
Abstract
Upon exit from the endoplasmic reticulum (ER), the nascent polypeptides of secretory proteins undergo sorting events. If properly folded, they are directly or indirectly recognized by the coat proteins of budding vesicles for forward transport, while unfolded or misfolded proteins are retained in the ER by a quality control mechanism. To gain insight into the interplay between ER export and ER quality control, we fused a secretory protein invertase to the C-terminus of mutated carboxypeptidase Y (CPY*), a model ER-associated degradation (ERAD) substrate in Saccharomyces cerevisiae. This substrate, designated CPY*-Inv, was largely exported from the ER, although it was fully recognized by the ERAD-related lectin, Yos9, and hence degraded by the ERAD when it remained in the ER. CPY*-Inv relied primarily on the p24 complex, a putative ER export receptor for invertase, for escape from ERAD, suggesting that the ERAD and the ER export of soluble secretory proteins are competitive.
Graphical Abstract
In the endoplasmic reticulum (ER), ERAD machineries and export receptors compete secretory proteins to determine whether they are degraded or exported from the ER.

Newly synthesized secretory proteins translocate into the endoplasmic reticulum (ER) where they undergo a sorting event, advancing to the Golgi apparatus or staying in the ER.Citation1,Citation2) Transmembrane proteins destined for the Golgi apparatus or beyond are incorporated into the budding vesicle via interactions between coat protein complex II (COPII) proteins and exit signals displayed on the cytosolic surface of cargo proteins. Loading of luminal proteins into the COPII vesicle relies on transmembrane cargo receptors whose cytosolic tails are associated with coat proteins. Although highly expressed proteins can be incorporated in the COPII vesicle by bulk flow export, those that do not contain ER exit signals mostly remain in the ER. Unfolded or unassembled proteins remain in the ER until they achieve the properly folded or oligomeric state through the ER-assisted folding (ERAF) pathway that consists of a variety of ER chaperones and foldases.Citation3) Despite a substantial commitment of ERAF, some proteins are terminally misfolded and eventually degraded by a multi-step disposal pathway called ER-associated degradation (ERAD).Citation4,Citation5) ERAD can be classified into three pathways according to the site of misfolding lesions. Transmembrane proteins with lesions on the cytoplasmic face, in the membrane-spanning domain, and in a luminal domain are degraded by the ERAD-C, ERAD-M, and ERAD-L pathways respectively.Citation6) Soluble secretory proteins are exclusively inspected by the ERAD-L checkpoint, by which misfolded proteins are recognized by ER lectins Yos9 and retrotranslocated to the cytoplasm in an ubiquitin ligase Hrd1-dependent manner for degradation.Citation7) One of the best-characterized ERAD-L substrates is mutated carboxypeptidase Y (CPY*), a mutant version of yeast carboxypeptidase Y (CPY), whose glycine residue at the 255th position is altered to arginine. Retention of misfolded proteins in the ER can be attributed to binding of ERAD machinery such as molecular chaperones and lectins to exposed hydrophobic patches and immature N-glycans on misfolded proteins, respectively.Citation6–Citation9) Proteins can be in equilibrium between the unfolded and the folded state, and the lower their thermodynamic stability, the higher the chance of exposing the hydrophobic regions where chaperones bind. Another aspect of ER retention of misfolded proteins is loss of the ER exit signal. The binding of chaperones to ER exit signals on proteins, whether inside or outside the ER, can prevent their recognition by corresponding receptors or the COPII export machinery. Otherwise, failure to acquire the native conformation can also lead to the destruction of three-dimensionally presented exit signals on the surface of a secretory protein. Since two mechanisms can occur simultaneously in the retention of a misfolded protein that has a simple single domain, it is difficult to determine which mechanism is dominant.
To date, the theory called FoldEx (folding for export) model has been proposed. It evaluates the efficiency of ER export of secretory proteins from the ER by combining protein folding energetics and a simplified Michaelis–Menten treatment of a protein homeostasis network consisting of the ERAF, ERAD, and ER export pathways.Citation10) Regarding the relationship between folding energetics and protein export, there is much experimental evidence that the efficiency of secretion of proteins is correlated with their thermodynamic stability and/or folding rate.Citation3,Citation11–Citation14) On the other hand, only a few examples are known for the kinetic study of protein sorting to the three pathways.Citation15–Citation17) Most of the substrates used in these analyses were artificial type I transmembrane proteins comprised of a luminal misfolded domain, a membrane-spanning domain, and a cytoplasmic tail with defined ER exit signals, which indicated that the ER export and ERAD are competitive.
In this study, we constructed a luminal multidomain substrate containing both folded and misfolded domains expressed in yeast cells at a physiological level. This substrate exhibited the characteristics of a potential ERAD substrate, but was exported from the ER without being a target of ERAD by the ER exit signal coded by the tertiary structure of the folded domain.
Materials and methods
Strains and media
The Saccharomyces cerevisiae strains used in this study are listed in Supplemental Table 1 (see doi:10.1080/09168451.2014.877185). Gene disruption was performed by the method described by Gueldener et al.Citation18) The entire open reading frame was replaced by the Escherichia coli kanr gene, Kluyveromyces lactis LEU2, or Schizosaccharomyces pombe his5+ with PCR primers containing approximately 50 nucleotides that were identical to the regions flanking the open reading frame. For PEP4 disruption, a DNA fragment of the pep4 allele partially disrupted by the LEU2 gene was used. A 9x myc tag was inserted in front of the ER retention signal (His-Asp-Glu-Leu) of Yos9, as described by Gauss et al.Citation19) Cells were grown at 30 °C in YPD (1% yeast extract, 2% peptone, and 2% glucose) or in synthetic complete (SC) medium (0.67% yeast nitrogen base without amino acids, 2% glucose, and 0.2% dropout mix), as described by Amberg et al.Citation20)
Plasmid construction
The plasmids used in this study are listed in Supplemental Table 2. Oligonucleotides 5′-AAAAGAATTCAAGGCTGCTAGCTCTGCATCAATGACAAACGAAACTAGC-3′ and 5′-ATAAGAATTCTTTTACTTCCCTTACTTGGA-3′ were used in the PCR procedure to obtain a DNA fragment coding for invertase that lacked signal sequence and termination codon. The fragment was ligated to an EcoRI-digested pRS316-3HA-ADHt to create pRS316-SUC2-3HA-ADHt (pKS2). Next, the prc1-1 allele, which codes for the mutant carboxypeptidase Y (CPY) whose glycine-255 was replaced with arginine, was PCR-amplified using primers 5′-ATCGGCGGCCGCATATGATGATACATATGTT-3′ and 5′-ACACACTAGTTAAGGAGAAACCACCGTGGA-3′, and pDN436 (a gift from Dr. D.T. Ng, National University of Singapore) as template DNA.Citation21) The wild-type PRC1 allele was amplified by PCR with the same primers. These fragments, both of which included the PRC1 promoter region (about 500 bp upstream of the start codon), were further ligated to a NotI/SpeI digested pKS2 to generate pKS3 (CPY*-Inv) and pKS4 (CPY-Inv), respectively. The glutamic acid residue at position 486 in the invertase moiety was replaced with lysine by site-directed mutagenesis on pKS3 with a QuikChange® Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA) to generate pKS5.
