
Abstract
MicroRNA (miRNA) identification was performed in Hemerocallis fulva by high-throughput sequencing in combination with bioinformatics prediction. A total of 14,843,184 and 16,072,575 RNA sequences were explored under normal and low temperature conditions, respectively. There was a significant difference in RNAs species and quantity between the two samples. Of all the miRNAs, 26 were significantly upregulated and 30 were significantly downregulated, while nine were either significantly upregulated or downregulated under low-temperature stress. Twenty-one highly expressed miRNA families were screened in at least six species. The number of miRNA families was very similar between monocotyledons and dicotyledons, and only a few were more frequently found in monocotyledons.
MicroRNAs (miRNAs), as endogenous non-coding small single-stranded RNAs of 16–29 nt, are the most widely studied at present.Citation1) Mature miRNAs display high evolutionary conservation. Recent studies have found that miRNAs are widely distributed in the genomes of animals, plants, fungi, and viruses, regulating gene expression related to growth, development, and diseases of organisms. In addition, miRNAs are functionally involved in many physiological and pathological processes of eukaryotes, including cell proliferation, differentiation, development, and apoptosis, serving as a novel regulation mode of small interfering RNA (siRNA) systems.Citation2) The miRNAs also play a pivotal role in the process of making adjustments and responses to environmental stresses in plants.Citation3,4)
The first miRNA, small lin-4 RNA, was identified in 1993 in a study of temporal control of the gene lin-14 in C. Elegans development,Citation5) and the first plant miRNA was found in 2002.Citation6) In plants, sRNAs are classified into miRNAs and siRNAs, according to their biogenesis and precursor structures.Citation7−Citation8) Many miRNAs have been discovered in plants, animals, and microorganisms. MiRNA data for many species, including Arabidopsis thaliana,Citation9) rice,Citation10) maize,Citation11) wheat,Citation12) and poplarCitation13) have been recorded on the miRBase website (http://www.mirbase.org/), and more data are continually being included and updated.
On this basis, deep sequencing of small RNA (smRNA), now emerging as a powerful experimental method, has been used to identify a substantial number of known and unknown, conservative and non-conservative miRNAs at the genomic level, and to construct gene expression profiles of smRNAs among different samples. Extended NGS has been applied to identify RNA and DNA viruses in tomato and squash.Citation14) A new computational algorithm has also been developed to allow for homology-independent discovery of viroids.Citation15) This provides an effective tool for analyzing smRNA functioning and allows broader application in exploring plant and animal miRNAs.
Abiotic stresses, such as drought, salinity, and cold, regulate the expression of thousands of genes in plants at both the transcriptional and post-transcriptional level. We are interested in exploring the potential roles of small regulatory RNAs in abiotic stress tolerance and gene regulation. High salinity, drought, and low temperature are three common environmental stress factors that seriously influence plant growth and development worldwide. Recently, miRNAs have emerged as a class of gene expression regulators linked to stress responses, but the relationship between miRNA expression and stress responses is just beginning to be explored.
The existence of stress-related elements in miRNA promoter regions provides further evidence confirming our results. These findings extend the current view about miRNA as ubiquitous regulators under stress conditions.
Cold stress is one of the most common abiotic factors affecting plants. It adversely affects plant growth, development, and spatial distribution. To determine the roles of miRNAs under cold stress in Populus tomentosa, two smRNA libraries from plantlets treated or not with cold (4 °C for 8 h) have been constructed. Hundred and forty-four conserved miRNAs belonging to 33 miRNA families and 29 new miRNAs belonging to 23 miRNA families were identified. Among these, 21 were downregulated and 9 were upregulated in response to cold stress.Citation16) This study indicated that Populus miRNAs play critical roles in the cold stress.
miRNAs play important roles in plant development and in responses to environmental stress. Male sterility in thermosensitive genic male sterile (TGMS) lines of wheat (Triticum aestivum) is strictly controlled by temperature. The early phase of anther development is especially susceptible to cold stress. The smRNA libraries, obtained from spike tissues of the TGMS line under cold and control conditions were deep sequenced, and 78 unique miRNA sequences were obtained from 30 families and small trans-acting interfering RNAs (tasiRNAs) derived from two TAS3 genes.Citation17) Twenty-six targets of 16 miRNA families and three targets of tasi-RNA were identified. By comparison of smRNA sequencing data-sets and real-time PCR results, six-miRNAs and one tasi-RNA (tasiRNA-ARF, for Auxin-Responsive Factor) as cold stress-responsive smRNAs in spike tissues of the TGMS line were identified.
