
Abstract
Cne1p is a yeast homolog of calnexin, which is a constituent of endoplasmic reticulum (ER)-associated protein quality control system in mammals. Cne1p may be involved in the degradation of misfolded lysozymes in Saccharomyces cerevisiae. To test this, c-Myc-tagged lysozymes were expressed in CNE1-deficient S. cerevisiae. The expression and secretion of an unstable lysozyme mutant G49N/D66H were enhanced and its intracellular localization was changed in the CNE1-deficient strain. Furthermore, when Cne1p was co-expressed with unstable lysozyme mutants (G49N/D66H, G49N/C76A, and K13D/G49N), its affinity to the misfolded mutant proteins was revealed by co-immunoprecipitation. The interaction with Cne1p was abrogated by the addition of tunicamycin, an inhibitor of N-glycosylation, indicating that N-linked carbohydrates might be necessary for protein binding to Cne1p. These results suggest that in yeasts, Cne1p interacts with misfolded lysozyme proteins possibly causing their retention in the ER and subsequent elimination via ER-associated degradation.
Graphical Abstract
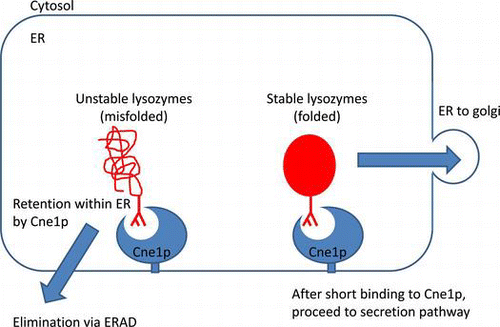
In yeasts, Cne1p interacts with misfolded lysozymes possibly causing their retention in the ER and subsequent elimination via ER-associated degradation.
Since proteins must be folded correctly to function properly, molecular chaperones are essential for cell protein quality control system associated with the endoplasmic reticulum (ER). Nevertheless, proteins are often misfolded. Accumulation of misfolded proteins in the cells is implicated in neurodegenerative diseases such as Huntington’s, Creutzfeldt–Jakob, and Alzheimer’s diseases.Citation1,2) Thus, protein quality control in the ER, where misfolded intermediates are retained is extremely important to eukaryotic organisms.Citation3–5) The ER-based selection system allows properly folded proteins with stable conformation to proceed to the secretion pathway, while the intermediates with unstable conformation are directed to the degradation pathway.Citation6)
It has been reported that calnexin is a major molecular chaperone transiently associated with numerous newly synthesized glycoproteins during their maturation in the ER.Citation7,8) In addition, calnexin has been characterized as a component of the ER quality control system, which retains the misfolded intermediates by binding to their oligosaccharide moieties until proper folding or degradation.Citation3–5) Genomic analysis and cDNA sequencing have revealed that the general structural organization of calnexin has been conserved through evolution of eukaryotic organisms. Cne1p is the homolog of calnexin in the yeast Saccharomyces cerevisiae, which has sequence and structure similarities to its mammalian counterpart. However, since Cne1p lacks a cytoplasmic tail and calcium-binding capacity,Citation6,9) there has been no consensus on whether Cne1p in yeasts has the same function as calnexin in mammals.