Cycloheximide chase assay
Cycloheximide (CHX) chase assay was performed as described previously.Citation22) In brief, after cells were grown to the middle-log phase (OD600 = 1.0–1.2) in SC medium at 30 °C, CHX was added to the medium at a final concentration of 0.2 mg/mL. Following incubation at 30 °C for the indicated durations, approximately 1.2 OD600 units of cells were harvested and suspended in 50 μL of SDS sample buffer (62.5 mm Tris–HCl pH 6.8, 2% SDS, 10% glycerol, 1 mM NaN3, 5% 2-mercaptoethanol, and 0.0025% bromophenol blue). To inactivate proteases, cells were rapidly boiled for 2 min and then lysed with glass beads. SDS sample buffer was added to the cell lysate to a final concentration of 0.02 OD600/μL. After centrifugation at 1000 × g for 2 min, the supernatant was collected and stored at −20 °C until loading to SDS-PAGE. When deglycosylation was necessary, 50 μL of endoglycosidase H (Endo H) solution (75 mM sodium citrate pH 5.5, and 200 units of Endo Hf from New England Biolabs Japan, Tokyo, Japan) was added to 25 μL of the above protein extract (equivalent to 0.5 OD600 units of cells) and then incubated at 37 °C overnight. After the addition of 25 μL of 4X SDS sample buffer (0.25 M Tris–HCl pH 6.8, 8% SDS, 40% glycerol, 20% 2-mercaptoethanol, and 0.01% bromophenol blue), the samples were boiled for 2 min and stored at −20 °C. For immunoblot analysis, 0.15 OD600 cell equivalents were resolved by SDS-PAGE, transferred to a PVDF membrane, and then probed with anti-CPY antiserum (Shibayagi, Shibukawa, Japan) and anti-3-phosphoglycerate kinase (PGK) antibody (Life Technologies, Carlsbad, CA). Immunoreactive protein bands were detected with horseradish peroxidase-conjugated goat anti-rabbit IgG (Bio-Rad laboratories, Hercules, CA) and chemiluminescence with Chemi-Lumi One L (Nacalai Tesque, Kyoto, Japan). The amounts of protein were quantified with ImageJ software (http://rsb.info.nih.gov/ij/), and were plotted with the amount at 0 min as 100%. The half-life (t1/2) of each substrate was calculated on the basis of semi-log plots. Error bars represent standard deviations of independent experiments repeated at least three times.
Protease sensitivity assay
YKS30 cells (prc1∆hrd1∆pep4∆) carrying pMH1 (CPY), pKS1 (CPY*), pKS4 (CPY-Inv), and pKS3 (CPY*-Inv) were grown to the early log phase (OD600 = approximately 1) in SC-Ura medium at 30 °C. Cells equivalent to 20 OD600 were spheroplasted in 250 μL of spheroplasting buffer (1.2 M sorbitol, 50 mM Tris–HCl, pH 7.5) containing 500 U of recombinant β-1,3 glucanase from Oerskovia xanthineolytica (Wako Pure Chemical Industries, Osaka, Japan) for 1 h at 30 °C. After washing with spheroplasting buffer, the spheroplasts were solubilized in 400 μL of 0.1 M sodium phosphate (pH 7.0) containing 1% Triton X-100 by incubation for 10 min on ice. The resulting cell lysate was divided into two aliquots of 200 μL each to which 50 μL of 20 μg/mL bovine trypsin (at a final concentration of 4 μg/mL) or 0.1 M sodium phosphate (pH 7.0) was added. After incubation at 37 °C, 50 μL (equivalent to 2 OD600) of the reaction mixture was removed, and the reaction was terminated by boiling in 2X SDS sample buffer (0.125 M Tris–HCl pH 6.8, 4% SDS, 20% glycerol, 2 mM NaN3, 10% 2-mercaptoethanol, and 0.005% bromophenol blue). Before SDS-PAGE, these samples were treated with Endo Hf as described above. For immunoblot analysis, 0.1 OD600 cell equivalents were used.
Co-immunoprecipitation
Preparation of microsomes and the subsequent native co-immunoprecipitation were performed as described previously, with slight modifications.Citation23) In brief, YTS346 cells harboring a plasmid expressing the 3HA-tagged CPY variant were grown to the early log phase, and those equivalent to 50 OD600 were harvested by centrifugation at 2000 × g for 2 min. After they were washed in distilled water, the cells were resuspended in 0.5 mL of lysis buffer (50 mM Tris–HCl pH 7.5, 150 mM sodium chloride, 2 mM EDTA, protease inhibitor cocktail from Roche Diagnostics Japan, Tokyo, Japan, 2 mM PMSF, 2 μg/mL of pepstatin A) and disrupted by bead beating with 0.5-mm zirconium beads on a Multi-beads shocker (Yasui Kikai, Osaka, Japan). Cell debris was removed by centrifugation at 800 × g over 5 min, and the resulting supernatant (0.4 mL) was pooled and combined with repeated bead-wash (0.3 mL × 2) to obtain total cell lysate (TCL). TCL (900 μL) were further subjected to ultracentrifugation (30,000 × g, 30 min) to obtain microsomes. The resulting pellet was resuspended in 0.5 mL of Tris-IP buffer (50 mM Tris–HCl pH 7.5, 150 mM sodium chloride, 2 mM EDTA, 1% Triton X-100, protease inhibitor cocktail, 2 mM PMSF, and 2 μg/mL of pepstatin A) and incubated on ice for 30 min. After ultracentrifugation (30,000 × g, 10 min), the supernatant was collected as a microsome fraction. For immunoprecipitation, 0.5 mL of Tris-IP buffer and 2 μL of anti-HA antibody (Santa Cruz Biotechnology, Santa Cruz, CA) were added to 0.4 mL of the supernatant and this was incubated at 4 °C for 2 h. The immunocomplex was then captured by adding 20 μL of protein A Sepharose (50% slurry, GE healthcare) and gentle rocking on a rocker at 4 °C for 30 min. The beads were washed three times with Tris-IP buffer, and proteins were eluted from the beads by incubation in SDS-sample buffer at 75 °C for 15 min. After centrifugation, the supernatant was subjected to immunoblot analysis with anti-CPY and anti-Kar2 (a gift from Dr. S. Nishikawa) antisera and anti-myc antibody (Santa Cruz Biotechnology, Santa Cruz, CA).
