
Abstract
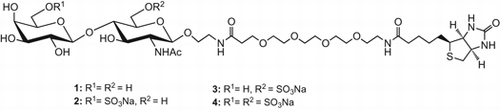
We synthesized four types of keratan and keratan sulfate repeating disaccharides containing non-sulfate, Galβ1-4GlcNAcβ, and three types of sulfates, Gal6Sβ1-4GlcNAcβ, Galβ1-4GlcNAc6Sβ, and Gal6Sβ1-4GlcNAc6Sβ in an efficient and stereo-controlled manner. These disaccharides were conjugated with biotin via a hydrophilic linker at the reducing terminal.
Graphical Abstract
Four types of biotinylated keratan sulfate repeating disaccharides containing non-sulfate were synthesized in an efficient and stereo-controlled manner.
Keratan sulfate (KS) is a member of the proteoglycan family that has a linear glycan chain composed of a repeating disaccharide unit, Galβ1-4GlcNAcβ. KS repeating oligosaccharides are often sulfated at O-6 of Gal and GlcNAc. A KS repeating disaccharide, the minimum unit of the KS oligosaccharide, has four different types of sulfation patterns, including the non-sulfate. KS oligosaccharides have recently been shown to have an inhibitory effect on axonal regrowth.Citation1) However, the relationship between the factors regulating neuronal actions and facile structures, including the sulfation patterns of KS oligosaccharides, in this interaction has not yet been elucidated.
A few studies have investigated the synthesis of KS oligosaccharides. In 1989, Ogawa’s group reported the synthesis of the KS tetrasaccharide, GlcNAc6Sβ1-3Gal6Sβ1-4GlcNAc6Sβ1-3Galβ,Citation2,Citation3) while Misra’s group described the synthesis of the octyl glycosides of N-acetyl lactosamine, Gal6Sβ1-4GlcNAcβ, and Galβ1-4GlcNAc6Sβ, in 2004.Citation4) However, these synthetic approaches are not suitable for preparing KS conjugates with four different types of sulfation patterns, including the non-sulfate.
We demonstrate in this study the efficient synthesis of KS disaccharides with three possible types of sulfation patterns, as well as one without the sulfate. The target KS disaccharides (1–4) were attached to biotin for biological use via a hydrophilic linker at the reducing terminal.
Results and discussion
Based on the retrosynthetic analysis depicted in Scheme , we attempted to synthesize the target compounds (1–4) by adopting a common disaccharide unit (18). Common disaccharide 18 was, respectively, protected with chemoselectively removable NAP and TBDPS at the position 6 of the Gal and GlcNAc residues.
Scheme 1. Retrosynthetic analysis for disaccharides 1–4.
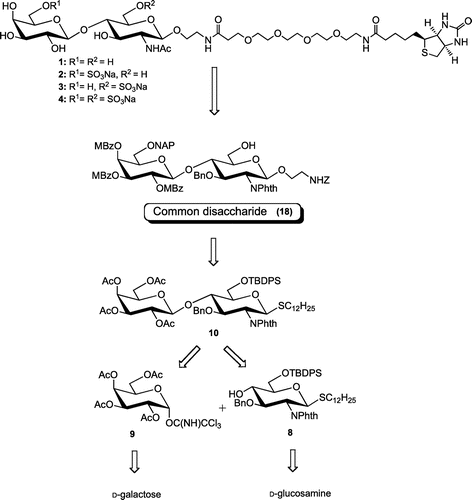
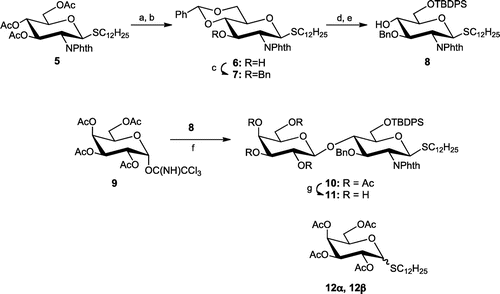
Scheme shows that all the acetyl groups in known thioglycoside 5 were removed by the Zemplén reaction. The triol was regiospecifically benzylidenated at the 4 and 6 positions with benzaldehyde dimethylacetal under acidic conditions to give 6 in a 78% yield in two steps. The resulting OH-3 of 6 was benzylated with NaH and BnBr to give 7 in an 86% yield. The benzylidene acetal of 7 was removed and the primary alcohol was selectively protected with TBDPS to afford 8 in a 69% yield in two steps. The glycosyl acceptor (8) was coupled with 2,3,4,6-tetra-O-acetyl-α-d-galactopyranosyl trichloroacetimidate (9) in the presence of TMSOTf at −20 °C. However, desired disaccharide 10 could only be obtained in a 2% yield under these reaction conditions. The dodecylthio group of 8 was transferred to the anomeric position of 9 to give 12α and 12β in respective 34 and 64% yields.
Scheme 2. Synthesis of disaccharide 11.
Note: Reagents and conditions: (a) NaOMe, MeOH; (b) PhCH(OMe)2, p-TsOH, 78% (2 steps); (c) BnBr, NaH, DMF, 0 °C-rt, 86%; (d) camphorsulfonic acid, CH2Cl2-MeOH; (e) TBDPSCl, imidazole, DMF, 69% (2 steps); (f) TMSOTf, CH2Cl2, −20 or −78 °C; (g) NaOMe, MeOH, 60% (2 steps).
Similar intermolecular transfer of the thio group to the anomeric position of the donor has been reported for glycosylation with a thioglycoside.Citation5–Citation7) In 2000, Yu’s group reported similar glycosylation with a trichloroacetimidate donor and an acceptor having thioglycoside in the presence of TMSOTf (0.05 eq. vs. the donor) at −10 °C to show intermolecular transfer of the dodecylthio group.Citation8) However, the coupling reaction at −78 °C afforded the desired disaccharide in a 92% yield without the formation of a side product.
We rationalized that intermolecular transfer of the dodecylthio group was due to activation of the sulfur atom on the dodecylthio group by the larger amount of TMSOTf (0.5 eq.) and higher temperature (−20 °C). We therefore adopted Yu’s conditions, lowered the reaction temperature to −78 °C, and decreased the amount of TMSOTf to 0.05 eq. We subsequently obtained desired disaccharide 10 without intermolecular transfer of the dodecylthio group; however, a portion of the corresponding isomer having an ortho-ester linkage was generated due to the lower acidity used (data not shown). We finally succeeded in suppressing the ortho-ester formation by increasing the amount of TMSOTf to 0.1 eq. Saponification then removed the four acetyl groups of 10 to give desired tetraol 11 in a 60% yield in two steps.
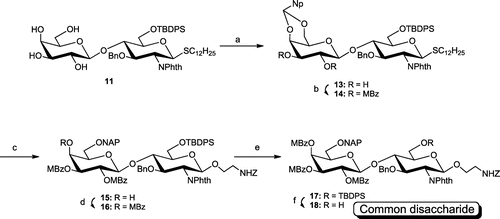
The tetraol (11) was 2-naphthylildenated at the 4 and 6 positions of the Gal residue with 2-naphthaldehyde under acidic conditions to give 13 in an 89% yield (Scheme ).Citation9) The resulting diol was protected with MBz to quantitatively give 14. Reductive ring opening of the naphthylidene acetal using BH3·NMe3, AlCl3, and a catalytic amount of H2O in THFCitation10) afforded 6-O-(2-naphthyl)methyl ether 15 in a 92% yield, without any formation of the regioisomer. The resulting OH-4 of 15 was methylbenzoylated to give 16 in a 98% yield. The disaccharide donor (16) was stereoselectively coupled to HO(CH2)2NHZ in the presence of NIS and TfOH to quantitatively give 17. We chemoselectively removed TBDPS to give common disaccharide 18 in a 79% yield.
Scheme 3. Synthesis of common disaccharide 18.