Hemerocallis sp., a perennation herbaceous plant, contains a variety of resistance genes against drought, high and low temperature, and salt stress, with good development potential and prospects. To the best of our knowledge, no studies on Hemerocallis sp. miRNA have been reported. In the current study, high-throughput sequencing (HiSeq) technology was employed to perform digital detection of smRNAs in Hemerocallis fulva, to identify miRNAs related to cold stress, and to discover novel miRNAs and their modes of under cold stress, to provide sufficient theoretical evidence for the exploration of stress-responsive genes, to determine the mechanism of stress-responsive proteins and relevant regulators during the expression process.
Materials and methods
Sample collection
Hongbaoshi, a cultivated variety of H. fulva, was used in this study. It was provided by the Plant Laboratory of Northeast Agricultural University. H. fulva were potted in an experimental field of Northeast Agricultural University. Plant showing good growth were selected and placed in growth chamber after 90 d of general management in the pots. The selected plants were uniformly treated for 14 d in growth chamber under a 12 h light/12 h dark photoperiod and 80% RH at 10 °C and then cold treated at –25 °C for 2 d. The plants growing at 10 °C under the same environmental condition were used as control. There were two biological replicates for each treatment. The roots were collected at 10 °C and at –25 °C. Root samples were rinsed with sterile water and dried with sterile paper, and then stored at –80 °C for subsequent experiments.
Extraction of total RNA
Total RNA was extracted from H. fulva with trizol. The concentration and purity of the miRNA were measured with a nucleic acid detector. Total RNA from two samples was evenly mixed, and RNA integrity was assessed by Agilent bioanalyzer (Agilent Technologies, Boston, MA).
Construction and sequencing of smRNA library
Total RNA was extracted from same amount of root sample mixture and prepared for construction of a smRNA library. Total RNA was separated on 15% denaturing polyacrylamide gels. smRNAs ranging from 17 to 24 nt in length were extracted, purified, and amplified by RT-PCR, and constitute smRNA and cDNA libraries for subsequent deep sequencing. Deep sequencing was conducted using HiSeq technology by BGI, Beijing.
Bioinformatic analysis of smRNA sequences
The extracted smRNA sequences were initially processed by removing the adaptor and the sequences of poor quality or showing contamination, and the sequence length distribution was measured statistically. The treated sequences were categorized and annotated to obtain the constituent data and expression levels for the various samples. The remaining smRNA sequences were statistically compared to rice miRNA sequences and other miRNA sequences, from which the contents of known miRNAs, and the distribution and expression of primary base and evolutionary degree were acquired.
Screening of differential expression of miRNA
The frequency of miRNA expression between samples at different temperatures (10 and –25 °C) was normalized to the same order. The normalized data between samples were compared statistically with regard to fold change and p values. Fold change of >1.5 at p < 0.01 was considered statistically significant, and the normalized data were used for statistical cartography.
Results
Assessment of RNA integrity
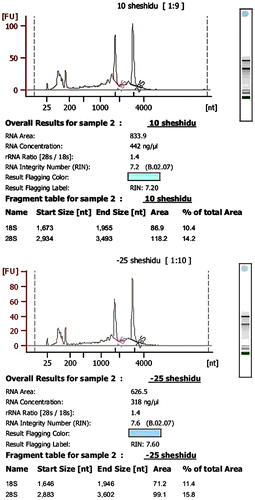
The results indicated that all samples contained 28S RNA peaks and that the ratio of the 28S/18S and RNA integrity numbers was relatively high, with a smooth baseline and no uplift. The sample showed no sign of degradation, and RNA integrity was well preserved, satisfying the standard and prepared for subsequent analysis (Fig. ).