Results
CPY*-Inv escaped the ERAD but underwent vacuolar degradation
To analyze the relationship between ER exit and ER retention of misfolded luminal proteins, we fused the yeast secretory protein invertase to the defined ERAD-L substrate CPY*. Invertase is secreted into the periplasmic space, and is extensively used to analyze protein traffic and the membrane topology of transmembrane proteins in yeast because it has the following useful properties as a reporter protein: (i) its folding state can be determined easily by measuring its enzymatic activity, (ii) it can be folded regardless of its subcellular localization, and (iii) transport of the invertase fusion protein along the secretory pathway can be monitored by checking the outer chain elongation of N-linked glycans that occur in the Golgi apparatus.Citation24) With our construct, invertase-3HA lacking the ER targeting sequence was fused to the C-termini of CPY and CPY* by a 14-amino acid linker to generate CPY-Inv and CPY*-Inv respectively (Fig. (A)). The chimeric genes were designed to be expressed under the control of the PRC1 (CPY gene) promoter from a low-copy plasmid. First, we introduced these plasmids to the wild-type strain (YKS12; suc2∆prc1∆) and measured invertase activity to determine whether the invertase domains within these chimeric proteins were correctly folded into the native conformation. The total invertase activities in the cells expressing CPY-Inv and CPY*-Inv were comparable to each other (19.0 ± 3.7 and 15.0 ± 5.5 units/OD cells, respectively), suggesting that fusion of the CPY or CPY* domain did not interfere with the folding of the invertase domain. Next we ran CHX chase assays and compared a turnover rate of CPY*-Inv with that of CPY*, a single domain ERAD-L substrate, to determine whether CPY*-Inv would be subjected to quality control. After the addition of CHX, cells were harvested at the indicated times in order to prepare cell extracts for immunoblot analysis with anti-CPY serum. It was possible that these proteins would undergo hyperglycosylation if they reached the medial Golgi,Citation25) and hence, we treated the cell extracts with Endo H to quantify the remaining proteins. We found that the two proteins were similar in degradation rates, with a half-life of 20–25 min in wild-type cells (Fig. (B) and (C)).
Fig. 1. CPY*-Inv escaped ERAD but underwent vacuolar degradation.
Note: (A) Schematic drawings of the fusion constructs used in this study. Invertase (Inv) is fused to the carboxyl termini of full-length CPY* and CPY to generate CPY*-Inv and CPY-Inv respectively. All constructs are expressed from the PRC1 promoter on a low-copy plasmid. (B) and (C) CHX chase assays were performed with wild-type (YKS12), hrd1∆ (YKS54), and pep4∆ (YKS28) cells expressing CPY* (B) or CPY*-Inv (C), as described in “Materials and methods.” The remaining proteins were analyzed by immunoblotting with anti-CPY antiserum and anti-3-phosphoglycerate kinase (PGK) antibody. Error bars represent standard deviations for three independent experiments. (D) Protein samples were prepared from the wild-type (YKS12, lanes 1 and 3) or pep4∆ cells (YKS28, lanes 2 and 4) harboring CPY-Inv (lanes 1 and 2) and CPY*-Inv (lanes 3 and 4) plasmid, treated with Endo H, and analyzed by immunoblotting with anti-CPY antiserum. (E) Protein samples were prepared from the pep4∆ cells (YKS28) expressing CPY* (lanes 1 and 2) or CPY*-Inv (lanes 3 and 4) with and without Endo H treatment, and were analyzed by immunoblotting with anti-CPY antiserum. (F) A CHX chase experiment for CPY*-Inv degradation in vps10∆ cells (YKS29) was done as described above.
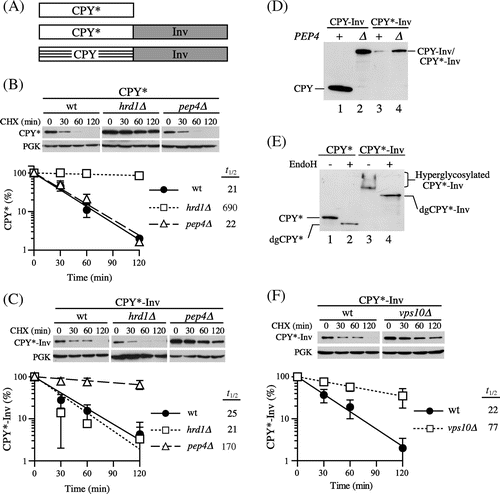
To determine whether the degradation of CPY*-Inv occurred via the ERAD, we tested cells lacking the Hrd1 protein, an E3 ubiquitin ligase involved in the ERAD of misfolded luminal proteins. As Fig. (C) shows, CPY*-Inv was unstable in the hrd1∆ cells, whereas CPY* was considerably stabilized in this strain as reported previously.Citation26) It has been reported that post-ER protein quality control mechanisms were responsible for a salvage pathway for the degradation of misfolded proteins that escaped from ERAD.Citation27) Overexpression of misfolded proteins is likely to cause saturation of the ERAD machinery, which leads to an overflow of them from the ER. In yeast, three such pathways have been reported for the degradation of misfolded luminal proteins: the HRD/DER-independent pathway (termed Hrd1-independent proteolysis or HIP), the vacuolar protein sorting (VPS) pathway, and autophagy.Citation25,Citation28,Citation29) Although the details of the mechanism of HIP are not known, at least Rsp5, another ubiquitin ligase, is required for the efficient degradation of CPY* overexpressed by a multicopy plasmid.Citation28) Hence, we examined to determine whether Rsp5 was involved in CPY*-Inv degradation. The cells containing the temperature-sensitive rsp5-1 allele, however, exhibited a rate of CPY*-Inv degradation identical to the wild-type strain at a nonpermissive temperature (Supplemental Fig. 1(A); t1/2 = 19 min), suggesting that Rsp5 is not required by the salvage pathway for CPY*-Inv degradation.