Note: Reagents and conditions: (a) 2-naphthaldehyde, p-TsOH·H2O, CH3CN, 89%; (b) MBzCl, DMAP, pyridine-CH2Cl2, quant.; (c) BH3·Me3 N, AlCl3, cat. H2O, THF, 92%; (d) MBzCl, DMAP, pyridine-CH2Cl2, 98%; (e) NIS, TfOH, MSAW300, Et2O-(CH2Cl)2, quant.; (f) 1 M tetra-n-butyl ammonium fluoride, AcOH, THF, 79%.
Scheme shows that the liberated primary position of 18 was protected with an acetyl group, and NAP on the Gal residue was chemoselectively removed with DDQ to give 19 in an 84% yield in two steps. NAP on 18 was similarly removed to give 20 in a 79% yield. The free hydroxyl groups of 19, 18, and 20 were sulfated with SO3·Me3N in DMF to respectively afford corresponding sulfates 21, 22, and 23 in 88%, quantitative and 90% yields. Phth, MBz, and Ac of 18, 21, 22, and 23 were removed with 1,3-diaminopropane. The generated free amine was acetylated with Ac2O and Et3N in MeOH. Bn, Z, and NAP were removed by hydrogenolysis in the presence of Pd-black. Finally, free amines on the aglycon were biotinylated to quantitatively give the target compounds (1–4). Selected 1H-NMR data for 1–4 are given in Table . The chemical shifts of H-6′ (2), H-6 (3), and of H-6 and 6′ (4), which were O-sulfated positions, were shifted down-field.
Scheme 4. Synthesis of target compounds 1–4.
Note: Reagents and conditions: (a) Ac2O, pyridine, quant.; (b) DDQ, cat. H2O, CH2Cl2-MeOH, 84% for 19 (2 steps), 78% for 20; (c) SO3· NMe3, DMF, 60 °C, 88% for 21, quant. for 22, 90% for 23; (d) 1,3-diaminopropane, EtOH, reflux; (e) Ac2O, Et3N, MeOH; (f) NaOH, MeOH (for 21, 22), 50% MeOH (for 23), reflux; (g) H2, Pd-black, aq. EtOH + dil. AcOH (for 18), aq. 2-PrOH + dil. AcOH (for 21, 22), dil. AcOH (for 23); (h) NHS-PEG4-biotin, 1 M Na3PO4, 0.15 M NaCl.
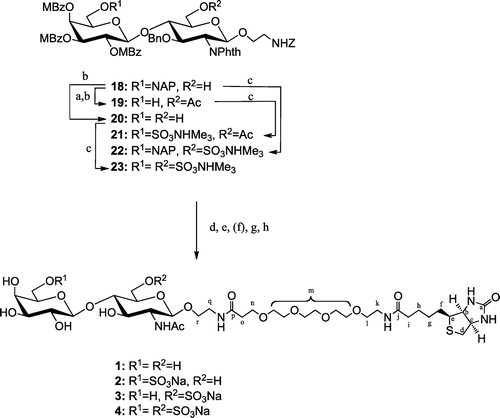
Table 1. Selected chemical shifts (ppm) for 1–4 by 600 MHz 1H-NMR.
Conclusion
In summary, we synthesized for the first time in an efficient and stereo-controlled manner four types of keratan and KS repeating disaccharides containing a non-sulfate, Galβ1-4GlcNAcβ, as well as three sulfates, Gal6Sβ1-4GlcNAcβ, Galβ1-4GlcNAc6Sβ, and Gal6Sβ1-4GlcNAc6Sβ, using a common disaccharide. These disaccharides were linked to biotin via a hydrophilic linker to successfully give the target compounds (1-4).
Experimental
General methods
1H-NMR assignments were confirmed by two-dimensional HH COSY experiments using Bruker ADVANCE II 600 MHz spectrometers. Silica gel chromatography and analytical TLC were performed in a column of Silica Gel 60 (Merck) and Silica Gel 60 N (spherical neutral; Kanto Kagaku). The gel for size-exclusion chromatography (Sephadex LH-20) was from GE Healthcare. Bond Elut was from Agilent Technologies. Molecular sieves were from GL Science and activated at 200 °C under reduced pressure prior to their use. All reactions in organic solvents were performed in a dry Ar-containing atmosphere. The organic phase of the reaction mixture was successively washed with aq. NaHCO3 and brine, and then dried over anhyd. MgSO4 by the usual work-up.
Dodecyl 4,6-O-benzylidene-2-deoxy-2-phthalimido-1-thio-β-d-glucopyranoside (6)
To a solution of 5 (5.57 g, 8.99 mmol) in MeOH (212 mL) was added 0.5 M NaOMe (9 mL) while stirring at rt. After 2 h, the reaction was quenched with Dowex 50W × 8 (H+ form), and the mixture was filtered. The volatiles were removed under reduced pressure to give a triol (4.27 g) which was dissolved in CH3CN (400 mL). To this solution, benzaldehyde dimethylacetal (2.6 mL, 17 mmol) and a catalytic amount of p-TsOH (pH < 2) were added, while stirring for 3 h. The reaction mixture was neutralized with Et3N. The volatiles were removed under reduced pressure, and the residue was treated in a column of silica gel (20:1–10:1 toluene-EtOAc) to give 6 (4.06 g) in a 78% yield in two steps. 1H-NMR δH (CDCl3): 8.01–7.26 (m, 10H, Ar H), 5.57 (s, 1H, PhCH), 5.39 (d, 1H, J1,2 = 10.62 Hz, H-1), 4.56 (br t, 1H, J = 9.57 Hz, H-3), 4.36 (dd, 1H, J5,6a = 5.04 Hz, Jgem = 10.44 Hz, H-6a), 4.31 (br t, 1H, J = 10.29 Hz, H-2), 3.80 (br t, 1H, J = 10.26 Hz, H-6b), 3.64 (m, 1H, H-5), 3.61 (br t, 1H, J = 9.15 Hz, H-4), 2.68 (m, 1H, 1/2SCH2), 2.59 (m, 1H, 1/2SCH2), 2.54 (s, 1H, 3-OH), 1.30–1.13 [m, 20H, SCH2(CH2)10CH3], 0.87 (t, 3H, J = 14.10 Hz, CH3). ESI-HRMS m/z [(M + Na)+]: calcd. for C33H43NO6SNa, 604.2703; found, 604.2690.
Dodecyl 3-O-benzyl-4,6-O-benzylidene-2-deoxy-2-phthalimido-1-thio-β-d-glucopyranoside (7)
To a suspension of NaH (205.1 mg, 4.70 mmol, 55%) in DMF (3.0 mL) was added 6 (1.37 g, 2.35 mmol) in dry DMF (24 mL) at 0 °C, and the mixture was stirred for 1 h. BnBr was then added. After 1 h, unreacted NaH was decomposed with MeOH, and the reaction mixture was quenched with aq. NH4Cl and diluted with EtOAc. The organic phase was washed with brine and dried over anhyd. MgSO4. The volatiles were removed under reduced pressure, and the residue was treated in a column of silica gel (7:1–4:1 n-hexane-EtOAc) to give 7 (1.36 g) in an 86% yield. 1H-NMR δH (CDCl3): 7.86–6.86 (m, 14H, Ar H), 5.63 (s, 1H, PhCH), 5.31 (d, 1H, J1,2 = 10.68 Hz, H-1), 4.79, 4.50 (ABq, 2H, J = 12.36 Hz, PhCH2), 4.44 (br t, 1H, J = 9.45 Hz, H-3), 4.41 (dd, 1H, J5,6a = 10.62 Hz, Jgem = 4.92 Hz, H-6a), 4.27 (br t, 1H, J = 10.29 Hz, H-2), 3.82 (br t, 1H, J = 10.02 Hz, H-5), 3.80 (br t, 1H, J = 9.09 Hz, H-4), 3.70 (dd, 1H, J5,6b = 9.66 Hz, H-6b), 2.64 (m, 1H, 1/2SCH2), 2.55 (m, 1H, 1/2SCH2), 1.51–1.13 [m, 20H, SCH2(CH2)10CH3], 0.87 (t, 3H, J = 14.16 Hz, CH3). ESI-HRMS m/z [(M + Na)+]: calcd. for C40H49NO6SNa, 694.3173; found, 694.3168.
