
Abstract
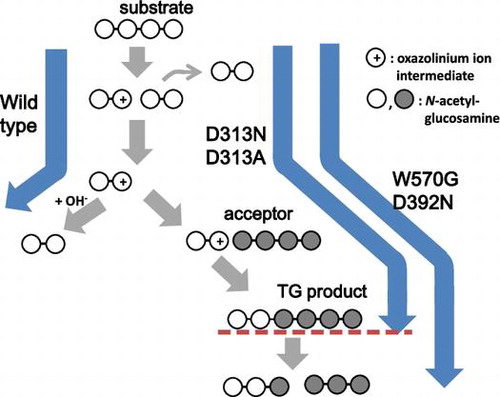
Enhancing the transglycosylation (TG) activity of glycoside hydrolases does not always result in the production of oligosaccharides with longer chains, because the TG products are often decomposed into shorter oligosaccharides. Here, we investigated the mutation strategies for obtaining chitooligosaccharides with longer chains by means of TG reaction catalyzed by family GH18 chitinase A from Vibrio harveyi (VhChiA). HPLC analysis of the TG products from incubation of chitooligosaccharide substrates, GlcNAcn, with several mutant VhChiAs suggested that mutant W570G (mutation of Trp570 to Gly) and mutant D392N (mutation of Asp392 to Asn) significantly enhanced TG activity, but the TG products were immediately hydrolyzed into shorter GlcNAcn. On the other hand, the TG products obtained from mutants D313A and D313N (mutations of Asp313 to Ala and Asn, respectively) were not further hydrolyzed, leading to the accumulation of oligosaccharides with longer chains. The data obtained from the mutant VhChiAs suggested that mutations of Asp313, the middle aspartic acid residue of the DxDxE catalytic motif, to Ala and Asn are most effective for obtaining chitooligosaccharides with longer chains.
Graphical Abstract
Mutations of Asp313, the middle aspartic acid residue of the DxDxE catalytic motif, of Vibrio harveyi GH18 chitinase enhanced the production of oligosaccharides with longer chains.
Chitinases (EC 3.2.1.14) are enzymes that hydrolyze chitin, an insoluble polysaccharide consisting of β-(1,4)-linked N-acetylglucosamine (GlcNAc) units and a major component of the shells of crustaceans, the exoskeletons of insects, and the cell walls of fungi.Citation1–3) Chitinases are classified into glycoside hydrolase family 18 (GH18) and family 19 (GH19), depending on the amino acid sequence identity of their catalytic domains and the mode of enzyme action.Citation4–9) In nature, degradation of insoluble chitin polymer by chitinases generates water-soluble chitooligosaccharide fragments.Citation10) Chitooligosaccharides, GlcNAcn (n, degree of polymerization or chain length), have various biological functions; for example, they can stimulate the plant immune system to respond to microbial infectionsCitation11–13) and can be used as antimicrobial agents.Citation14) However, the biological activities of chitooligosaccharides are most efficient, when the chain lengths are more than five or six.Citation14,15) Usually, chemical synthesis of chitooligosaccharides with such longer chains is cumbersome and costly due to the selective protection and subsequent manipulation of various monosaccharide donors and acceptors.Citation16,17) Therefore, enzymatic synthesis employing the transglycosylation (TG) activity of chitinases may serve as a better biological tool for a large-scale production of such biologically active compounds.
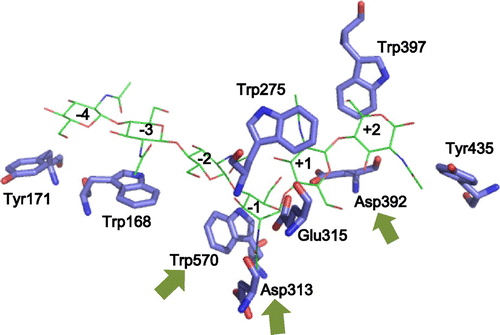
TG reaction catalyzed by GH18 chitinases usually takes place through two steps.Citation18–20) In the first step, the glycosidic oxygen is protonated by a catalytic acid to cleave the β-1,4-glycosidic linkage and to form the oxazolinium ion intermediate, in which the C1 carbon of the −1 sugar is stabilized by anchimeric assistance of the sugar N-acetamido group. In the second step, the oxazolinium ion intermediate is attacked by a water molecule from the β-side, leading to hydrolysis with net retention of anomeric form. When a water molecule is outcompeted by another acceptor, such as carbohydrates, TG reaction takes place, resulting in the formation of a glycosidic linkage and yielding longer chain chitooligosaccharides instead. Chitinases from various sources have been reported to potentially catalyze TG reaction. For examples, a chitinase from Nocardia orientalis was reported to convert GlcNAc4 substrate to GlcNAc6 under high ammonium sulfate concentration.Citation21) Recently, Serratia proteamaculans chitinase D (SpChiD) showed high TG activity with GlcNAc3–6 substrates generating GlcNAc7–13 products, which were hydrolyzed into smaller GlcNAcn after 90 min of the reaction.Citation22) Mutations of some amino acids located close to the catalytic cleft were found to enhance TG activity in various GH18 chitinases, such as Serratia marcescens chitinase A and chitinase B.Citation19,20) Similar enhancement of TG activity was reported for the mutants of Bacillus circulans WL-12 chitinase A1, those of Trichoderma harzanium chitinase 42,Citation23) and those of S. proteamaculans chitinase D (SpChiD).Citation24) Such mutants displayed higher TG activity, whereas their hydrolytic activity was dramatically diminished. From those studies on the TG reaction catalyzed by family GH18 chitinases, it is obvious that enhancing the TG activity of chitinases does not always result in the production of chitooligosaccharides with longer chains, because the TG products are often decomposed into shorter oligosaccharides. In this study, we investigated the mutation strategies for obtaining chitooligosaccharides with longer chains by means of enzymatic TG reaction using family GH18 chitinase A from Vibrio harveyi (VhChiA). Mutations were introduced into Asp313, Asp392, and Trp570, each of which is responsible for sugar residue binding at subsites −2, −1, +1, and +2, as seen from the crystal structure of VhChiA shown in Fig. . We found that mutations of Asp313, the middle aspartic acid residue of the catalytic motif DxDxE, to Ala and Asn are most effective for obtaining chitooligosaccharides with longer chains.