Next, we tested the pep4∆ strain to determine whether CPY*-Inv is degraded in the vacuole. Since the PEP4 gene encodes for vacuolar proteinase A, a key enzyme in the maturation of several vacuolar proteases, the pep4∆ strain lacks most vacuolar protease activities.Citation30) A CHX chase assay with pep4∆ cells revealed that CPY*-Inv but not CPY* was remarkably stabilized in this strain (Fig. (B) and (C)). In cells expressing CPY-Inv, the CPY moiety was liberated and processed to its mature form in a PEP4-dependent manner (Fig. (D), lanes 1 and 2). On the other hand, the CPY* moiety did not appear in the presence or the absence of PEP4, while the total amount of CPY*-Inv was significantly decreased in the wild-type cells as compared with the pep4∆ cells (Fig. (D), lanes 3 and 4). These results indicate that the decrease in CPY*-Inv was not due to cleavage at the linker between CPY* and invertase, but to degradation of the CPY* domain.
As described above, there are two possible pathways for the delivery of misfolded proteins to the vacuole for degradation, the VPS and autophagic pathways. It is known that ER stress triggers autophagy by which the protein aggregates accumulating in the ER can be taken up by autophagosomes without passing through the Golgi apparatus.Citation31–Citation33) On the other hand, cargo proteins for the VPS pathway are delivered to the vacuole via the Golgi apparatus.Citation34) Because invertase is hyperglycosylated during transit through the Golgi apparatus,Citation35) analysis of the glycosylation state might make it possible to determine which pathway is responsible for the vacuolar delivery of CPY*-Inv. Hence, extracts from pep4∆ cells were treated with and without Endo H and subjected to immunoblot analysis with anti-CPY antiserum. In the cells expressing CPY*-Inv, a smear signal of CPY*-Inv was seen (Fig. (E), lane 3). It was converged into a single band when treated with Endo H (Fig. (E), lane 4), indicating that CPY*-Inv was hyperglycosylated. In addition, we found that CPY*-Inv was stabilized in the cells lacking Vps10, a type I transmembrane sorting receptor for various vacuolar hydrolases and cycles between the trans-Golgi and endosomes (Fig. (F); t1/2 = 77 min), but not in the atg1∆ cells, which lack the autophagic pathway (Supplemental Fig. 1(B); t1/2 = 18 min). Taken together, these results indicate that CPY*-Inv transited to the Golgi apparatus, followed by delivery of it to the vacuole via the VPS pathway.
CPY*-Inv as a potential ERAD-L substrate
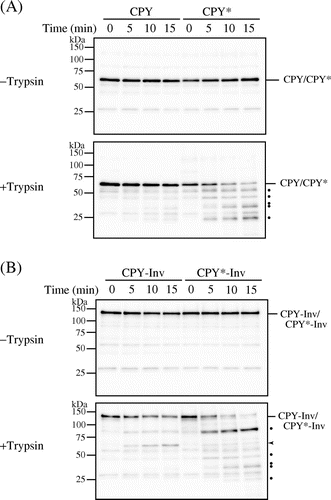
There are two possibilities as to the efficient exit of CPY*-Inv from the ER: Either the fusion of invertase assisted a proper folding of the CPY* domain, or the invertase domain acted as an ER exit signal for CPY*-Inv. Accordingly, first we compared the folding states of the CPY domains within CPY-Inv and CPY*-Inv by monitoring sensitivity to exogenously added protease in vitro. Spheroplasts were prepared from prc1∆hrd1∆pep4∆ cells expressing wild-type CPY, CPY*, CPY-Inv, or CPY*-Inv, lysed with Triton X-100, and treated with trypsin. After the protein samples were treated with Endo H, the remaining CPY and its derivatives were analyzed by immunoblot probed with anti-CPY antiserum. Under this condition, properly folded CPY was highly resistant to exogenously added trypsin during the time course, while misfolded CPY* was rapidly degraded (Fig. (A)). CPY-Inv was also resistant to trypsin treatment, although small amounts of the protein of sizes corresponding to the CPY moiety appeared (Fig. (B), arrowhead). This protein might have been generated by a scission at the linker, which contained a lysine residue, between CPY and invertase. Tryptic digestion of CPY*-Inv decreased the amount of it and produced a band pattern similar to CPY*, indicating that the CPY* domain was degraded. These results suggest that the CPY* domain of CPY*-Inv was misfolded, similarly to the single domain protein CPY*, and that a fusion of invertase to CPY* did not contribute to the correct folding of CPY*.
Fig. 2. CPY*-Inv but not CPY-Inv was sensitive to exogenously added protease.
Note: Cell lysates were prepared from prc1∆hrd1∆ pep4∆ (YKS30) cells expressing CPY (A), CPY* (A), CPY-Inv (B), or CPY*-Inv (B), and then incubated with or without trypsin for the indicated times, as described in “Materials and methods.” After Endo H treatment, samples were analyzed by SDS-PAGE, followed by immunoblot analysis with anti-CPY antiserum. Dots show tryptic digests, and arrowhead indicates the size corresponding to the CPY moiety.
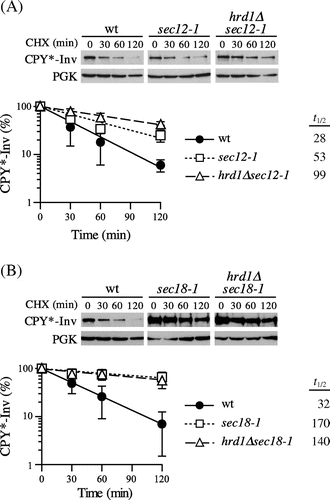
Next, we examined to determine whether CPY*-Inv was targeted to ERAD when it was intensively retained in the ER by blocking ER-to-Golgi transport. We utilized temperature-sensitive alleles of the SEC12 and SEC18 genes, which are necessary for COPII-coated vesicle formation and its fusion to the cis-Golgi, respectively.Citation36,Citation37) After inactivation at a restrictive temperature of 37 °C, CHX was added to monitor the degradation of CPY*-Inv. As shown in Fig. (A), only a slight stabilization of CPY*-Inv was observed in the sec12-1 cells (t1/2 = 53 min) as compared to the wild-type cells. When ERAD was impaired by disrupting the HRD1 gene in the sec12-1 cells, CPY*-Inv was further stabilized (t1/2 = 99 min), indicating that CPY*-Inv was targeted to the ERAD-L. In contrast, CPY*-Inv was significantly stabilized in the sec18-1 cells (t1/2 = 170 min), but there was no additive effect on CPY*-Inv degradation due to the deletion of HRD1 (Fig. (B); t1/2 = 140 min), suggesting that CPY*-Inv was sequestered from the ERAD machinery by efficient incorporation of it into the COPII vesicles, preventing its degradation by ERAD.
Fig. 3. Blockage of ER exit directed CPY*-Inv to ERAD.