Dodecyl 3-O-benzyl-6-O-tert-butyldiphenylsilyl-2-deoxy-2-phthalimido-1-thio-β-d-glucopyranoside (8)
To a solution of 7 (5.12 g, 7.62 mmol) in CH2Cl2 (48 mL) and MeOH (48 mL), a catalytic amount of camphorsulfonic acid was added while stirring for 4 d. The reaction was quenched with Et3N. The volatiles were removed under reduced pressure, and the residue was treated in a column of silica gel (4:1–1:1 toluene-EtOAc) to give a corresponding 4,6-diol (4.10 g) which was dissolved in DMF (100 mL). Imidazole (1.06 g, 15.5 mmol) and TBDPSCl (2.0 mL, 7.7 mmol) were added to the solution while stirring for 5 d. The reaction mixture was diluted with EtOAc. The organic phase was treated as described for general methods, and the residue was then treated in a column of silica gel (10:1–3:1 n-hexane-EtOAc) to give 8 (4.35 g) in a 69% yield (two steps) as a syrup. 1H-NMR δH (CDCl3): 7.90–6.90 (m, 19H, Ar H), 5.23 (d, 1H, J1,2 = 10.38 Hz, H-1), 4.78, 4.55 (ABq, 2H, J = 12.24 Hz, PhCH2), 4.29 (dd, 1H, J2,3 = 10.26 Hz, J3,4 = 8.46 Hz, H-3), 4.21 (br t, 1H, J = 10.35 Hz, H-2), 3.98 (dd, 1H, J5,6a = 4.56 Hz, Jgem = 10.62 Hz, H-6a), 3.92 (m, 2H, H-4, 6b), 3.60 (m, 1H, H-5), 3.07 (d, 1H, J4,OH = 2.28 Hz, OH-4), 2.62 (m, 1H, 1/2SCH2), 2.52 (m, 1H, 1/2SCH2), 1.43 (m, 2H, SCH2CH2), 1.30–1.13 [m, 18H, SC2H4(CH2)9CH3], 1.09 [s, 9H, C(CH3)3], 0.86 (t, 3H, J = 14.22 Hz, CH3). ESI-HRMS m/z [(M + Na)+]: calcd. for C49H63NO6SSiNa, 844.4038; found, 844.4026.
Dodecyl β-d-galactopyranosyl-(1→4)-3-O-benzyl-6-O-tert-butyldiphenylsilyl-2-deoxy-2-phthalimido-1-thio-β-d-glucopyranoside (11)
Method A: A solution of 8 (495.2 mg, 544.7 μmol) and 2,3,4,6-tetra-O-acetyl-α-d-galactopyranosyl trichloroacetimidate (9, 668.9 mg, 1.36 mmol) in CH2Cl2 (55 mL) was stirred over AW300 molecular sieves (1.44 g) for 30 min at rt. To this solution, TMSOTf (173 μL, 679 μmol) was added at −20 °C for 2.5 h. The reaction was quenched with aq. NaHCO3 and diluted with CHCl3. The insoluble materials were filtered through Celite. The organic phase was treated as describe for general methods, and the crude mixture was then treated in an LH-20 gel permeation column (1:1 CHCl3-MeOH) and silica gel column (6:1–5:1 n-hexane-EtOAc) to give 10 (9.7 mg, 2%), dodecyl thioglycosides (12α, 101.0 mg) and (12β, 184.3 mg) in respective 34 and 64% yields.
Method B: 4A molecular sieves (2.93 g) were added to a solution of 8 (2.81 g, 3.42 mmol) and 9 (2.53 g, 5.13 mmol) in CH2Cl2 (233 mL), and the mixture was stirred for 30 min at rt. To this mixture TMSOTf (46 μL, 0.26 mmol) was added while stirring at −78 °C for 2 h. The reaction was treated in the same way as that described for Method A. The residue was purified through a column of silica gel (15:1–5:1 toluene-EtOAc) to give 10 (4.26 g). The disaccharide (10) was diluted in MeOH (160 mL), and 0.5 m NaOMe (3.4 mL) was added to the solution while stirring at rt. After 4 h, the reaction was quenched with Dowex 50Wx8 (H+ form), and the mixture was filtered. The volatiles of the filtrate were removed under reduced pressure, and the residue was directly treated in a column of silica gel to give 11 (2.02 g, 60% in two steps). Compound 11. 1H-NMR δH (CDCl3): 7.83–6.88 (m, 19H, Ar H), 5.20 (d, 1H, J1,2 = 10.50 Hz, H-1), 4.90, 4.50 (ABq, 2H, J = 12.18 Hz, PhCH2), 4.76 (d, 1H, J1′,2′ = 7.80 Hz, H-1′), 4.42 (br t, 1H, J = 9.48 Hz, H-3), 4.28 (br t, 1H, J = 8.94 Hz, H-4), 4.27 (br t, 1H, J = 10.43 Hz, H-2), 4.22 (dd, 1H, J5,6a = 2.04 Hz, Jgem = 11.04 Hz, H-6a), 4.03 (d, 1H, H-6b), 3.94 (d, 1H, J3′,4′ = 2.76 Hz, H-4′), 3.75 (m, 2H, H-6′ab), 3.69 (br t, 1H, J = 8.61 Hz, H-2′), 3.57 (br d, 1H, J = 9.66 Hz, H-5), 3.41 (dd, 1H, J2′,3′ = 9.42 Hz, H-3′), 3.34 (br t, 1H, J = 4.11 Hz, H-5′), 2.65 (m, 1H, 1/2SCH2), 2.58 (m, 1H, 1/2SCH2), 1.47 (m, 2H, SCH2CH2), 1.28–1.11 [m, 18H, SC2H4(CH2)9CH3], 1.08 [s, 9H, C(CH3)3], 0.87 (t, 3H, J = 14.16 Hz, CH3). ESI-HRMS m/z [(M + Na)+]: calcd. for C55H73NO11SSiNa, 1006.4566; found, 1006.4549.
Dodecyl 2,3,4,6-tetra-O-acetyl-1-thio-α-d-galactopyranoside (12α)
1H-NMR δH (CDCl3): 5.71 (d, 1H, J3,4 = 5.52 Hz, H-4), 4.45 (d, 1H, J1,2 = 3.18 Hz, H-1), 5.26 (dd, 1H, J2,3 = 10.86 Hz, H-3), 4.22 (dd, 1H, H-2), 4.59 (br t, 1H, J = 6.44 Hz, H-5), 4.11 (m, 2H, H-6ab), 2.56 (m, 1H, 1/2SCH2), 2.49 (m, 1H, 1/2SCH2), 2.15, 2.08, 2.05, 2.00 (4s, each 3H, 4Ac), 1.58 (m, 2H, SCH2CH2), 1.38–1.23 [m, 18H, SC2H4(CH2)9CH3], 0.88 (t, 3H, J = 14.04 Hz, CH3). ESI-HRMS m/z [(M + Na)+]: calcd. for C26H44O9SNa, 555.2598; found, 555.2585.
Dodecyl 2,3,4,6-tetra-O-acetyl-1-thio-β-d-galactopyranoside (12β)
1H-NMR δH (CDCl3): 5.43 (d, 1H, J3,4 = 3.30 Hz, H-4), 5.24 (br t, 1H, J = 9.96 Hz H-2), 5.05 (dd, 1H, J2,3 = 9.66 Hz, H-3), 4.48 (d, 1H, J1,2 = 10.02 Hz, H-1), 4.16 (dd, 1H, J5,6b = 6.60 Hz, Jgem = 11.34 Hz, H-6b), 4.11 (dd, 1H, J5,6a = 6.60 Hz, H-6a), 3.93 (t, 1H, H-5), 2.64–2.73 (m, 2H, SCH2), 2.16, 2.07, 2.05, 2.00 (4s, each 3H, 4Ac), 1.38–1.25 [m, 20H, SCH2(CH2)10CH3], 0.88 (t, 3H, J = 14.04 Hz, CH3). ESI-HRMS m/z [(M + Na)+]: calcd. for C26H44O9SNa, 555.2598; found, 555.2598.