Lysozyme from chicken egg whites has been well studied with respect to the structural and functional properties. We have been studying lysozyme structure and function using a variety of stable and unstable lysozyme mutants.Citation10–13) To investigate the function of Cne1p, we introduced the N-X-T/S sequence, which is the N-glycosylation signal in eukaryotic cells, by substituting Gly49 with asparagine, and we constructed the G49N lysozyme.Citation14) We have also generated other lysozyme mutants: K13D with the destabilized a-helix 5-15, C76A, which lacks the C76-C94 disulfide bridge, and D66H with the disrupted network of hydrogen bonds that stabilizes the β domain.Citation12) We have reported that the G49N lysozyme was stable, whereas the K13D, C76A, and D66H mutants were thermodynamically unstable.Citation11,12)
Previously, we reported that the secretion of unstable glycosylated lysozyme mutants (G49N/D66H, G49N/C76A, K13D/G49N) was enhanced in the CNE1-deficient S. cerevisiae compared to that in the wild-type yeasts. In contrast, the secretion of stable glycosylated mutants (G49N, G49N/S91T) and non-glycosylated lysozyme (wild-type, S91T), as well as unstable non-glycosylated mutants (D66H, C76A, K13D) was almost the same in the wild-type and CNE1-deficient yeasts.Citation11,12) These results suggest that the unstable glycosylated lysozyme mutants might be degraded in the wild-type S. cerevisiae through Cne1p. In this study, we expressed stable and unstable c-Myc-tagged glycosylated lysozyme mutants in wild-type and CNE1-deficient S. cerevisiae and compared their expression levels and intracellular localization. We also investigated the interaction of these mutant proteins with Cne1p. The results of this study suggest that unstable lysozyme mutants expressed in yeasts might be retained in the ER through interaction with Cne1p.
Materials and methods
Bacterial strains and plasmids
Escherichia coli XL-1 blue (recA1 endA1 gyrA96 thi-1 hsdR17 supE44 relA1 lac [F′ proAB lacIqZΔM15 Tn10 (tetr)]) (Amersham BioScience, Buckinghamshire, UK) was used as a host strain in all cloning experiments. The S. cerevisiae diploid strains W303-1b (Mata ade2-1 can1-100 ura3-1 leu2-3, 112 trp1-1 his3-11, 15) and W303-1b cne1::Leu2 were provided by Dr Parlati, McGill University, Canada.Citation6) The E. coli-yeast shuttle vector pYG100 was provided by Dr K. Matsubara of the Osaka University. The cloning vectors pUC18 and pBluescript II (SK+) were purchased from Toyobo (Osaka, Japan), and the pT7Blue-T vector used to clone PCR products was purchased from Takara Bio Inc. (Otsu, Japan). The Pichia pastoris expression vector pPICZα-A (Invitrogen, Carlsbad, USA) was used to add the α-factor signal sequence, and c-Myc, and His tags to the lysozyme gene. The yeast-E. coli shuttle vectors pRS426 and pRS314 were provided by Dr R. Akada, Department of Applied Chemistry and Chemical Engineering, the Yamaguchi University.
Media and growth conditions
E. coli was grown in low-salt LB medium (1% tryptone, 0.5% yeast extract, and 0.5% NaCl) supplemented with 50 μg/mL carbenicillin for selection of transformants. S. cerevisiae was grown at 30 °C in YPD medium (2% tryptone, 1% yeast extract, and 2% glucose). For clone selection and protein secretion, the wild-type yeast cells carrying pRS426/lysozyme were grown at 30 °C in yeast minimum medium (JMM) supplemented with 40 μg/mL of adenine, 60 μg/mL of leucine, 40 μg/mL of tryptophan, and 20 μg/mL of histidine. The CNE1-deficient cells carrying pRS426/lysozyme were grown at 30 °C in JMM supplemented with 40 μg/mL of adenine, 40 μg/mL of tryptophan, and 20 μg/mL of histidine. The CNE1-deficient cells carrying both pRS426/lysozyme and pRS314/CNE1 were grown at 30 °C in JMM supplemented with 40 μg/mL of adenine and 20 μg/mL of histidine.