Note: (A) Wild-type (YKS12), sec12-1 (YKS57), and hrd1∆sec12-1 (YKS61) cells containing CPY*-Inv plasmid were grown to early log phase at a permissive temperature. After incubation of the cells at a nonpermissive temperature for 30 min, CHX was added and aliquots were removed at the indicated times for immunoblot analysis, as described in Fig. (C). (B) Wild-type (YKS12), sec18-1 (YKS62), and hrd1∆sec18-1 (YKS78) cells containing CPY*-Inv plasmid were analyzed as described above.
Invertase domain acted as an ER exit signal
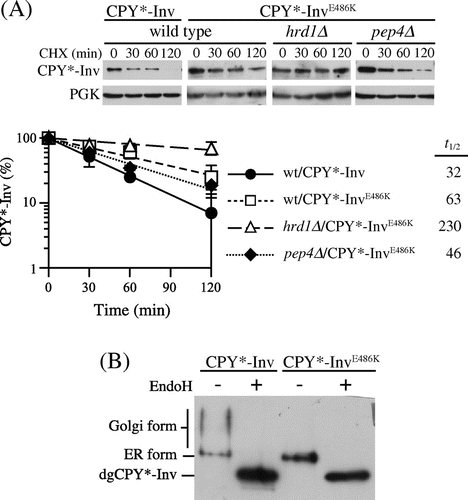
In the paradigm of protein sorting in vesicular trafficking, a receptor for soluble cargo proteins directly or indirectly binds to coat proteins of budding vesicles, and this ensures the incorporation of cargo proteins into the vesicle.Citation38) Although the ER exit signal within invertase has not yet been identified, it has been reported that glutamate-486 was involved in its exit from the ER.Citation39,Citation40) This amino acid residue is highly conserved among members of glycoside hydrolase family 32 (GH32), which includes not only invertases but also other fructofuranosidases such as inulinase and levanase from bacteria, fungi, and plants. This suggests that the conserved glutamate residue has important roles in the GH32 family of enzymes. The crystal structure of an invertase from Thermotoga maritima, a hyperthermophilic bacterium, revealed that this amino acid residue resided on a β-sandwich module, whose function remains unknown.Citation41) Replacement of the glutamate-486 residue with lysine in yeast invertase caused retention of it in the ER without noticeable degradation.Citation39,Citation40) Hence, we decided to introduce this mutation into CPY*-Inv to test the possibility that the invertase domain can function as the ER exit signal for CPY*-Inv. When CPY*-InvE486 K was expressed in wild-type cells, it exhibited slight stabilization as compared with CPY*-Inv (Fig. (A), t1/2 = 63 min). It was further stabilized with deletion of the HRD1 gene (Fig. (A); t1/2 = 230 min), unlike pep4∆ cells (Fig. (A), t1/2 = 46 min). In addition, analysis of the N-glycosylation state revealed that the E486 K mutant underwent less hyperglycosylation than CPY*-Inv, suggesting that the E486 K variant was retained in the ER (Fig. (B)). Together, these results indicate that ER retention of CPY*-Inv by the E486 K mutation in the invertase domain directed it to the ERAD.
Fig. 4. Invertase domain in CPY*-Inv acted as an ER exit signal.
Note: (A) CHX chase experiments were run to monitor the degradation of CPY*-InvE486 K in the wild-type (YKS12), hrd1∆ (YKS54), and pep4∆(YKS28) cells, as described in Fig. (C). (B) pep4∆ cells (YKS28) expressing CPY*-Inv or CPY*-InvE486 K were grown to early log phase, and cell extracts were prepared. Samples were then analyzed by immunoblotting with anti-CPY antiserum.
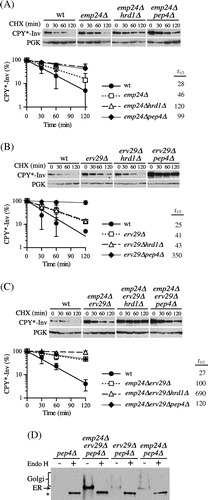
Although the E486 K mutation is unlikely to destabilize invertase,Citation40) we cannot exclude the possibility that it was retained in the ER by quality control mechanisms due to a conformational change in the invertase domain rather than a loss of interactions with the corresponding receptors.Citation39) Accordingly, we tried to decrease the ER-to-Golgi transport of CPY*-Inv by deleting the genes coding for the corresponding receptors. Previous studies have suggested that ER-to-Golgi transport of invertase is mediated largely by the p24 complex, comprised of conserved type 1 transmembrane proteins, including Emp24, Erv25, Erp1, and Erp2.Citation42,Citation43) Hence, we deleted the EMP24 gene to determine whether the p24 complex is involved in exit of CPY*-Inv from the ER. As Fig. (A) shows, the loss of Emp24 increased the half-life of CPY*-Inv up to approximately 50 min. Its degradation was further inhibited not only in the emp24∆pep4∆ (t1/2=99 min) but also in the emp24∆hrd1∆ cells (t1/2=120 min). These results suggest that the ER–Golgi transport of CPY*-Inv was retarded by loss of the p24 complex, which partially switched its site of disposal from the vacuole to the ER. Because the degradation of CPY*-Inv was partially dependent on the vacuole in the emp24∆ cells, we assumed that there might be other sorting receptors responsible for its exit from the ER. It has been reported that Erv29, the COPII-localized transport receptor for CPY, is also required for post-ER degradation of CPY* when it is overexpressed.Citation25,Citation28) Hence, we hypothesized that Erv29 can recognize the CPY* domain suppressing the defect in the ER–Golgi transport of CPY*-Inv caused by deletion of EMP24. To address this question, we deleted the ERV29 gene from emp24∆ cells, and found that the half-life of CPY*-Inv was significantly increased up to 100 min in the double mutant. Degradation was hardly seen in the emp24∆erv29∆hrd1∆ triple mutant, while there was no obvious difference in the turnover rate between the emp24∆erv29∆ and the emp24∆erv29∆pep4∆ mutants (Fig. (C), t1/2 = 100–120 min). Moreover, CPY*-Inv was highly accumulated in the ER in the emp24∆erv29∆ cells, as estimated by its glycosylation state (Fig. (D)). Although it is possible that loss of these receptors affected the proper folding of the invertase domain in the ER rather than the ER–Golgi transport of CPY*-Inv, the total invertase activities in the emp24∆erv29∆ cells expressing CPY*-Inv were similar to that in wild-type cells (19.0 ± 3.7 and 19.5 ± 0.7 units/OD cells for the wild-type and the emp24∆erv29∆ cells, respectively), suggesting that the invertase domains were correctly folded and that they preserved the function of the ER exit signal. Furthermore, this suggests that both Emp24 and Erv29 can function as receptors for CPY*-Inv and that their defect retained CPY*-Inv in the ER lumen directing it to ERAD. In view of the fact that CPY*-Inv turnover was dependent on PEP4 in the erv29∆ single mutant (Fig. (B)), Erv29 might play a minor role, but can partially suppress the defect in ER–Golgi transport in the emp24∆ cells.