Dodecyl 4,6-O-(2-naphthylidene)-β-d-galactopyranosyl-(1→4)-3-O-benzyl-6-O-tert-butyldiphenylsilyl-2-deoxy-2-phthalimido-1-thio-β-d-glucopyranoside (13)
To a solution of 11 (1.18 g, 1.20 mmol) in CH3CN, 2-naphthaldehyde (374.8 mg, 2.40 mmol) and p-TsOH (22.8 mg, 0.12 mmol) were added while stirring for 2 h. The reaction mixture was neutralized with Et3N. The volatiles were removed under reduced pressure, and the residue was treated in a column of silica gel (4:1–2:1 toluene-EtOAc) to give 13 (1.20 g) in an 89% yield which was used without further purification. 1H-NMR δH (CDCl3): 7.84–6.80 (m, 26H, Ar H), 5.67 (s, 1H, NpCH), 5.21 (d, 1H, J1,2 = 10.56 Hz, H-1), 5.10, 4.64 (ABq, 2H, J = 12.24 Hz, PhCH2), 4.85 (d, 1H, J1′,2′ = 7.74 Hz, H-1′), 4.45 (dd, 1H, J2,3 = 10.02 Hz, J3,4 = 8.82 Hz, H-3), 4.40 (d, 1H, Jgem = 12.36 Hz, H-6′a), 4.34 (t, 1H, H-4), 4.29 (br d, 2H, J = 10.20 Hz, H-2,6a), 4.20 (d, 1H, J3′,4′ = 3.66 Hz, H-4′), 4.04 (d, 1H, Jgem = 12.36 Hz, H-6b), 4.01 (d, 1H, H-6′b), 3.79 (t, 1H, H-2′), 3.54 (m, 2H, H-5, 3′), 3.36 (s, 1H, H-5′), 2.66 (m, 1H, 1/2SCH2), 2.58 (m, 2H, 1/2SCH2, OH-2′), 2.51 (d, 1H, J3′,OH = 9.36 Hz, OH-3′), 1.52–1.30 [m, 20H, SCH2(CH2)10CH3], 1.10 [s, 9H, C(CH3)3], 0.87 (t, 3H, J = 14.16 Hz, CH3). ESI-HRMS m/z [(M + Na)+]: calcd. for C66H79NO11SSiNa, 1144.5035; found, 1144.5013.
Dodecyl 2,3-di-O-(4-methyl)benzoyl-4,6-O-(2-naphthylidene)-β-d-galactopyranosyl-(1→4)-3-O-benzyl-6-O-tert-butyldiphenylsilyl-2-deoxy-2-phthalimido-1-thio-β-d-glucopyranoside (14)
Compound 13 (1.10 g, 0.98 mmol) was dissolved in CH2Cl2 (5.0 mL) and pyridine (30 mL). p-Toluoylchloride (312 μL, 2.35 mmol) and a catalytic amount of DMAP were added to the solution while stirring for 5 d. The reaction mixture was diluted with CHCl3. The organic phase was successively washed with 1 m HCl, aq. NaHCO3 and brine, and dried over anhyd. MgSO4. The volatiles were removed under reduced pressure, and the residue was treated in a column of silica gel (10:1 toluene-EtOAc) to quantitatively give 14 (1.35 g). 1H-NMR δH (CDCl3): 7.85–6.75 (m, 34H, Ar H), 5.91 (dd, 1H, J1′,2′ = 8.04 Hz, J2′,3′ = 10.44 Hz, H-2′), 5.65 (s, 1H, NpCH), 5.29 (dd, 1H, J3′,4′ = 3.66 Hz, H-3′), 5.25 (d, 1H, H-1′), 5.14, 4.72 (ABq, 2H, J = 12.30 Hz, PhCH2), 5.12 (d, 1H, J1,2 = 10.26 Hz, H-1), 4.60 (d, 1H, H-4′), 4.53 (d, 1H, Jgem = 12.36 Hz, H-6′a), 4.47 (dd, 1H, J3,4 = 8.40 Hz, J4,5 = 9.72 Hz, H-4), 4.32 (dd, 1H, J2,3 = 10.26 Hz, H-3), 4.26 (t, 1H, H-2), 4.10 (d, 1H, H-6b′), 3.92 (d, 1H, Jgem = 10.56 Hz, H-6a), 3.83 (d, 1H, H-6b), 3.57 (s, 1H, H-5′), 3.23 (d, 1H, H-5), 2.62 (m, 1H, 1/2SCH2), 2.53 (m, 1H, 1/2SCH2), 2.32, 2.29 (2 s, each 3H, 2MePh), 1.48–1.18 [m, 20H, SCH2(CH2)10CH3], 1.14 [s, 9H, C(CH3)3], 0.87 (t, 3H, J = 14.22 Hz, CH3). ESI-HRMS m/z [(M + Na)+]: calcd. for C82H91NO13SSiNa, 1380.5873; found, 1380.5852.
Dodecyl 2,3-di-O-(4-methyl)benzoyl-6-O-(2-naphthylmethyl)-β-d-galactopyranosyl-(1→4)-3-O-benzyl-6-O-tert-butyldiphenylsilyl-2-deoxy-2-phthalimido-1-thio-β-d-glucopyranoside (15)
To a solution of 14 (1.35 g, 0.98 mmol) in THF (37 mL) was added BH3·NMe3
(427.3 mg, 5.88 mmol) while stirring for 30 min. To this mixture, AlCl3 (786.6 mg, 5.88 mmol) and H2O (1 drop) were added while stirring overnight. The reaction was quenched with aq. NH4Cl and diluted with EtOAc. The organic phase was successively washed with aq. NaHCO3 and brine, and dried over anhyd. MgSO4. The volatiles were removed under reduced pressure, and the residue was treated in an LH-20 gel permeation column (1:1 CHCl3-MeOH) to give 15 (1.23 g) in a 92% yield. 1H-NMR δH (CDCl3): 7.83–6.79 (m, 34H, Ar H), 5.80 (dd, 1H, J1′,2′ = 8.04 Hz, J2′,3′ = 10.32 Hz, H-2′), 5.21 (dd, 1H, J3′,4′ = 3.18 Hz, H-3′), 5.25 (d, 1H, H-1′), 5.11 (d, 1H, J1,2 = 10.02 Hz, H-1), 4.94, 4.60 (ABq, 2H, J = 12.30 Hz, ArCH2), 4.71, 4.67 (ABq, 2H, J = 12.18 Hz, ArCH2), 4.44 (dd, 1H, J3,4 = 8.40 Hz, J4,5 = 9.72 Hz, H-4), 4.42 (dd, 1H, J4′,OH = 3.84 Hz, H-4′), 4.32 (br t, 1H, J = 10.20 Hz, H-3), 4.26 (br t, 1H, J = 10.20 Hz, H-2), 3.76 (m, 5H, H-6ab,5′,6′ab), 3.22 (d, 1H, H-5), 2.73 (d, 1H, OH-4′), 2.60 (m, 1H, 1/2SCH2), 2.52 (m, 1H, 1/2SCH2), 2.35, 2.29 (2 s, each 3H, 2MePh), 1.47–1.06 [m, 20H, SCH2(CH2)10CH3], 1.06 [s, 9H, C(CH3)3], 0.86 (t, 3H, J = 14.28 Hz, CH3). ESI-HRMS m/z [(M + Na)+]: calcd. for C82H93NO13SSiNa, 1382.6029; found, 1382.6003.