Fig. 1. Assessment of RNA Integrity in H. fulva at low temperature.
Type and quantitative analysis of smRNA segments of H. fulva
With MATLAB software, 14,843,184 and 16,072,575 RNA sequences were identified under at 10 and –25 °C representing 14,064,385 (94.88%) and 15,309,725 (95.38%) smRNA segments, respectively, as shown in Table .
Table 1. Sequence type, quantity, and percentage of smRNAs in H. fulva at various temperatures.
Analysis of the miRNA base characteristics of H. fulva
Based on the rice miRNA database, the miRNA constitute and primary base distributions of miRNAs of H. fulva of various lengths were analyzed statistically, and the accuracy of the predicted outcomes was evaluated.
As shown in Fig. , smRNAs (18–30 nt in length) of H. fulva showed an extreme bias towards primary base (U). The smRNAs on a given site showed bias to a certain base (Fig. ). The smRNAs obtained in this study were qualified for subsequent analysis as mature miRNA characteristics and high reliability.
Fig. 2. Primary nucleotide bias of MiRNA fragments (18–30 nt).
Note: (A) First nucleotide bias of miRNA at 10 °C. (B) First nucleotide bias of miRNA at –25 °C. □ G;
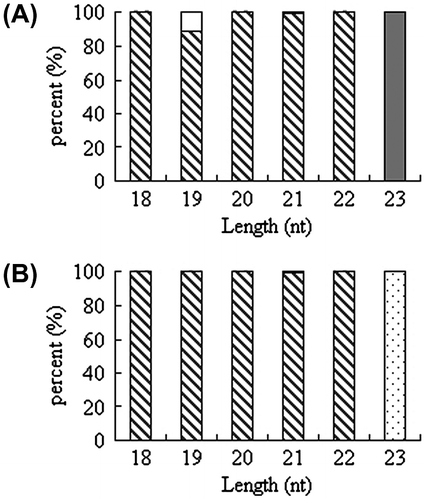
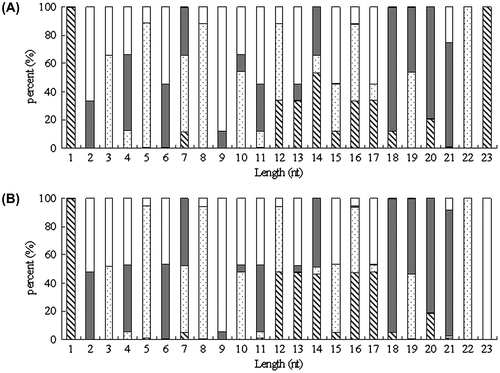
Fig. 3. Nucleotide bias of MiRNA fragments at the various site.
Note: (A) Nucleotide bias at 10 °C. (B) Nucleotide bias at –25 °C. □ G;
miRNA expression profile of H. fulva
After classification, annotation, and screening of sRNA, 99,000 miRNAs were screened from room temperature samples (10 °C) and 110,245 miRNAs from low temperature samples (–25 °C). According to the miRBase website (http://www.mirbase.org/), there are 543 known miRNA in rice. As illustrated in Table , 120 and 103 miRNAs identified from H. fulva at 10 and –25°C, respectively, were also found in rice.
Table 2. Statistical analysis of known miRNAs of H. fulva.
The expression levels of conservative miRNAs identified in two group samples were analyzed according to frequency as detected by Solexa sequencing. Seventeen miRNA sequences were relatively highly expressed, among which miR166 showed the highest frequency, followed by miR166a and miR166b. Under cold stress, the expression frequency of smRNA exceeded 10,000 times in three families, 1,000–10, 000 times in four families, and 2–100 times in at least 17 families. miR403, miR472, and miR479, which showed preferences of occurrence in monocotyledon, were not detected in either of the two samples (Supplemental 1; see Biosci. Biotechnol. Biochem. http://dx.doi.org/10.1080/09168451.2014.878214).