Fig. 5. CPY*-Inv escaped the ERAD via the p24 complex and Erv29.
Note: (A) CHX chase assays were performed in wild-type (YKS12), emp24∆ (YKS48), emp24∆hrd1∆ (YKS49), and emp24∆pep4∆ cells (YKS50) containing the CPY*-Inv plasmid. (B) CHX chase assays were performed in wild-type (YKS12), erv29∆ (YKS44), erv29∆hrd1∆ (YKS47), and erv29∆pep4∆ (YKS52) cells containing the CPY*-Inv plasmid. (C) CHX chase assays were performed in wild-type (YKS12), erv29∆emp24∆ (YKS59), hrd1∆erv29∆emp24∆ (YKS74), and pep4∆erv29∆emp24∆ cells (YKS75) containing the CPY*-Inv plasmid. (D) pep4∆ (YKS28), pep4∆erv29∆emp24∆ (YKS75), erv29∆pep4∆ (YKS52), and emp24∆pep4∆ cells (YKS50) expressing CPY*-Inv were grown to early log phase, and cell extracts were prepared. After the cell extracts were treated with Endo H or not treated, samples were analyzed by immunoblotting with anti-CPY antiserum.
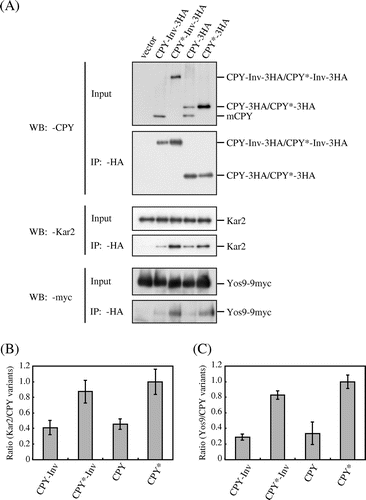
Although the invertase domain acted as the ER exit signal for the escape of CPY*-Inv from the ERAD, it is also possible that fusion of invertase to CPY* prevented this chimera from remaining in the ER with reduced recognition of the misfolded domain by the ER chaperones. To test this hypothesis, we examined the interaction between CPY*-Inv and ER chaperones Kar2 and Yos9 by means of native co-immunoprecipitation experiments. Crude membranes from hrd1∆emp24∆erv29∆ cells expressing Yos9-9myc and 3 × HA-tagged CPY variants were lysed with Triton X-100 and subjected to immunoprecipitation with anti-HA antibody. Even without ER cargo receptors Emp24 and Erv29 a fraction of CPY-3HA was converted to mature forms (mCPY), which were not precipitated with anti-HA antibody, probably because the 3 × HA tags were trimmed by vacuolar proteases. Small amounts of Kar2 and Yos9-9myc were co-precipitated with CPY-3HA, which might have corresponded to the ER form. Although CPY-Inv (tagged with 3 × HA) was hardly detected in the crude membrane fraction (Input), it was readily precipitated with anti-HA antibody and co-precipitated these chaperones to an extent similar to CPY-3HA (Fig. ). Both CPY*-3HA and CPY*-Inv co-precipitated more chaperones than CPY-3HA and CPY-Inv, implying that the misfolded CPY* domains associated with the chaperones with higher affinity than the CPY domain with proper folding.
Fig. 6. The association of Kar2 and Yos9 with CPY*-Inv.
Note: (A) Microsomes were prepared from YTS346 cells expressing CPY-3HA, CPY*-3HA, CPY-Inv, or CPY*-Inv and subjected to native co-immunoprecipitation with anti-HA antibody. Microsomes (Input) and immunoprecipitates (IP: α-HA) were analyzed by immunoblotting with anti-CPY (IB: α-CPY) and anti-Kar2 (IB: α-Kar2) antisera and anti-myc antibody (IB: α-myc). (B) and (C) The relative amounts of Kar2 (B) and Yos9-myc (C) co-precipitated with CPY variants were quantified. The amount of Kar2 or Yos9-myc co-precipitated with CPY* was set to 1. Error bars represent standard deviations for three independent experiments.
CPY*-Inv was efficiently associated with Yos9 (Fig. ) and was degraded by ERAD when retained in the ER (Figs. ). Hence, we proceeded to determine whether Yos9 indeed contributed to ERAD of CPY*-Inv with the CPY*-InvE486 K variant, which was retained in the ER and targeted to ERAD (Fig. ). As expected, the degradation of CPY*-InvE486 K was severely impaired in the yos9∆ cells (Supplemental Fig. ). Taken together, these results suggest that the CPY* domain was recognized by Yos9 for the degradation of ER-retained CPY*-Inv, and the fusion of invertase did not interfere with recognition of the CPY* domain by the ERAD components.