Dodecyl 2,3,4-tri-O-(4-methyl)benzoyl-6-O-(2-naphthylmethyl)-β-d-galactopyranosyl-(1→4)-3-O-benzyl-6-O-tert-butyldiphenylsilyl-2-deoxy-2-phthalimido-1-thio-β-d-glucopyranoside (16)
To a solution of 15 (152.2 mg, 111.8 μmol) in CH2Cl2 (1.5 mL) and pyridine (1.0 mL) p-toluoylchloride (18 μL, 0.13 mmol) and a catalytic amount of DMAP were added while stirring for 6 d. The reaction mixture was treated in the same way for the synthesis of 14. The crude mixture was treated in an LH-20 gel permeation column (1:1 CHCl3-MeOH) to give 16 (161.2 mg) in a 98% yield. 1H-NMR δH (CDCl3): 7.90–6.90 (m, 38H, Ar H), 5.95 (d, 1H, J3′,4′ = 3.54 Hz, H-4′), 5.76 (dd, 1H, J1′,2′ = 8.04 Hz, J2′,3′ = 10.38 Hz, H-2′), 5.47 (dd, 1H, H-3′), 5.25 (d, 1H, H-1′), 5.12 (d, 1H, J1,2 = 10.14 Hz, H-1), 4.98, 4.69 (ABq, 2H, J = 11.88 Hz, ArCH2), 4.67, 4.48 (ABq, 2H, J = 11.58 Hz, ArCH2), 4.46 (br t, 1H, J = 9.06 Hz, H-4), 4.34 (dd, 1H, J2,3 = 10.14 Hz, J3,4 = 8.28 Hz, H-3), 4.30 (t, 1H, H-2), 4.08 (br t, 1H, J = 6.39 Hz, H-6′a), 3.85 (br d, 2H, H-6ab), 3.66 (dd, 1H, J5′,6′b = 9.66 Hz, Jgem = 5.70 Hz, H-6′b), 3.57 (dd, 1H, J5′,6′a = 7.68 Hz, H-5′), 3.24 (br d, 1H, J = 9.78 Hz, H-5), 2.63 (m, 1H, 1/2SCH2), 2.55 (m, 1H, 1/2SCH2), 2.30, 2.30, 2.29 (3 s, each 3H, 3MePh), 1.52–1.12 [m, 20H, SCH2(CH2)10CH3], 1.08 [s, 9H, C(CH3)3], 0.87 (t, 3H, J = 14.28 Hz, CH3). ESI-HRMS m/z [(M + Na)+]: calcd. for C90H99NO14SSiNa, 1500.6448; found, 1500.6411.
2-(N-Benzyloxycarbonyl)aminoethyl 2,3,4-tri-O-(4-methyl)benzoyl-6-O-(2-naphthylmethyl)-β-d-galactopyranosyl-(1→4)-3-O-benzyl-6-O-tert-butyldiphenylsilyl-2-deoxy-2-phthalimido-β-d-glucopyranoside (17)
AW 300 molecular sieves (993.2 mg) were added to a solution of 16 (819.1 mg, 553.9 μmol) and benzyl N-(2-hydroxyethyl)carbamate (162.2 mg, 830.9 μmol) in (CH2Cl)2 (8.5 mL) and Et2O (29 mL), and the mixture was stirred for 30 min at rt. To this mixture, NIS (249.8 mg, 1.11 mmol) and TfOH (15 μL, 0.16 mmol) were added while stirring for 6 h at rt. The reaction mixture was treated in the same way as that for the synthesis of 11. The crude mixture was treated in a column of silica gel (20:1–15:1 toluene-EtOAc) to quantitatively give 17 (867.6 mg). 1H-NMR δH (CDCl3): 7.77–6.74 (m, 43H, Ar H), 5.95 (d, 1H, J3′,4′ = 3.36 Hz, H-4′), 5.76 (dd, 1H, J1′,2′ = 8.04 Hz, J2′,3′ = 10.38 Hz, H-2′), 5.47 (dd, 1H, H-3′), 5.27 (d, 1H, H-1′), 5.05 (d, 1H, J1,2 = 8.58 Hz, H-1), 4.98, 4.67 (ABq, 2H, J = 11.97 Hz, ArCH2), 4.89 (m, 3H, NH, ArCH2), 4.68, 4.48 (ABq, 2H, J = 12.24 Hz, ArCH2), 4.46 (br t, 1H, J = 9.18 Hz, H-4), 4.28 (dd, 1H, J2,3 = 10.80 Hz, J3,4 = 8.82 Hz, H-3), 4.17 (dd, 1H, H-2), 4.07 (br t, 1H, J = 6.84 Hz, H-5′), 3.89 (br d, 1H, J = 10.38 Hz, H-6a), 3.81 (br d, 1H, J = 11.40 Hz, H-6b), 3.70 (m, 1H, 1/2OCH2), 3.67 (dd, 1H, J5′,6′a = 5.52 Hz, Jgem = 9.54 Hz, H-6′a), 3.58 (dd, 1H, J5′,6′b = 7.68 Hz, H-6′b), 3.45 (m, 1H, 1/2OCH2), 3.24 (m, 3H, H-5, NHCH2), 2.28 (s, 9H, 3MePh), 1.09 [s, 9H, C(CH3)3]. ESI-HRMS m/z [(M + Na)+]: calcd. for C88H86N2O17SiNa, 1493.5588; found, 1493.5549.
2-(N-Benzyloxycarbonyl)aminoethyl 2,3,4-tri-O-(4-methyl)benzoyl-6-O-(2-naphthylmethyl)-β-d-galactopyranosyl-(1→4)-3-O-benzyl-2-deoxy-2-phthalimido-β-d-glucopyranoside (18)
Compound 17 (867.6 mg, 553.9 μmol) was dissolved in THF (4.0 mL) and AcOH (320 μL, 5.55 mmol). To this mixture, 1 m TBAF (2.8 mL, 2.8 mmol) was added while stirring for 13 d. The reaction mixture was diluted with CHCl3. The organic phase was washed with brine and dried over MgSO4. The volatiles were removed under reduced pressure, and the residue was treated in an LH-20 gel permeation column (1:1 CHCl3-MeOH) to give 18 (538.4 mg) in a 79% yield. 1H-NMR δH (CDCl3): 7.83–6.75 (m, 33H, Ar H), 5.91 (d, 1H, J3′,4′ = 3.24 Hz, H-4′), 5.91 (dd, 1H, J1′,2′ = 7.98 Hz, J2′,3′ = 10.38 Hz, H-2′), 5.56 (dd, 1H, H-3′), 5.06 (d, 1H, J1,2 = 8.34 Hz, H-1), 5.06 (s, 1H, NH), 5.04 (d, 1H, H-1′), 4.97, 4.67 (ABq, 2H, J = 12.45 Hz, ArCH2), 4.89 (m, 2H, ArCH2), 4.59, 4.43 (ABq, 2H, J = 12.36, Hz, ArCH2), 4.30 (br t, 1H, J = 9.04 Hz, H-3), 4.15 (br t, 1H, H-5′), 4.12 (m, 1H, H-2), 4.04 (br t, 1H, J = 9.27 Hz, H-4), 3.70 (br s, 2H, H-6ab), 3.34 (m, 1H, 1/2OCH2), 3.55 (m, 3H, H-6′ab, 1/2OCH2), 3.28 (m, 2H, H-5, 1/2NCH2), 3.16 (m, 1H, 1/2NCH2), 2.37, 2.32, 2.29 (3 s, each 3H, 3MePh), 1.99 (br s, 1H, OH-6). ESI-HRMS m/z [(M + Na)+]: calcd. for C77H68N2O17Na, 1255.4410; found, 1255.4380.