Compared to miRNA database of all plants from miRBase, our results indicate that expression of the same miRNA family varied between different samples. Expression of the miRNAs of H. fulva differed under normal and low temperatures. Specifically, the miRNAs normally highly expressed at low temperature were often only slightly expressed and sometimes not expressed at all at low temperature. Conversely, the miRNAs which were slightly expressed or not expressed at all low temperature were highly expressed room temperature, indicating that the miRNA expression of H. fulva shows a specific temporal sequence (Supplemental 2).
Significance analysis of miRNA expression of H. fulva
In Fig. , each dot represents a miRNA. The X-axis represents the expression level of the miRNA of H. fulva at room temperature, and the Y-axis represents the expression level of the miRNA of H. fulva at cold temperature. A red dot indicates significant upregulation of miRNA expression, a green dot shows significant downregulation of miRNA expression, and a blue dot indicates no significant change as miRNA expression as between the two groups.
Fig. 4. Significance analysis of known MiRNAs at 10 °C (S10A) and –25 °C (S25A).
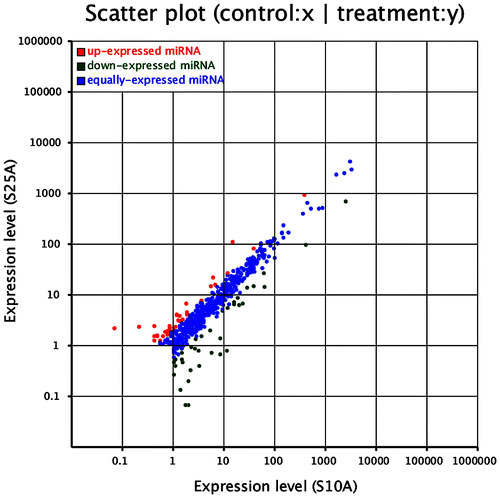
The miRNAs expression of H. fulva was statistically analyzed to evaluate further the expression differences in known miRNAs and determine whether the expression of miRNA as between two samples was significantly different.
The expression levels of a minor proportion of miRNAs were upregulated or downregulated, while a majority of the miRNAs did not change dramatically after temperature stress. Twenty-six miRNAs were significantly upregulated low-temperature stress, 30 were significantly downregulated, and nine were significantly upregulated, or downregulated.
Family profile of the RNAs in H. fulva
Species for which the genome sequencing was almost known were selected to analyze the evolutionary degree of known miRNAs of H. fulva. We found that the numbers of most miRNA families in H. fulva were similar as between monocotyledon and dicotyledon, and only a few were more frequently seen in monocotyledon.
Twenty-one miRNA families are highly expressed in at least six species, nine of which are highly conserved and found at least in 20 species, seven detected in 16–20 species, and five detected in 6–15 species (Supplemental 3).
Discussion
Zhang et al.Citation18) have reported that the miRNA of soybean has an obvious cytosine preference (61%) at N19, suggesting that the function of this miRNA is related to pre-miRNA shear and RISC assembly, consistently with previous studies.Citation19) In the present study, the mature miRNA sequences located at different sites showed an apparent bias towards the base, and showed an extreme bias towards the primary base Uracil (U), which facilitates the binding of miRNA to ARGONAUTE1,Citation20) we speculate that miRNAs of H. fulva play important roles in its growth and differentiation.