Discussion
The major role of the ER quality control is to prevent the delivery of aberrant proteins showing cytotoxicity along the secretory pathway. To achieve this, cells must retain abnormal proteins in the ER and remove them. CPY*, a single-domain model substrate for ERAD-L, is degraded primarily by the Hrd1-dependent pathway (Fig. (B)),Citation26) but the fusion of a soluble secretory protein invertase as a fully folded domain directed CPY* to forward transport, which was largely mediated by the p24 complex, a putative ER-to-Golgi cargo receptor for invertase (Fig. ). These results indicate that misfolded proteins can exit from the ER if the interaction between the ER export signal and the corresponding receptor is fairly well preserved. Since impairment of the receptor-mediated ER exit of CPY*-Inv resulted in degradation of it via the Hrd1-dependent pathway (Figs. (A) and (C)), it is likely that active ER retention by the ERAD machinery and receptor-mediated ER export compete for secretory proteins in the ER. This is confirmed by the fact that an increased concentration of CPY* within the ER lumen caused by a deficiency in the ERAD pathway or overexpression of CPY* increases the forward transport of CPY*.Citation15,Citation25,Citation44)
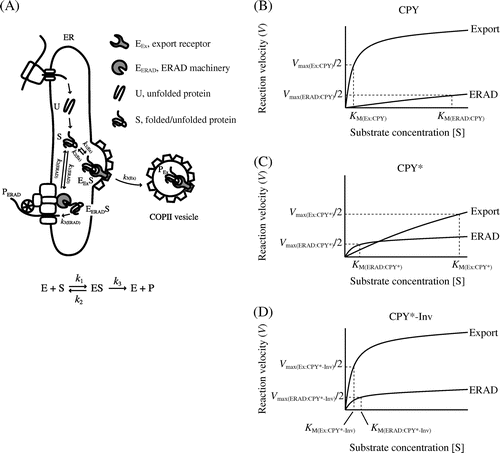
Our data based on two luminal substrates, CPY* and CPY*-Inv, appear to accord with the FoldEx theory for evaluating the efficiency of secretory protein export from the ER based on enzymatic Michaelis–Menten treatment.Citation10) The FoldEx model considers each of the three pathways (ERAF, ERAD, and ER export) as an enzyme reaction, with the secretory proteins as substrates. As for enzymatic kinetics, the rate of each pathway (V) is determined by rate constants (k) and Michaelis constants specific to each pathway (KM) as well as the concentrations of the components of each pathway (as enzymes; [E]) and folded/misfolded proteins (as substrates; [S]) (Fig. (A)). In enzymatic kinetics, the Michaelis constant represents the affinity between an enzyme and its substrate. Likewise, the binding affinity between folded or misfolded proteins and the machinery of ERAD and ER export might be key to determining the dominant pathway. Compared to fully folded CPY, the binding affinity of CPY* to export receptors, Erv29, might decrease (KM(Ex:CPY) < KM(Ex:CPY*)) and increase for ERAD components, such as molecular chaperone Kar2/BiP and lectin Yos9 (KM(ERAD:CPY) > KM(ERAD:CPY*) in Fig. (B) and (C)).Citation7,Citation45) Therefore, ERAD dominates when the concentration of CPY* in the ER is sufficiently low (Fig. (C)). Conversely, overexpression (equal to increased concentration) of CPY* is likely to saturate the ERAD machinery, resulting in the export of CPY* to the Golgi apparatus and beyond (Fig. (C)).Citation25,Citation28) As discussed by Wiseman et al.Citation10) since the concentrations of ERAD and the export machinery, which affect the reaction velocity of a pathway, differ by cell type and culture conditions, the export efficiency of given certain misfolded protein can be changed. The fusion of invertase as an ER-export signal to CPY* might increase its affinity to another export receptor, the p24 complex (KM(Ex:CPY*-Inv) < KM(Ex:CPY*) in Fig. (C) and (D)), thereby increasing its export to a level greater than that of the ERAD pathway, thereby facilitating the escape of CPY* from the ERAD pathway. When the ER export of CPY*-Inv was impaired by a sec12 mutation (VEx:CPY*-Inv ≈ 0), loss of the ER exit signal on invertase (increased KM(Ex:CPY*-Inv)), or deletion of export receptor genes ([E]Ex:CPY*-Inv ≈ 0), its turnover became Hrd1-dependent (Figs. ), indicating competition between the ERAD pathway and ER export.
Fig. 7. Michaelis–Menten plots relate rates of ER export/ERAD to the concentrations of folded and misfolded secretory proteins.
Note: (A) A simplified FoldEx model for protein export. Unfolded (U) ensembles are converted to the folded or misfolded states (S) via ERAF. The S ensembles bind to ER export and ERAD machineries (EEx and EERAD, respectively) with specific rate constants (k1 and k2). Finally, the S ensembles are incorporated into COPII vesicles (PEx) or are retro-translocated to the cytosol (PERAD). The relationships between velocity of ER export or ERAD and the concentrations of folded or misfolded secretory proteins in the ER are adapted to Michaelis–Menten kinetics ((B) CPY; (C) CPY*; and (D) CPY*-Inv). Note that the graphs do not reflect actual values from experiments.
Recently, fusion proteins in which folded CPY or unfolded CPY* were fused tandemly (CPY-CPY, CPY-CPY*, CPY*-CPY, and CPY*-CPY*) were used to analyze protein metabolism in the ER.Citation9) Although both CPY*-CPY and CPY*-CPY* were targeted to the ERAD, CPY-CPY* exited from the ER, similarly to the CPY-CPY protein, thereby escaping from the ERAD. This was due to decreased association of CPY-CPY* with Yos9 as well as a concomitant recognition of the folded CPY domain as an ER exit signal by the export receptor, Erv29. In the case of CPY*-Inv, it exited from the ER, although it remained associated with Yos9 as efficiently as a single domain CPY* protein (Fig. ). This indicates that its incorporation into COPII vesicles through interaction between the invertase domain and the p24 complex (the cargo receptor) is predominant over the ER retention mediated by Yos9.
Fusion protein technology has been applied to various organisms to achieve efficient production of recombinant proteins,Citation46–Citation49) but the roles of fusion proteins vary. Some are used as purification tags and some are to improve solubility and folding. In eukaryotic microbes, including yeasts and filamentous fungi, highly expressed secretory proteins from host cells are fused as career proteins to foreign proteins to improve production of them. Although a well-established effect of protein fusion in eukaryotic microbes is stabilization of foreign mRNA, it is thought that protein fusion also contributes to the folding of foreign proteins.Citation46,Citation47) The present study, however, indicates that the career protein fusion protected CPY* from ER quality control by promoting receptor-mediated exit of it from the ER rather than by assisting the folding of it. CPY*-Inv reached the Golgi apparatus recognized by the vacuolar sorting receptor Vps10 for vacuolar degradation.
In yeast, misfolded proteins that reach the cis-Golgi are recognized and retrieved by the ER by an unknown mechanism.Citation50) When a substantial amount of misfolded proteins reaches the cis-Golgi, the retrieval mechanism can be saturated and hence they can transit instead to the trans-Golgi where they undergo vacuolar sorting for degradation.Citation51–Citation53) Although they are secreted if not sorted to the vacuoles, it is possible that they are degraded at the cell surface or undergo endocytic degradation.Citation52,Citation54) In order to avoid unnecessary degradation of recombinant proteins and to increase their yield, it might be important to regulate protein traffic and protein quality control by engineering the host cells and proteins to be expressed.
Supplemental material
The supplemental material for this paper is available at http://dx.doi.org/10.1080/09168451.2014.877185.
Acknowledgments
We thank Dr. Davis T. Ng and Dr. Shuh-Ichi Nishikawa for providing plasmids, and antibodies.
Funding
This work was supported by MEXT KAKENHI [grant number 17780249], [grant number 20059002]; JSPS KAKENHI [grant number 19580391].
Notes
Abbreviations: ER, endoplasmic reticulum; COPII, coat protein complex II; ERAF, ER-assisted folding; ERAD, ER-associated degradation; FoldEx, folding for export; CPY, carboxypeptidase Y; CHX, cycloheximide; Endo H, endoglycosidase H; PGK, 3-phosphoglycerate kinase; HIP, Hrd1-independent proteolysis; VPS, vacuolar protein sorting.