2-(N-Benzyloxycarbonyl)aminoethyl 2,3,4-tri-O-(4-methyl)benzoyl-β-d-galactopyranosyl-(1→4)-6-O-acetyl-3-O-benzyl-2-deoxy-2-phthalimido-β-d-glucopyranoside (19)
To a solution of 18 (130.6 mg, 105.9 μmol) in CH2Cl2 (2.0 mL), pyridine (90 μL) and Ac2O (90 μL) were added while stirring overnight. The volatiles were removed under reduced pressure. To the residue in CH2Cl2 (2.1 mL) and MeOH (0.6 mL), DDQ (24.0 mg, 106 μmol) and H2O (1 drop) were added while stirring for 3 d. The reaction mixture was diluted in CHCl3. The organic phase was treated as described for the general methods. The residue was treated in an LH-20 gel permeation column (1:1 CHCl3-MeOH) to give 19 (84.1 mg) with an 84% yield in two steps. 1H-NMR δH (CDCl3): 7.93–6.80 (m, 26H, Ar H), 5.87 (dd, 1H, J1′,2′ = 7.92 Hz, J2′,3′ = 10.38 Hz, H-2′), 5.72 (d, 1H, J3′,4′ = 3.30 Hz, H-4′), 5.50 (dd, 1H, H-3′), 5.07 (d, 1H, J1,2 = 8.58 Hz, H-1), 5.07 (br s, 1H, NH), 4.95, 4.90 (ABq, 2H, J = 12.24 Hz, PhCH2), 4.89, 4.52 (ABq, 2H, J = 12.06 Hz, PhCH2), 4.86 (d, 1H, H-1′), 4.35 (m, 1H, H-6a), 4.31 (m, 1H, H-3), 4.16 (m, 1H, H-6b), 4.11 (m, 1H, H-2), 3.92 (m, 2H, H-4,5′), 3.64 (m, 1H, 1/2OCH2), 3.53 (m, 2H, H-5, 1/2OCH2), 3.46 (m, 1H, H-6′a), 3.36 (m, 1H, H-6′b), 3.20 (m, 2H, NCH2), 2.54 (br s, 1H, 6′-OH), 2.38, 2.37, 2.28 (3 s, each 3H, 3MePh), 2.01 (s, 3H, Ac). ESI-HRMS m/z [(M + Na)+]: calcd. for C63H62N2O18Na, 1157.3890; found, 1157.3862.
2-(N-Benzyloxycarbonyl)aminoethyl 2,3,4-tri-O-(4-methyl)benzoyl-β-d-galactopyranosyl-(1→4)-3-O-benzyl-2-deoxy-2-phthalimido-β-d-glucopyranoside (20)
To a solution of 18 (77.0 mg, 62.4 μmol) in CH2Cl2 (1.3 mL) and MeOH (0.4 mL), DDQ (16.8 mg, 74.9 μmol) and H2O (1 drop) were added while stirring overnight. The reaction mixture was treated in the same way as that for the synthesis of 19, and the residue was treated in an LH-20 gel permeation column (1:1 CHCl3-MeOH) to give 20 (52.9 mg) in a 78% yield. 1H-NMR δH (CDCl3): 7.95–6.78 (m, 26H, Ar H), 5.85 (dd, 1H, J1′,2′ = 7.95 Hz, J2′,3′ = 10.32 Hz, H-2′), 5.77 (d, 1H, J3′,4′ = 3.06 Hz, H-4′), 5.59 (dd, 1H, H-3′), 5.24 (br t, J = 5.46 Hz, NH), 5.16 (d, 1H, J1,2 = 7.62 Hz, H-1), 5.15 (d, 1H, H-1′), 4.92, 4.65 (ABq, 2H, J = 12.12 Hz, PhCH2), 4.82, 4.66 (ABq, 2H, J = 12.36 Hz, PhCH2), 4.27 (m, 1H, H-3), 4.21 (m, 1H, H-2), 4.11 (m, 1H, H-6′a), 3.85 (br d, 1H, J = 12.18 Hz, H-6a), 3.75 (br s, 1H, H-6b), 3.51 (m, 3H, H-5′,6′b, 1/2OCH2), 3.33 (br d, 1H, J = 6.12 Hz, H-4), 3.20 (m, 1H, 1/2NCH2), 3.10 (m, 1H, 1/2NCH2), 2.89 (br s, 1H, H-5), 2.38, 2.33, 2.28 (3 s, each 3H, 3MePh). ESI-HRMS m/z [(M + Na)+]: calcd. for C61H60N2O17Na, 1115.3784; found, 1115.3760.
O-Sulfation of 19, 18 and 20
The starting material was dissolved in DMF (2.0 mL per 50 mg of the starting material). To this solution, SO3·Me3N (20 equiv. per hydroxyl group) was added while stirring at 60 °C for 1–3 h. The reaction mixture was treated in an LH-20 gel permeation column (1:1 CHCl3-MeOH) to give the corresponding sulfate (21, 22, and 23) in respective 88%, quantitative and 90% yields.
2-(N-Benzyloxycarbonyl)aminoethyl 2,3,4-tri-O-(4-methyl)benzoyl-6-O-sulfo-β-d-galactopyranosyl-(1→4)-6-O-acetyl-3-O-benzyl-2-deoxy-2-phthalimido-β-d-glucopyranoside, trimethylamine salt (21)
1H-NMR δH (CDCl3): 7.91–6.85 (m, 26H, Ar H), 5.93 (br s, 1H, H-4′), 5.78 (dd, 1H, J1′,2′ = 7.92 Hz, J2′,3′ = 10.32 Hz, H-2′), 5.46 (dd, 1H, J3′,4′ = 2.94 Hz, H-3′), 5.06 (d, 2H, J1,2 = 12.06 Hz, H-1, NH), 5.00, 4.59 (ABq, 2H, J = 12.06 Hz, PhCH2), 4.94, 4.90 (ABq, 1H, J = 12.30 Hz, PhCH2), 4.92 (d, 1H, H-1′), 4.39 (m, 1H, H-5′), 4.33 (d, 1H, Jgem = 11.28 Hz, H-6b), 4.27 (m, 2H, H-3,6′a), 4.17 (dd, 1H, J5,6a = 4.32 Hz, H-6a), 4.15 (m, 1H, H-6′b), 3.96 (m, 2H, H-2,4), 3.62 (m, 1H, 1/2CH2), 3.53 (m, 1H, 1/2CH2), 3.48 (m, 2H, H-5,5′), 3.21 (br s, 2H, CH2), 2.77 (t, 9 H, NMe3), 2.37, 2.32, 2.29 (3 s, each 3H, 3MePh), 2.05 (s, 3H, Ac). ESI-HRMS m/z [(M + Na)+]: calcd. for C63H61N2O21SNaCitation1,Citation2259.3277; found, 1259.3248.
2-(N-Benzyloxycarbonyl)aminoethyl 2,3,4-tri-O-(4-methyl)benzoyl-6-O-(2-naphthylmethyl)-β-d-galactopyranosyl-(1→4)-3-O-benzyl-2-deoxy-2-phthalimido-6-O-sulfo-β-d-glucopyranoside, trimethylamine salt (22)
1H-NMR δH (CD3OD): 7.96–6.56 (m, 33H, Ar H), 5.84 (d, 1H, J3′,4′ = 3.36 Hz, H-4′), 5.64 (dd, 1H, J1′,2′ = 7.80 Hz, J2′,3′ = 10.26 Hz, H-2′), 5.56 (dd, 1H, H-3′), 5.37 (d, 1H, H-1′), 5.01 (d, 1H, J1,2 = 8.52 Hz, H-1), 4.89, 4.47 (ABq, 1H, J = 11.64 Hz, ArCH2), 4.69 (s, 2H, ArCH2), 4.61, 4.36 (ABq, 2H, J = 12.18 Hz, ArCH2), 4.35 (d, 1H, Jgem = 10.50 Hz, H-6a), 4.33 (br t, J = 6.60 Hz, H-5′), 4.17 (dd, 1H, J2,3 = 10.50 Hz, J3,4 = 9.18 Hz, H-3), 4.09 (br t, 1H, J = 9.42 Hz, H-4), 4.03 (d, 1H, H-6b), 3.92 (dd, 1H, H-2), 3.60 (m, 1H, 1/2CH2), 3.55 (dd, 1H, J5′,6′a = 5.82 Hz, Jgem = 9.54 Hz, H-6′a), 3.49 (dd, 1H, J5′,6′b = 7.50 Hz, H-6′b), 3.37 (m, 2H, 1/2CH2, H-5), 3.30 (m, 2H, CH2), 2.77 (s, 9 H, NMe3), 2.27, 2.18, 2.15 (3 s, each 3H, 3MePh). ESI-HRMS m/z [(M + Na)+]: calcd. for C72H68N2O20SNa, 1357.3798; found, 1357.3782.