MiR160 controls the formation of root-cap cells, mainly by regulating negatively the expression of ARF10 and ARF16.Citation21) In addition, miR160 directs NAC1 mRNA to cleavage NAC1 mRNA, and suppresses expression of the NAC1 protein. With two mutants formed by miR164a and miR164b, expression of NAC1 was upregulated and the number of lateral roots was significantly increased. By contrast, overexpression of miR164 led to a decrease in the expression of NAC1 mRNA and the number of lateral root in wild-type A. thaliana.Citation22) The miR164 mutation interrupts the auxin signaling pathway in plants and upregulates the expression of NAC1 mRNA, and this enhances the resistance of plants. Overexpression of miR164 downregulates the expression of NAC1 mRNA, and improves the resistance of plants.Citation23) In the present study, we found that both osa-miR160 and osa-miR164 family members possesed the characteristics mentioned above. Thus, both osa-miR160 and osa-miR164 play active roles in improving the resistance of H. fulva against to stress.
Osa-miR156, obtained in this study, had the highest expression abundance in rice, at up to 30,481. Low temperature is a vernalization process that leads plants from vegetative growth to reproductive growth. Our results also verified that tae-miR156 is a key regulatory miRNA regulating the plant life cycle and flowering induction.Citation24) In the current study, expression of osa-miR156 was upregulated 30,474-fold under cold stress. Hence, we assume that osa-miR156 can regulate the mass accumulation of cold tolerance-related target genes, improving the resistance of H. fulva to low temperature.
This study indicates that miRNA, as a smRNA molecule, plays multiple roles in regulating many biological processes of plants. With environmental alteration, the expression profile of miRNA changes so as to exert a profound and complex effect upon plant defense responses.
On one hand, the expression of miRNA was upregulated under cold stress, and this downregulated the expression of downstream target genes. These genes might be negative regulators in the resistance of plants environmental stresses. On the other hand, the expression of certain miRNAs was downregulated under environmental stress, which instead upregulated the target genes. Genes act as positive regulators in the resistance of environmental stress.
Our results provide evidence towards understanding the regulatory mechanism of stress-responsive protein synthesis, screening of pivotal regulating genes, alternative metabolic pathways, and related genes of H. fulva.
Supplemental material
The supplemental material for this paper is available at http://dx.doi.org/10.1080/09168451.2014.878214.
Supplemental Tables 1-3
Download PDF (116.6 KB)Funding
This study was supported by the Science and Technology Project of the Heilongjiang Forestry Industry General Bureau [sgzjY2010026] and the Key Laboratory for Agricultural Biological Functional Genes of Northeast Agricultural University [NSGJ2012-10].
Notes
Abbreviations: NGS, Next-generation sequencing.
Related Research Data
References
- Zhang B, Stellwag EJ, Pan X. Large-scale genome analysis reveals unique features of microRNAs. Gene. 2009;443:100–109.10.1016/j.gene.2009.04.027
- He L, Hannon GJ. MicroRNAs: small RNAs with a big role in gene regulation. Nat. Rev. Genet. 2004;5:522–531.10.1038/nrg1379
- Jones-Rhoades MW, Bartel DP. Computational identification of plant microRNAs and their targets, including a stress-induced miRNA. Mol. Cell. 2004;14:787–799.10.1016/j.molcel.2004.05.027
- Sunkar R, Kapoor A, Zhu JK. Posttranscriptional induction of two Cu/Zn superoxide dismutase genes in Arabidopsis is mediated by downregulation of miR398 and important for oxidative stress tolerance. Plant Cell. 2006;18:2051–2065.10.1105/tpc.106.041673
- Lee RC, Feinbaum RL, Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. 1993;75:843–854.10.1016/0092-8674(93)90529-Y
- Llave C, Kasschau KD, Rector MA, Carrington C. Endogenous and silencing associated small RNAs in plants. Plant Cell. 2002;14:1605–1619.10.1105/tpc.003210
- Jin H, Zhu JK. How many ways are there to generate small RNAs. Mol. Cell. 2010;38:775–777.10.1016/j.molcel.2010.06.004
- Vazquez F, Legrand S, Windels D. The biosynthetic pathways and biological scopes of plant small RNAs. Trends Plant Sci. 2010;15:337–345.10.1016/j.tplants.2010.04.001
- Sunkar R, Zhu JK. Novel and stress-regulated microRNAs and other small RNAs from Arabidopsis. Plant Cell. 2004;16:2001–2019.10.1105/tpc.104.022830
- Sunkar R, Girke T, Jain PK, Zhu JK. Cloning and characterization of microRNAs from rice. Plant Cell. 2005;17:1397–1411.10.1105/tpc.105.031682
- Erica M, Luca G, Mario EP. Characterization of five microRNA families in maize. J. Exp. Bot. 2006;57:2601–2612.