References
- Barlowe C. Trends Cell Biol. 2003;13:295–300.
- Gürkan C, Stagg SM, Lapointe P, Balch WE. Nat. Rev. Mol. Cell Biol. 2006;7:727–738.
- Sekijima Y, Wiseman RL, Matteson J, Hammarström P, Miller SR, Sawkar AR, Balch WE, Kelly JW. Cell. 2005;121:73–85.
- Ellgaard L, Molinari M, Helenius A. Science. 1999;286:1882–1888.
- Kostova Z, Wolf DH. EMBO J. 2003;22:2309–2317.
- Nakatsukasa K, Brodsky JL. Traffic. 2008;9:861–870.
- Denic V, Quan EM, Weissman JS. Cell. 2006;126:349–359.
- Ellgaard L, Helenius A. Nat. Rev. Mol. Cell Biol. 2003;4:181–191.
- Izawa T, Nagai H, Endo T, Nishikawa S. Mol. Biol. Cell. 2012;23:1283–1293.
- Wiseman RL, Powers ET, Buxbaum JN, Kelly JW, Balch WE. Cell. 2007;131:809–821.
- Kowalski JM, Parekh RN, Wittrup KD. Biochemistry. 1998;37:1264–1273.
- Tatara Y, Yoshida T, Ichishima E. Biosci. Biotechnol. Biochem. 2005;69:2101–2108.
- Kumita JR, Johnson RJ, Alcocer MJ, Dumoulin M, Holmqvist F, McCammon MG, Robinson CV, Archer DB, Dobson CM. FEBS J. 2006;273:711–720.
- Geiger R, Gautschi M, Thor F, Hayer A, Helenius A. J. Biol. Chem. 2011;286:5813–5822.
- Kincaid MM, Cooper AA. Mol. Biol. Cell. 2007;18:455–463.
- Pagant S, Kung L, Dorrington M, Lee MC, Miller EA. Mol. Biol. Cell. 2007;18:3398–3413.
- Wang S, Ng DT. Mol. Biol. Cell. 2010;21:1153–1165.
- Gueldener U, Heinisch J, Koehler GJ, Voss D, Hegemann JH. Nucleic Acids Res. 2002;30:23e.
- Gauss R, Trautwein M, Sommer T, Spang A. Yeast. 2005;22:1–12.
- Amberg DC, Burke D, Strathern JN. Methods in yeast genetics 2005: a Cold Spring Harbor Laboratory course manual. 2005 ed. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 2005; p. 230.
- Ng DT, Spear ED, Walter P. J. Cell Biol. 2000;150:77–88.
- Plemper RK, Egner R, Kuchler K, Wolf DH. J. Biol. Chem. 1998;273:32848–32856.
- Xie W, Kanehara K, Sayeed A, Ng DT. Mol. Biol. Cell. 2009;20:3317–3329.
- Darsow T, Odorizzi G, Emr SD. Methods Enzymol. 2000;327:95–106.
- Spear ED, Ng DT. Mol. Biol. Cell. 2003;14:2756–2767.
- Bordallo J, Plemper RK, Finger A, Wolf DH. Mol. Biol. Cell. 1998;9:209–222.
- Arvan P, Zhao X, Ramos-Castaneda J, Chang A. Traffic. 2002;3:771–780.
- Haynes CM, Caldwell S, Cooper AA. J. Cell Biol. 2002;158:91–102.
- Kruse KB, Brodsky JL, McCracken AA. Mol. Biol. Cell. 2006;17:203–212.
- Jones EW, Zubenko GS, Parker RR. Genetics. 1982;102:665–677.
- Bernales S, McDonald KL, Walter P. PLoS Biol. 2006;4:e423.
- Kamimoto T, Shoji S, Hidvegi T, Mizushima N, Umebayashi K, Perlmutter DH, Yoshimori T. J. Biol. Chem. 2006;281:4467–4476.
- Yorimitsu T, Nair U, Yang Z, Klionsky DJ. J. Biol. Chem. 2006;281:30299–30304.
- Mullins C, Bonifacino JS. Bioessays. 2001;23:333–343.
- Esmon B, Novick P, Schekman R. Cell. 1981;25:451–460.
- Graham TR, Emr SD. J. Cell Biol. 1991;114:207–218.
- Schekman R, Orci L. Science. 1996;271:1526–1533.
- Baines AC, Zhang B. Trends Biochem. Sci. 2007;32:381–388.
- Preuss D, Mulholland J, Kaiser CA, Orlean P, Albright C, Rose MD, Robbins PW, Botstein D. Yeast. 1991;7:891–911.
- McCracken AA, Werner ED, Powell MJ, Kruse KB, Brodsky JL. Yeast. 2000;16:49–55.
- Alberto F, Bignon C, Sulzenbacher G, Henrissat B, Czjzek M. J. Biol. Chem. 2004;279:18903–18910.
- Schimmoller F, Singer-Kruger B, Schroder S, Kruger U, Barlowe C, Riezman H. EMBO J. 1995;14:1329–1339.
- Marzioch M, Henthorn DC, Herrmann JM, Wilson R, Thomas DY, Bergeron JJ, Solari RC, Rowley A. Mol. Biol. Cell. 1999;10:1923–1938.
- Taxis C, Vogel F, Wolf DH. Mol. Biol. Cell. 2002;13:1806–1818.
- Quan EM, Kamiya Y, Kamiya D, Denic V, Weibezahn J, Kato K, Weissman JS. Mol. Cell. 2008;32:870–877.
- Romanos MA, Scorer CA, Clare JJ. Yeast. 1992;8:423–488.
- Gouka RJ, Punt PJ, van den Hondel CA. Appl. Microbiol. Biotechnol. 1997;47:1–11.
- Esposito D, Chatterjee DK. Curr. Opin. Biotechnol. 2006;17:353–358.
- Idiris A, Tohda H, Kumagai H, Takegawa K. Appl. Microbiol. Biotechnol. 2010;86:403–417.
- Vashist S, Kim W, Belden WJ, Spear ED, Barlowe C, Ng DT. J. Cell Biol. 2001;155:355–368.
- Busca R, Martinez M, Vilella E, Pognonec P, Deeb S, Auwerx J, Reina M, Vilaro S. J. Biol. Chem. 1996;271:2139–2146.
- Hong E, Davidson AR, Kaiser CA. J. Cell Biol. 1996;135:623–633.
- Holkeri H, Makarow M. FEBS Lett. 1998;429:162–166.
- Kang HA, Kim SJ, Choi ES, Rhee SK, Chung BH. Appl. Microbiol. Biotechnol. 1998;50:187–192.