Construction of the cloning vector carrying GPD promoter and terminator
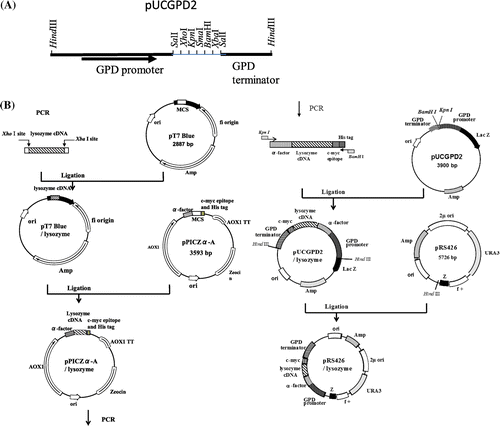
To express the gene of interest under the glyceraldehyde-3-phosphate dehydrogenase (GPD) promoter, the pUCGPD2 vector was constructed. For this, pYG100 was digested with HindIII and the 1.2-kb fragment including GPD promoter and terminator was purified and digested with SalI. The resultant 1.1-Kb fragment carrying the GPD promoter and the 0.1-Kb fragment carrying the GPD terminator were subcloned into pUC18 and pBluescript II (SK+), respectively, to construct the plasmids pUC18/GPD-promoter and pBluescript/GPD-terminator. After digestion of pBluescript/GPD-terminator with EcoRI and KpnI, the 1.1-Kb fragment was inserted into EcoRI and KpnI sites of the pUC18/GPD-promoter. The resultant plasmid designated as pUCGPD2 had multiple cloning sites between the GPD promoter and the terminator (Fig. (A)).
Fig. 1. Restriction map of pUCGPD2 (A) and construction of the expression plasmids for c-Myc-tagged lysozymes (B).
Cloning and expression of c-Myc-tagged chicken lysozyme mutants in yeasts
The expression plasmids containing c-Myc-tagged lysozyme mutants were constructed as shown in Fig. (B). The primers used in this study are listed in Fig. . For the construction of the c-Myc-tagged lysozyme expression plasmids, three plasmids, pUCGPD2 containing GPD promoter and terminator, pPICZα-A containing α-factor signal sequence and c-Myc epitope, and pRS426 as an S. cerevisiae-specific vector, were used.
Fig. 2. Primers used in this study.

The cDNAs of mutant lysozymes (G49N, D66H/G49N, G49N/C76A, and K13D/G49N) cloned into pYG100 earlierCitation11,12) were used as templates for the amplification of the mutant lysozyme genes with primers A and B. The PCR products were subcloned into the pT7Blue-T vector. The resultant pT7Blue/lysozyme plasmids were digested with XhoI and XbaI and subcloned into pPICZα-Α between the α-factor signal sequence and c-Myc sequence. The resultant pPICZα-A/lysozyme plasmids were used as templates for amplification of the mutant lysozyme genes containing α-factor and c-Myc sequences with primers C and D. The PCR products were subcloned into pUCGPD2 under the GPD promoter and the plasmids were digested with HindIII to obtain the c-Myc-tagged-lysozyme gene under the GPD promoter, which was inserted into the pRS426 vector.
The resultant plasmids (pRS426/lysozyme) were introduced into S. cerevisiae W303-1b and W303-1b cne1::Leu2 strains by the lithium acetate method. Clones with the highest lysozyme activity were selected and expanded by incubation in 3 mL of JMM for two days at 30 °C with shaking. The cultures were transferred to a 500-mL flask containing 100 mL JMM and incubated for another two days under the same conditions.
Fluorescent immunostaining of lysozymes in S. cerevisiae
Yeast cells in mid-log phase were harvested and fixed with formaldehyde (final concentration 3.7%). The cells were washed with 0.1 M potassium phosphate buffer (pH 7.5) and treated with zymolyase. Spheroplast cells were resuspended in PBS and 10 μL of cell suspension was placed on the poly-L-lysine-coated slides. After 5 min, the slides were washed thrice with PBS and fixed in cold methanol (−20 °C) for 6 min and in cold acetone (−20 °C) for 30 s. After blocking with 3% BSA in PBS for 30 min at room temperature (RT), the slides were incubated with anti-c-Myc antibody for 60 min at RT followed by the incubation with FITC-labeled anti-mouse IgG. All fluorescence images were generated using a Carl Zeiss LSM510 scanning laser confocal microscope.