- Yao YY, Guo GG, Ni ZF, Sunkar R, Du JK, Zhu JK, Sun QX. Cloning and characterization of microRNAs from wheat (Triticum aestivum L.). Genome Biol. 2007;8:1–15.
- Lu SF, Sun YH, Shi R, Clark C, Li LG, Chiang VL. Novel and mechanical stress-responsive microRNAs in populus trichocarpa that are absent from Arabidopsis. Plant Cell. 2005;17:2186–2203.10.1105/tpc.105.033456
- Hagen C, Frizzi A, Kao J, Jia L, Huang M, Zhang Y, Huang S. Using small RNA sequences to diagnose, sequence, and investigate the infectivity characteristics of vegetable-infecting viruses. Arch. Virol. 2011;156:1209–1216.10.1007/s00705-011-0979-y
- Wu Q, Wang Y, Cao M, Pantaleo V, Burgyan J, Li WX, and Ding SW. Homology independent discovery of replicating pathogenic circular RNAs by deep sequencing and a new computational algorithm. Proc. Natl. Acad. Sci. USA. 2012;109:3938–3943.
- Chen L, Zhang YY, Ren YY, Xu JC, Zhang ZY, Wang YW. Genome-wide identification of cold-responsive and new microRNAs in Populus tomentosa by high-throughput sequencing. Biochem. Biophys. Res. Commun. 2012;417:892–896.10.1016/j.bbrc.2011.12.070
- Tang ZH, Zhang LP, Xu CG, Yuan SH, Zhang FT, Zheng YL, Zhao CP. Uncovering small RNA-mediated responses to cold stress in a wheat thermosensitive genic male-sterile line by deep sequencing. Plant Physiol. 2012;159:721–738.10.1104/pp.112.196048
- Zhang BH, Pan XP, Stellwag EJ. Identification of soybean microRNAs and their targets. Planta. 2008;229:161–182.10.1007/s00425-008-0818-x
- Puzey JR, Kramer EM. Identification of conserved Aquilegia coerulea microRNAs and their targets. Gene. 2009;488:46–56.10.1016/j.gene.2009.08.005
- Mi SJ, Cai T, Hu YG, Chen YM, Hodges E, Ni FR, Wu L, Li S, Zhou HY, Long CZ, Chen S, Hannon GJ, Qi YJ. Sorting of small RNAs into Arabidopsis ARGONAUTE complexes is directed by the 5′terminal nucleotide. Cell. 2008;133:116–127.10.1016/j.cell.2008.02.034
- Wang JW, Wang LJ, Mao YB, Cai WJ, Xue HW, Chen XY. Control of root cap formation by microRNA-targeted auxin response factors in Arabidopsis. Plant Cell. 2005;17:2204–2216.10.1105/tpc.105.033076
- Guo HS, Xie Q, Fei JF, Chua NH. MicroRNA directs mRNA cleavage of the transcription factor NAC1 to down regulate auxin signals for Arabidopsis lateral root development. Plant Cell. 2005;17:1376–1386.10.1105/tpc.105.030841
- Liu PP, Montgomery TA, Fahlgren N. Repression of auxin response factor 10 by microRNA164 is critical for seed germination and post-germination stages. Plant J. 2007;52:133–146.10.1111/j.1365-313X.2007.03218.x
- Wu G, Park MY, Conway SR, Wang JW, Weigel D, Poethig RS. The sequential action of miR156 and miR172 regulates developmental timing in Arabidopsis. Cell. 2009;138:750–759.10.1016/j.cell.2009.06.031