature. 8:394–431.
- Helgason T, Fitter AH, Young JPW. 1999. Molecular diversity of arbuscular mycorrhizal fungi colonising Hyacinthoides non-scripta (bluebell) in a seminatural woodland. Mol Ecol. 8:659–666. doi: 10.1046/j.1365-294x.1999.00604.x
- Hempel S, Renker C, Buscot F. 2007. Differences in the species composition of arbuscular mycorrhizal fungi in spore, root and soil communities in a grassland ecosystem. Environ Microbiol. 9:1930–1938. doi: 10.1111/j.1462-2920.2007.01309.x
- Kohout P, Sudová R, Janousková M, Ctvrtlíková M, Hejda M, Pánková H, Slavíková R, Štajerová K, Vosátka M, Sýkorová Z. 2014. Comparison of commonly used primer sets for evaluating arbuscular mycorrhizal fungal communities: is there a universal solution? Soil Biol Biochem. 68:482–493. doi: 10.1016/j.soilbio.2013.08.027
- Kowalchuk GA, De Souza FA, van Veen JA. 2002. Community analysis of arbuscular mycorrhizal fungi associated with Ammophila arenaria in Dutch coastal sand dunes. Mol Ecol. 11:571–581. doi: 10.1046/j.0962-1083.2001.01457.x
- Krüger M, Stockinger H, Krüger C, Schüßler A. 2009. DNA-based species level detection of Glomeromycota: one PCR primer set for all arbuscular mycorrhizal fungi. New Phytol. 183:212–223. doi: 10.1111/j.1469-8137.2009.02835.x
- Lee J, Lee S, Yong JPW. 2008. Improved PCR primers for the detection and identification of arbuscular mycorrhizal fungi. FEMS Microbiol Ecol. 65:339–349. doi: 10.1111/j.1574-6941.2008.00531.x
- Li LF, Li T, Zhang Y, Zhao ZW. 2009. Molecular diversity of arbuscular mycorrhizal fungi and their distribution patterns related to host-plants and habitats in a hot and arid ecosystem, southwest China. FEMS Microbiol Ecol. 3:418–427.
- Lumini E, Orgiazzi A, Borriello R, Bonfante P, Bianciotto V. 2010. Disclosing arbuscular mycorrhizal fungal biodiversity in soil through a land-use gradient using a pyrosequencing approach. Environ Microbiol. 12:2165–2179.
- Öpik M, Metsis M, Daniell TJ, Zobel1 M, Moora1 M. 2009. Large-scale parallel 454 sequencing reveals host ecological group specificity of arbuscular mycorrhizal fungi in a boreonemoral forest. New Phytol. 18:424–434. doi: 10.1111/j.1469-8137.2009.02920.x
- Öpik M, Zobel M, Canter JJ, Davison J, Facelli JM, Hiiesalu I, Jairus T, Kalwij JM, Koorem K, Leal ME. 2013. Global sampling of plant roots expands the described molecular diversity of arbuscular mycorrhizal fungi. Mycorrhiza. 23:411–430. doi: 10.1007/s00572-013-0482-2
- Pivato B, Mazurier S, Lemanceau P, Siblot S, Berta G, Mougel C, Van Tuinen D. 2007. Medicago species affect the community composition of arbuscular mycorrhizal fungi associated with roots. New Phytol. 176:197–210. doi: 10.1111/j.1469-8137.2007.02151.x
- Santos JC, Finlay RD, Tehler A. 2006. Molecular analysis of arbuscular mycorrhizal fungi colonising a semi-natural grassland along a fertilisation gradient. New Phytol. 172:159–168. doi: 10.1111/j.1469-8137.2006.01799.x
- Sato K, Suyama Y, Saito M, Sugawara K. 2005. A new primer for discrimination of arbuscular mycorrhizal fungi with polymerase chain reaction-denature gradient gel electrophoresis. Grassl Sci. 51:179–181. doi: 10.1111/j.1744-697X.2005.00023.x
- Schloss PD, Handelsman J. 2005. Introducing DOTUR, a computer program for defining operational taxonomic units and estimating species richness. Appl Environ Microbiol. 71:1501–1506. doi: 10.1128/AEM.71.3.1501-1506.2005
- Schwarzott D, Schüßler A. 2001. A simple and reliable method for SSU rRNA gene DNA extraction, amplification, and cloning from single AM fungal spores. Mycorrhiza. 10:203–207. doi: 10.1007/PL00009996
- Simon LM, Lalonde TD, Bruns TD. 1992. Specific amplification of 18S fungal ribosomal genes from vesicular arbuscular endomycorrhizal fungi colonising roots. Appl Environ Microbiol. 58:291–295.
- Smith SE, Read DJ. 2008. Mycorrhizal symbiosis. San Diego, CA: Academic.
- Stein C, Rißmann C, Hempel S, Renker C, Buscot F, Prati D, Auge H. 2009. Interactive effects of mycorrhizae and a root hemiparasite on plant community productivity and diversity. Oecologia. 159:191–205. doi: 10.1007/s00442-008-1192-x
- Tamura K, Dudley J, Nei M, Kumar S. 2007. MEGA4: molecular evolutionary genetics analysis (MEGA) software version 4.0. Mol Biol Evol. 24:1596–1599. doi: 10.1093/molbev/msm092
- Vandenkoornhuyse P, Ridgway KP, Watson IJ, Fitter AH, Young JPW. 2003. Co-existing grass species have distinctive arbuscular mycorrhizal communities. Mol Ecol. 12:3085–3095. doi: 10.1046/j.1365-294X.2003.01967.x
- Wang YT, Huang YL, Qiu Q, Xin GR, Yang ZY, Shi SH. 2011. Flooding greatly affects the diversity of arbuscular mycorrhizal fungi communities in the roots of wetland plants. PLoS ONE. 6(9). doi:10.1371/journal.pone.0024512
- Zhang LD, Zhang JL, Christie P, Li XL. 2008. Pre-inoculation with arbuscular mycorrhizal fungi suppresses root knot nematode (Meloidogyne incognita) on cucumber (Cucumis sativus). Biol Fert Soils. 45:205–211. doi: 10.1007/s00374-008-0329-8