Construction of FLAG-tagged Cne1p expression plasmid
The CNE1 gene flanked with the 19-amino-acid N-terminal signal sequence and C-terminal FLAG epitope was amplified from S. cerevisiae W303-1b genomic DNA using primers E and F. The PCR product encoding FLAG-tagged truncated Cne1p was subcloned into the pT7Blue-T vector. The resultant plasmid pT7Blue/CNE1 was digested with KpnI and XbaI and subcloned into pUCGPD2 containing the GPD promoter. After the digestion of pUCGPD2/CNE1 with HindIII, the GPD promoter-containing Cne1p fragment was isolated and subcloned into the centromere vector pRS314 partially digested with HindIII. The resultant plasmid pRS314/CNE1 was used for the expression of FLAG-tagged Cne1p.
Co-immunoprecipitation of mutant lysozymes with Cne1p
CNE1-deficient S. cerevisiae strains expressing c-Myc-tagged lysozyme mutants and FLAG-tagged soluble CNE1 were grown at 30 °C in YPD medium until mid-log phase. An N-glycosylation inhibitor tunicamycin (Wako Pure Chemical Industries, Ltd., Osaka, Japan) was added to YPD medium at 5 μg/mL. The cells were harvested, washed with PBS, and lysed with CelLytic Y buffer (Sigma-Aldrich, St. Louis, USA).
Cne1p-containing protein complexes were precipitated using the FLAG immunoprecipitation kit (Sigma-Aldrich). Briefly, agarose beads with the immobilized anti-FLAG antibody were added to cell lysates. After incubation at 4 °C for 1 min, agarose beads were precipitated by centrifugation, washed, and Cne1p-associated proteins were eluted by the addition of 3 × FLAG peptide. The eluted proteins were separated by SDS polyacrylamide gel electrophoresis, transferred onto PVDF membrane (Immobilon-P, Merck Millipore, Darmstadt, Germany), and analyzed by western blotting. For the detection of c-Myc-tagged lysozyme, the membranes were incubated with anti-c-Myc monoclonal antibody (NACALAI TESQUE, Inc., Kyoto, Japan) diluted 1:2,000 with 1% BSA-containing PBS. For the detection of FLAG-tagged Cne1p, anti-FLAG-antibody (Sigma-Aldrich) was used at a dilution of 1:5,000 in 1% BSA-containing PBS. HRP-labeled anti-mouse IgG (Sigma-Aldrich) was used as a secondary antibody at a dilution of 1:80,000 with 1% BSA-containing PBS. Immunoreactivity was detected using the enhanced chemiluminescence method (ECL system, Amersham) and autoradiography films (Hyperfilm-ECL, Amersham).
Results and discussion
Unstable lysozymes mutants are degraded in the presence of Cne1p
Previously, we reported that the secretion of unstable lysozyme mutants was enhanced in CNE1-deficient S. cerevisiae compared to very low secretion in the wild-type strain,Citation11,12) suggesting that in the wild-type yeasts, the unstable lysozyme mutants might be degraded through interaction with Cne1p. It is thought that calnexin is a component of the protein quality control system, which retains misfolded intermediates in the ER through the interaction with N-linked oligosaccharides until proper folding or degradation. Therefore, we suggest that Cne1p, a yeast homolog of calnexin, also functions by retaining the misfolded intermediates of the unstable glycosylated lysozyme mutants in the ER until elimination by ERAD.