2-(N-Benzyloxycarbonyl)aminoethyl 2,3,4-tri-O-(4-methyl)benzoyl-6-O-sulfo-β-d-galactopyranosyl-(1→4)-3-O-benzyl-2-deoxy-2-phthalimido-6-O-sulfo-β-d-glucopyranoside, bistrimethylamine salt (23)
1H-NMR δH (CD3OD): 7.95–6.75 (m, 26H, Ar H), 5.83 (d, 1H, J3′,4′ = 3.18 Hz, H-4′), 5.67 (dd, 1H, J1′,2′ = 7.92 Hz, J2′,3′ = 10.26 Hz, H-2′), 5.56 (dd, 1H, H-3′), 5.40 (d, 1H, H-1′), 5.01 (d, 1H, J1,2 = 8.58 Hz, H-1), 4.92, 4.57 (ABq, 1H, J = 12.00 Hz, PhCH2), 4.69 (s, 2H, PhCH2), 4.39 (br t, 1H, J = 6.63 Hz, H-5′), 4.35 (br d, 1H, J = 9.30 Hz, H-6a), 4.14 (m, 2H, H-3,6′a), 4.01 (m, 3H, H-4,6b,6′b), 3.86 (dd, 1H, J2,3 = 10.44 Hz, H-2), 3.57 (m, 1H, 1/2OCH2), 3.36 (m, 1H, 1/2OCH2), 3.35 (d, 1H, J5,6a = 9.96 Hz, H-5), 2.99 (t, 2H, NCH2), 2.82 (s, 18 H, 2NMe3), 2.25, 2.18, 2.17 (3 s, each 3H, 3MePh). ESI-HRMS m/z [(M + Na)+]: calcd. for C61H58N2O23S2Na1, 3319.2559; found, 1319.2536.
Biotinylated KS (0S) disaccharide (1)
To a solution of 18 (57.2 mg, 46.4 μmol) in EtOH (2.4 mL) was added 1,3-diaminopropane (232 μL, 2.8 mmol). The reaction mixture was heated overnight under reflux, and then the volatiles were removed under reduced pressure. To the residue in MeOH (5.0 mL), Ac2O (0.2 mL) and Et3N (0.2 mL) were added while stirring overnight. The volatiles were removed again, and the residue was subjected to reverse-phase chromatography in a Bond Elut C8 column (0–90% MeOH). The product was dissolved in 50% aq. EtOH (2.0 mL) and then hydrogenated in the presence of a catalytic amount of Pd-black in an H2 atomosphere, while stirring for 2 d. H2O (1.0 mL) and AcOH (1 drop) were added to the reaction mixture, and the reaction was continued for 4 d. The insoluble materials were removed on Celite, and the volatiles were removed under reduced pressure. The product having free amine at the end of the linker was dissolved in 1 M Na3PO4 and 0.15 M NaCl (1.5 mL). To this solution, NHS-PEG4-biotin (60.4 mg, 99.7 μmol) was added while stirring overnight. The reaction mixture was treated in an LH-20 gel permeation column (H2O) to quantitatively give 1 from 18. 1H-NMR δH (D2O, selected): 4.51 (dd, 1H, Jb,c = 7.92 Hz, Jc,d = 4.86 Hz, H-c), 4.48 (d, 1H, J1,2 = 8.37 Hz, H-1), 4.38 (d, 1H, J1′,2′ = 7.86 Hz, H-1′), 4.33 (dd, 1H, Jb,e = 4.50 Hz, H-b), 4.00 (m, 1H, H-r), 3.90 (dd, 1H, J5′,6′a = 2.16 Hz, Jgem = 12.18 Hz, H-6′a), 3.84 (d, 1H, J3′,4′ = 3.36 Hz, H-4′), 3.79 (m, 1H, H-r), 3.75 (dd, 1H, J5′,6′b = 5.10 Hz, H-6′b), 3.75–3.60 (m, 2H, H-6ab), 3.67 (m, 2H, H-2,3), 3.58 (dd, 1H, J2′,3′ = 9.90 Hz, H-3′), 3.52 (m, 3H, H-4,5,5′), 3.45 (dd, 1H, H-2′), 3.24 (m, 1H, H-e), 3.17 (m, 2H, H-q), 2.90 (dd, 1H, Jgem = 13.02 Hz, H-d), 2.69 (d, 1H, H-d′), 1.96 (s, 3H, NAc). ESI-HRMS m/z [(M + Na)+]: calcd. for C37H65N5O18SNa, 922.3938; found, 922.3925.
Biotinylated KS (6′S) disaccharide (2)
To a solution of 21 (80.9 mg, 65.4 μmol) in EtOH (3.5 mL) was added 1,3-diaminopropane (290 μL, 3.9 mmol). The reaction mixture was heated overnight under reflux, and then the volatiles were removed under reduced pressure. To the residue in MeOH (2.3 mL), Ac2O (0.3 mL) and Et3N (0.3 mL) were added while stirring overnight. The volatiles were removed again, and the residue was subjected to reverse-phase chromatography in a Bond Elut C8 column (0–90% MeOH). To a solution of the product in MeOH (0.6 mL) was added 0.5 m NaOH (250 μL). The reaction mixture was heated under reflux for 9 h and then quenched with 1% AcOH. The volatiles were removed under reduced pressure, and the residue was purified through a Bond Elut C8 column (0–90% MeOH) as already described. The product was dissolved in 50% aq. 2-PrOH (1.0 mL) and then hydrogenated in the presence of a catalytic amount of Pd-black in an H2 atomosphere while stirring for 1 d. H2O (1.0 mL) and AcOH (1 drop) were added to the reaction mixture, and the reaction was continued for 2 d. The insoluble materials were removed on Celite, and the volatiles were removed under reduced pressure. The product was dissolved in 1 m Na3PO4 and 0.15 m NaCl (0.8 mL). To this solution, NHS-PEG4-biotin (27.7 mg, 47.1 μmol) was added while stirring overnight. The reaction mixture was treated in an LH-20 gel permeation column (H2O) to quantitatively give 2 from 21. 1H-NMR δH (D2O, selected): 4.45 (m, 2H, H-c, 1), 4.37 (d, 1H, J1′,2′ = 8.37 Hz, H-1′), 4.27 (m, 1H, H-b), 4.13 (d, 1H, Jgem = 4.74 Hz, H-6′a), 3.96 (d, 1H, H-6′b), 3.75 (m, 1H, H-2), 3.74–3.59 (m, 2H, H-6ab), 3.57 (m, 1H, H-2′), 3.36 (m, 1H, H-e), 2.85 (dd, 1H, Jc,d = 5.10 Hz, Jgem = 13.02 Hz, H-d), 2.63 (d, 1H, H-d′), 1.90 (s, 3H, NAc). ESI-HRMS m/z [(M + Na)+]: calcd. for C37H64N5O21S2Na2, 1024.3325; found, 1024.3331.