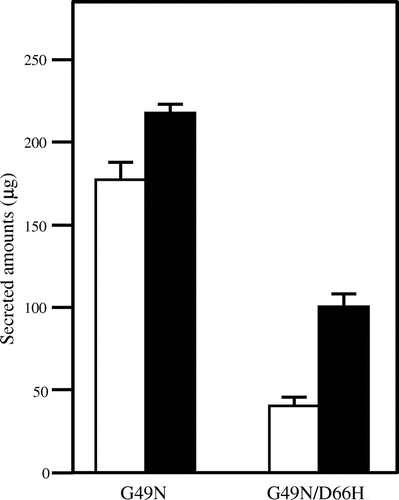
To observe the intracellular expression of the mutated lysozyme proteins, we constructed the plasmids carrying c-Myc-tagged mutant lysozyme genes and transferred them into CNE1-deficient and wild-type yeasts. A stable G49N glycosylated lysozyme mutant and an unstable G49N/D66H glycosylated amyloid lysozyme mutant were used as references. To confirm that the expression of the lysozymes carrying the N-terminal c-Myc-tag was the same as that of non-tagged lysozymes, we compared their secretion. As shown in Fig. , the secretion of c-Myc-tagged G49N/D66H lysozyme by the CNE1-deficient strain was higher than that in the wild-type strain, whereas the secretion of c-Myc-tagged G49N lysozyme was almost the same, which is consistent with our previous results using non-tagged lysozymes. Furthermore, we reported that the addition of some peptides such as hydrophobic sequence and serine-rich sequence did not affect their structures. These results suggest that in the presence of Cne1p the unstable c-Myc-tagged proteins might be degraded in the same manner as the unstable non-tagged proteins and confirmed that the addition of the c-Myc tag to lysozyme did not affect its secretion in yeasts. Therefore, we used c-Myc-tagged lysozymes in the following experiments.
Fig. 3. Secretion of the c-Myc-tagged Mutant Lysozymes G49N and G49N/D66H Expressed in the Wild Type (white column) and CNE1-deficient (black column) S. cerevisiae.
Note: Data are presented as mean ± standard deviation (n = 3).
Lysozyme expression in yeasts was analyzed by immunostaining with c-Myc antibody detected by fluorescence microscopy. As shown in Fig. (A), the intracellular expression of the G49N/D66H lysozyme was increased in the CNE1-deficient strain, whereas it was very low in the wild-type yeasts, suggesting Cne1p-dependent degradation of the G49N/D66H protein in the wild-type strain. In contrast, in the CNE1-deficient yeasts, the misfolded mutant proteins might have escaped Cne1p-based protein quality control system and proceeded to the secretion pathway, resulting in the increase of the intracellular and secreted unstable lysozymes in the CNE1-deficient strain. The increase in the intracellular expression of the stable (G49N) lysozyme, though not as significant as that of the unstable protein, was also detected in the CNE1-deficient strain, suggesting that even stable lysozymes might be subjected to degradation by ERAD system because of overexpression.
Fig. 4. Immunofluorescence staining of the mutant lysozymes G49N and G49N/D66H expressed in wild type and CNE1-deficient S. cerevisiae using Anti-cMyc epitope antibody.
Notes: (A) Low magnification (×100); (B) high magnification (×1000).
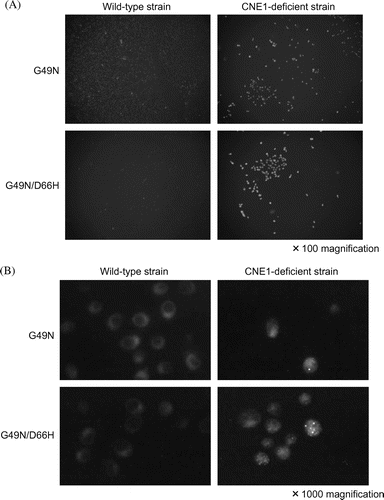
To observe the intracellular localization of stable (G49N) and unstable (G49N/D66H) mutated lysozymes, high-power microscopy was used (Fig. (B)). In the wild-type strain, G49N and G49N/D66H lysozymes were detected around the nucleus, most likely in the ER, suggesting the interaction with Cne1p and retention in the ER. However, in the CNE1-deficient strain, the localization pattern of the G49N/D66H protein was different, probably because the absence of Cne1p compromised protein quality control allowing the misfolded intermediates to avoid retention in the ER and proceed to secretion or to another Cne1p-independent degradation pathway.
Recently, we found that the expression of the unstable mutant lysozyme G49N/D66H in P. pastoris resulted in the induction of autophagy and that the inhibition of autophagy by wortmannin enhanced the secretion of the mutant protein (our unpublished results). Therefore, in CNE1-deficient S. cerevisiae, a portion of the misfolded G49N/D66H lysozyme might be forcibly secreted, while the remaining protein might enter the autophagy/vacuole degradation pathway. In mammals, two ERAD pathways, ubiquitin/proteasome and autophagy/lysosome, have been found to degrade misfolded proteins accumulated in the ER. Our results also suggest the existence of alternative ERAD pathways such as autophagy/vacuole pathway in S. cerevisiae.
Cne1p interaction with mutant lysozymes in vivo
To investigate whether the unstable lysozyme mutants interacted with Cne1p, the soluble Cne1p was expressed in CNE1-deficient yeasts. To ensure Cne1p solubility, C-terminal transmembrane and cytoplasmic regions in the CNE1 gene were substituted with a FLAG tag and the modified CNE1 gene was expressed in the CNE1-deficient strain together with the c-Myc tagged lysozyme mutants. As shown in Fig. (A), after the immunoprecipitation with the anti-FLAG antibody, a 55-kDa protein was detected in all strains by western blotting using the anti-FLAG antibody. The protein molecular weight corresponded to that of Cne1p with truncated C-terminal region, suggesting that the truncated Cne1p was expressed in all strains. As shown in Fig. (B), a 25-kDa protein was detected in the strain expressing the unstable lysozyme mutant (G49N/D66H) together with the truncated Cne1p, whereas no bands were detected in the strain expressing the stable mutant (G49N) and the truncated Cne1p.
Fig. 5. Co-immunopreciptation of c-Myc-tagged mutant lysozymes and FLAG-tagged soluble Cne1p.
Notes: After immunoprecipitation with anti-FLAG antibody, western blotting was performed using anti-FLAG (A) or anti-c-Myc (B) antibody. Lane 1, soluble Cne1p; lane 2, soluble Cne1p + G49N/D66H lysozyme; lane 3, soluble Cne1p + G49N lysozyme.
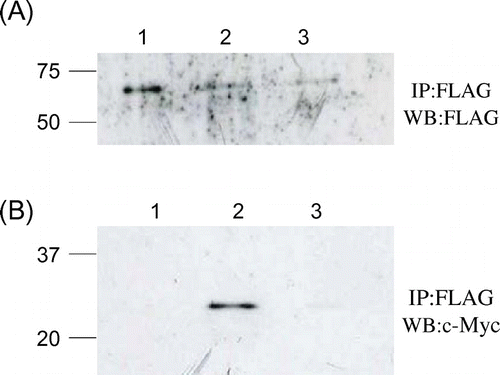
The molecular weight of 25 kDa correspond to the recombinant protein comprising lysozyme (about 14.4 kDa), N-terminal pro-α-factor (about 7.0 kDa), and C-terminal c-Myc and His tags (about 2.6 kDa). Prepro-α-factor should be processed before the translocation to the ER, whereas pro-α-factor signal should remain in the ER and processed by Kex2 peptidase upon the translocation to the Golgi compartment.Citation15) Given that the proteins with pro-α factor signal have been detected, these results suggest that the interaction of Cne1p with the unstable mutant glycosylated lysozymes might occur in the ER lumen.