Biotinylated KS (6S) disaccharide (3)
To a solution of 22 (64.1 mg, 46.7 μmol) in EtOH (2.7 mL) was added 1,3-diaminopropane (234 μL, 2.8 mmol). The reaction mixture was heated overnight under reflux, and then the volatiles were removed under reduced pressure. To the residue in MeOH (1.5 mL) were added Ac2O (0.2 mL) and Et3N (0.2 mL) while stirring overnight. The volatiles were removed again, and the residue was subjected to reverse-phase chromatography in a Bond Elut C8 column (0–90% MeOH). To a solution of the product in MeOH (0.6 mL) was added 0.5 M NaOH (250 μL). The reaction mixture was heated under reflux for 6 h and then quenched with 1% AcOH. The volatiles were removed under reduced pressure, and the residue was purified through Bond Elut C8 (0–90% MeOH) as already described. The product was dissolved in 50% 2-PrOH (1.0 mL) and then hydrogenated in the presence of a catalytic amount of Pd-black in an H2 atmosphere while stirring for 2 d. H2O (1.0 mL) and AcOH (1 drop) were added to the reaction mixture, and the reaction was continued for 4 d. The insoluble materials were removed on Celite, and the volatiles were removed under reduced pressure. The product having free amine at the end of the linker was dissolved in 1 M Na3PO4 and 0.15 M NaCl (0.8 mL). To this solution, NHS-PEG4-biotin (9.9 mg, 18.5 μmol) was added while stirring overnight. The reaction mixture was treated in an LH-20 gel permeation column (H2O) to quantitatively give 3 from 22. 1H-NMR δH (D2O, selected): 4.54 (m, 1H, H-c), 4.51 (m, 2H, H-1,1′), 4.38 (m, 1H, H-b), 4.35 (m, 1H, H-6a), 4.27 (br s, 1H, H-6b), 3.86 (br d, 1H, J3′,4′ = 3.48 Hz, H-4′), 3.85–3.58 (m, 2H, H-6′ab),3.70 (m, 1H, H-2), 3.68 (m, 1H, H-3), 3.62 (m, 1H, H-3′), 3.63 [dd, 1H, J1′,2′, J2′,3′ = 9.87, 7.98 Hz (reversible), H-2′], 3.28 (m, 1H, H-e), 2.91 (dd, 1H, Jc,d = 4.95 Hz, Jgem = 13.02 Hz, H-d), 2.71 (d, 1H, H-d′), 1.96 (s, 3H, NAc). ESI-HRMS m/z [(M + Na)+]: calcd. for C37H64N5O21S2Na2, 1024.3325; found, 1024.3325.
Biotinylated KS (6,6′-diS) disaccharide (4)
To a solution of 23 (67.1 mg, 49.0 μmol) in EtOH (2.8 mL) was added 1,3-diaminopropane (245 μL, 2.9 mmol). The reaction mixture was heated overnight under reflux, and then the volatiles were removed under reduced pressure. To the residue in MeOH (1.5 mL), Ac2O (0.2 mL) and Et3N (0.2 mL) were added while stirring overnight. The volatiles were removed again, and the residue was subjected to Bond Elut C8 reverse-phase chromatography (0–90% MeOH). To a solution of the product in 50% aq. MeOH (1.5 mL) was added 0.5 M NaOH (200 μL). The reaction mixture was heated under reflux for 8 h and then quenched with 1% AcOH. The volatiles were removed under reduced pressure, and the residue was purified through Bond Elut C8 (0–90% MeOH) as already described. The product was dissolved in H2O (1.5 mL) and AcOH (1 drop) and then hydrogenated in the presence of a catalytic amount of Pd-black in an H2 atmosphere while stirring for 4 d. The insoluble materials were removed on Celite, and the volatiles were removed under reduced pressure. The product having free amine at the end of the linker was dissolved in 1 M Na3PO4 and 0.15 M NaCl (0.3 mL). To this was added NHS-PEG4-biotin (2.6 mg, 4.6 μmol) while stirring overnight. The reaction mixture was treated in an LH-20 gel permeation column (H2O) to quantitatively give 4 from 23. 1H-NMR δH (D2O, selected): 4.54 (dd, 1H, Jb,c = 7.92 Hz, Jc,d = 4.86 Hz, H-c), 4.49 (d, 1H, J1,2 = 7.92 Hz, H-1), 4.47 (d, 1H, J1′,2′ = 7.86 Hz, H-1′), 4.35 (m, 2H, H-b,6a), 4.24 (m, 1H, H-6b), 4.10 (m, 1H, H-6′a), 3.94 (d, 1H, J3′,4′ = 3.06 Hz, H-4′), 3.93 (dd, 1H, H-6′b), 3.84 (m, 1H, H-r), 3.70 (m, 1H, H-3), 3.69 (br t, 1H, J = 5.94 Hz, H-2), 3.63 (m, 1H, H-3′), 3.48 (dd, 1H, J2′,3′ = 10.02 Hz, H-2′), 3.33 (m, 2H, H-q), 3.27 (m, 1H, H-e), 2.93 (dd, 1H, Jgem = 13.02 Hz, H-d), 2.72 (d, 1H, H-d′), 1.94 (s, 3H, NAc). ESI-HRMS m/z [(M + Na)+]: calcd. for C37H63N5O24S3Na1, 3126.2713; found, 1126.2700.
Acknowledgments
This study was supported by grant aid for scientific research in innovative areas (23110003) from MEXT, Japan. N.T. is grateful for the JSPS Research Fellowship for Young Scientists. We thank Mrs. Mayumi Ikenari (Research Center for Bioscience and Technology, Division of Instrumental Analysis, Tottori University) for performing the ESI-HRMS measurements.
Notes
Abbreviations: KS, keratan sulfate; NAP, 2-naphthylmethyl; TBDPS, tert-butyldiphenylsilyl; TMSOTf, trimethylsilyl trifluoromethanesulfonate; MBz, 4-methylbenzoyl; Z, benzyloxycarbonyl; Bn, benzyl; NIS, N-iodosuccinimide; TfOH, trifluoromethanesulfonic acid; DDQ, 2,3-dichloro-5,6-dicyano-1,4-benzoquinone; Phth, phthaloyl; DMAP; dimethylaminopyridine; THF, tetrahydrofurane; MS4A, molecular sieves 4A.
Related Research Data
References
- Imagama S, Sakamoto K, Tauchi R, Shinjo R, Ohgomori T, Ito Z, Zhang H, Nishida Y, Asami N, Takeshita S, Sugiura N, Watanabe H, Yamashita T, Ishiguro N, Matsuyama Y, Kadomatsu K. Keratan sulfate restricts neural plasticity after spinal cord injury. J. Neurosci. 2011;31:17091–17102.
- Kobayashi M, Yamazaki F, Ito Y, Ogawa T. A synthetic approach to karatan sulfate I: synthesis of trisulfated glycotetraose. Tetrahedron Lett. 1989;30:4547–4550.
- Kobayashi M, Yamazaki F, Ito Y, Ogawa T. A regio- and stereo-controlled synthesis of β-D-GlcpNAc6SO3-(1→3)-β-D-Galp6SO3-(1→4)-β-D-GlcpNAc6SO3-(1→3)-D-Galp, a linear acidic glycan fragment of keratan sulfate I. Carbohydr. Res. 1990;201:51–67.
- Misra AK, Agnihotri G, Madhusudan SK, Tiwari P. Practical synthesis of sulfated analogs of lactosamine and sialylated lactosamine derivatives. J. Carbohydr. Chem. 2004;23:191–199.
- Zhu T, Boons G-J. Intermolecular aglycon transfer of ethyl thioglycosides can be prevented by judicious choice of protecting groups. Carbohydr. Res. 2000;329:709–715.
- Li Z, Gildersleeve JC. Mechanistic studies and methods to prevent aglycon transfer of thioglycosides. J. Am. Chem. Soc. 2006;128:11612–11619.
- Belot F, Jacquinet JC. Intermolecular aglycon transfer of a phenyl 1-thiogalactosaminide derivative under trichloroacetimidate glycosylation conditions. Carbohydr. Res. 1996;290:79–86.
- Yu H, Yu B, Wu X, Hui Y, Han X. Synthesis of a group of diosgenyl saponins with combined use of glycosyl trichloroacetimidate and thioglycoside donors. J. Chem. Soc., Perkin Trans. 1. 2000;1445–1453.
- Fujiwara K, Goto A, Sato D, Ohtaniuchi Y, Tanaka H, Murai A, Kawai H, Suzuki T. Convergent synthesis of the ABCDE-ring part of ciguatoxin CTX3C. Tetrahedron Lett. 2004;45:7011–7014.
- Borbás A, Szabó ZB, Szilágyi L, Bényei A, Lipták A. Dioxane-type (2-naphthyl)methylene acetals of glycosides and their hydrogenolytic transformation into 6-O- and 4-O-(2-naphthyl)methyl (NAP) ethers. Tetrahedron. 2002;58:5723–5732.