Cne1p interacts with unstable mutant lysozymes
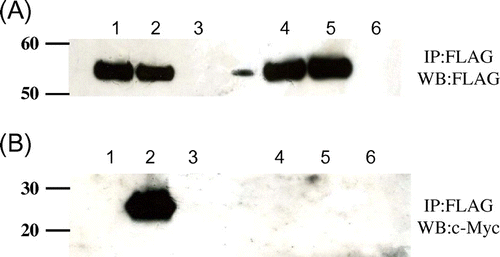
To determine whether Cne1p interacted with other unstable lysozyme proteins, two of them were co-expressed with the truncated Cne1p. These glycosylated mutant lysozymes, one containing a destabilized a-helix 5-15 (K13D/G49N) and the other lacking a C76-C94 disulfide bridge (G49N/C76A), have been previously shown to be thermodynamically unstable.Citation12) After expressing these mutants with Cne1p, we performed co-immunoprecipitation experiments using the anti-FLAG antibody. As shown in Fig. (A), Cne1p was expressed and immunoprecipitated by the anti-FLAG antibody in all tested strains. In addition, a 25-kDa c-Myc-tagged protein was detected in the strains expressing unstable lysozyme mutants (G49N/D66H, G49N/C76A, K13D/G49N) by western blotting, whereas no c-Myc-reactive band was detected in the stable lysozyme (G49N)-expressing yeasts (Fig. (B)). These results suggest that the unstable lysozyme proteins might interact with Cne1p. Furthermore, the G49N/D66H mutant lysozyme was precipitated more efficiently compared to the G49N/C76A and K13D/G49N mutants, indicating stronger interaction with Cne1p.
Fig. 6. Co-immunopreciptation of various c-Myc-tagged unstable mutant lysozymes and FLAG-tagged soluble Cne1p.
Notes: After immunoprecipitation with anti-FLAG antibody, western blotting was performed using anti-FLAG (A) or anti-c-Myc antibody (B) antibody. Lane 1, soluble Cne1p + G49N lysozyme; lane 2, soluble Cne1p + G49N/D66H lysozyme; lane 3, soluble Cne1p + G49N/C76A lysozyme; lane 4, soluble Cne1p + K13D/G49N lysozyme.

Previously, we reported that the G49N/D66H lysozyme had lower thermodynamic stability than other mutant glycosylated lysozymes and that the secretion of this mutant lysozyme was very low because of its aggregability.Citation11) The substitution D66H destroys the network of hydrogen bonds that stabilizes the β domain, resulting in a large and concerted movement of the β sheet and long loop within the β domain away from each other.Citation16) These data suggest that the folding of the unstable G49N/D66H lysozyme in the ER would be a slow process during which it would interact with Cne1p. Taken together, these results suggest that unstable proteins may interact with Cne1p in the ER and that the affinity to Cne1p correlates with protein instability.
Cne1p interacts with unstable glycosylated lysozyme through N-linked carbohydrates
It has been reported that in mammalian cells, calnexin and its soluble homolog calreticulin interact with the target proteins through N-linked sugar moieties.Citation17) To determine whether Cne1p interacted with N-glycosylated proteins, we treated yeasts with tunicamycin, an inhibitor of N-glycosylation. As shown in Fig. (B), a 25-kDa protein was detected in the strain co-expressing Cne1p and the unstable lysozyme G49N/D66H mutant. However, the treatment with tunicamycin blocked the interaction of the G49N/D66H mutant protein with Cne1p.
Fig. 7. Co-immunopreciptation of c-Myc-tagged mutant lysozymes and FLAG-tagged soluble Cne1p before and after treatment with tunicamycin.
Notes: After immunoprecipitation with anti-FLAG antibody, western blotting was performed using anti-FLAG (A) or anti-c-Myc (B) antibody. Lanes 1 and 4, soluble Cne1p + G49N lysozyme; lanes 2 and 5, soluble Cne1p + G49N/D66H lysozyme; lanes 3 and 6, G49N/D66H lysozyme. Lanes 1–3, in the absence of tunicamycin; lanes 4–6, in the presence of tunicamycin.
Previously, we reported that the secretion of the stable non-glycosylated lysozyme (wild-type S91T) and unstable non-glycosylated CNE1 mutants (D66H, C76A, K13D) was almost the same,Citation11,12) suggesting that only glycosylated variants were degraded through the interaction with Cne1p. As has been described above, in the ER calnexin interacts with misfolded intermediates through their N-glycosylated sugar chains, suggesting that the calnexin yeast homolog Cne1p may interact with misfolded proteins through the same mechanism. Our results show that tunicamycin, an inhibitor of N-glycosylation in the ER,Citation18) blocked Cne1p interaction with G49N/D66H lysozyme (Fig. ), confirming this hypothesis. Thus, Cne1p can interact only with glycosylated unstable mutant lysozymes.
In conclusion, our results suggest that Cne1p in S. cerevisiae interacts with misfolded intermediates of N-glycosylated proteins and retains them in the ER until proper folding, indicating that Cne1p in yeasts exhibits the same functional mechanism as calnexin in mammalian cells.
Acknowledgments
We thank Ms T. Uchiyamada, Ms Y. Sugyo, and Ms Y. Tanaka for their supports in the establishment of experimental methods for expression of c-Myc-tagged lysozymes. We are grateful to Dr I. Miyakawa (Graduate School of Science and Engineering, Yamaguchi University) for providing fluorescence microscopy.
Notes
Abbreviations: ER, endoplasmic reticulum; ERAD, ER-associated degradation; GPD, glyceraldehyde-3-phosphate dehydrogenase; PBS, phosphate-buffered saline; BSA, bovine serum albumin.
References
- Muchowski PJ. Neuron. 2002;35:9–12.10.1016/S0896-6273(02)00761-4
- Sakahira H, Breuer P, Hayer-Hartl MK, Hartl FU. Proc. Nat. Acad. Sci. 2002;99:16412–16418.10.1073/pnas.182426899
- Hammond C, Braakman I, Helenius A. Proc. Nat. Acad. Sci. USA. 1994;91:913–917.10.1073/pnas.91.3.913
- Jackson MR, Cohen-Doyle MF, Peterson PA, Williams DB. Science. 1994;263:384–387.10.1126/science.8278813
- Ware FE, Vassilakos A, Peterson PA, Jackson PA, Williams DB. J. Biol. Chem. 1995;270:4697–4704.
- Parlati F, Dominguez M, Bergeron JJM, Thomas DY. J. Biol. Chem. 1995;270:244–253.
- Ou WJ, Cameron PH, Thomas DY, Bergeron JJM. Nature. 1993;364:771–776.10.1038/364771a0
- Letourneur O, Sechi S, Willete-Brown J, Robertson MW, Kinet JP. J. Biol. Chem. 1995;270:8249–8256.
- Jakob CA, Burda PS, te Heesen S, Aebi M, Roth J. Glycobiology. 1998;8:155–164.10.1093/glycob/8.2.155
- Kato A, Nakamura S, Ban M, Azakami H, Yutani K. Biochim. Biophys Acta. 2000;1481:88–96.10.1016/S0167-4838(00)00123-0
- Song Y, Azakami H, Shamima B, He J, Kato A. FEBS Lett. 2002;512:213–217.10.1016/S0014-5793(02)02258-5
- Song Y, Sata J, Saito A, Usui M, Azakami H, Kato A. J. Biochem. 2001;130:757–764.10.1093/oxfordjournals.jbchem.a003046
- Harada A, Yagi H, Saito A, Azakami H, Kato A. Biosci. Biotechnol. Biochem. 2007;71:2952–2961.
- Nakamura S, Takasaki H, Kobayashi K, Kato A. J. Biol. Chem. 1993;268:12706–12712.
- Chaudhuri B, Latham SE, Helliwell SB, Seeboth P. Biochem. Biophys. Res. Commun. 1992;183:212–219.10.1016/0006-291X(92)91630-9
- Booth DR, Sunde M, Bellotti V, Robinson CV, Hutchinson WL, Fraser PE, Hawkins PN, Dobson CM, Radford SE, Blake CF, Pepys MB. Nature. 1997;385:787–793.10.1038/385787a0
- Williams DB. J. Cell Sci. 2006;119:615–623.10.1242/jcs.02856
- Jannatipour M, Callejo M, Parodi AJ, Armstrong J, Rokeach LA. Biochemistry. 1998;37:17253–17261.10.1021/bi981785c