Fig. 1. Superimposition of the active site structure of ligand-free wild-type VhChiA and VhChiA E315M mutant complexed with GlcNAc6 (only GlcNAc6 shown as green; PDB code, 3B9A).
Notes: GlcNAc-binding subsites are indicated by integers based on the nomenclature suggested by Davies et al.Citation39). The amino acid residues presented as the stick model are important for chitooligosaccharide binding. The structure of ligand-free wild-type VhChiA was obtained from the PDB database (PDB code, 3B8S)Citation30) and displayed by the program PyMol (www.pymol.org/). The arrows indicate the mutation targets.
Materials and methods
Materials
Chitooligosaccharides, GlcNAc2–6, were produced by acid hydrolysis of chitin,Citation25) and purified by gel-filtration column of Gcl-25 m (JNC Co., Tokyo). Colloidal chitin was prepared from crab chitin by the method of Hsu and Lockwood.Citation26) Ni-NTA agarose resin was purchased from Bio-Rad Laboratories (Hercules, USA), and HiPrep 16/60 Sephacryl S-100 resin was from GE Healthcare. Other reagents were of analytical grade and commercially available.
Mutation targets
To enhance the TG activity, two strategies were proposed: (1) enhancing the acceptor-binding abilityCitation27) and (2) suppressing the attack of a nucleophilic water molecule to the transition state.Citation20,28) Since the acceptor-binding site (+1 and + 2) of wild-type enzymes are evolutionarily optimized for efficiently accepting their natural substrates, the mutations introduced into the acceptor-binding site usually reduce the acceptor-binding ability. Thus, mutations for enhancing the binding ability are quite difficult. In the former studies,Citation19,29) mutations were introduced into the glycon-binding site (−2 and −1) to suppress the sugar-binding ability of the negatively numbered subsites. The suppression of the sugar-binding to the negatively numbered subsites relatively enhances the binding ability toward the positively numbered subsites (acceptor-binding site). In fact, mutation of Trp167 (subsite −3) of S. marcescens chitinase A enhanced the TG activity.Citation19) Here, we tried to mutate Trp570, which is responsible for the sugar-residue binding at subsites −1 and −2 (Fig. ),Citation30,31) to glycine. Asp392, which is supposed to be responsible for the acceptor-binding at subsites+1 and+2,Citation30) was also mutated to asparagine, which may facilitate hydrogen bonding interaction with the sugar residue. In addition to these mutations, we mutated the middle aspartic acid residue (Asp313) in the DxDxE catalytic motif, because this mutation was reported to significantly enhance the TG activity of S. marcescens chitinases.Citation20,23)
Production and purification of the wild type and mutates VhChiA
Four VhChiA mutants W570G, D392N, D313A, and D313N were generated by PCR-based site-directed mutagenesis as described previously.Citation31,32) The recombinant wild-type VhChiA and its mutants were highly expressed in E. coli M15 cells as described by Pantoom et al.Citation33) For purification, the IPTG-induced cells were collected by centrifugation, re-suspended in 40 mL of 20 mM Tris-HCl buffer, pH 8.0, containing 150 mM NaCl, and then lysed on ice using an Ultrasonic disruptor with a 1.5 cm-diameter probe. The supernatant obtained after centrifugation at 12,000 rpm for 40 min was applied to a Ni-NTA agarose affinity column (Bio-Rad Laboratories, Hercules, CA, USA), washed thoroughly with 5 and 20 mM imidazole, and then eluted with 250 mM imidazole prepared in 20 mM Tris-HCl, pH 8.0, and 150 mM NaCl. The eluted fractions were further purified by gel filtration chromatography on a HiPrep 16/60 Sephacryl S-100 HR column connected to an FPLC purifier system (GE Healthcare). After SDS-PAGE analysis, the chitinase-containing fractions were pooled, then dialyzed with 20 mM phosphate buffer at pH 7.0, and concentrated using the Vivaspin-20 ultrafiltration membrane concentrator (Mr 10,000 cutoff, Vivascience AG, Hannover, Germany). A final protein concentration was determined by UV absorbance at 280 nm, using the extinction coefficient obtained from the equation proposed by Pace et al.Citation34)
Time-course study of TG reaction by quantitative HPLC
A reaction mixture (100 μL) contained chitooligosaccharide substrate (6.8 mM GlcNAc4, 5.5 mM GlcNAc5, or 4.6 mM GlcNAc6), VhChiA (5 μM of wild-type, W570G, or D392N, 16 μM of D313A, or 8 μM of D313N), and 20 mM phosphate buffer, pH 7.0. The reaction mixture was incubated at 40 °C, and then an aliquot of (10 μL) was transferred to a new microcentrifuge tube containing 10 μL of 0.1 M NaOH to terminate the enzymatic reaction at various times of incubation. To determine the enzymatic products, the resultant solution was immediately applied onto a gel filtration column of TSK-GEL G2000PW (7.5 mm × 600 mm) connected with a Hitachi L-7000 HPLC system (Hitachi Koki Co., Ltd, Tokyo, Japan). Elution was conducted with a Milli-Q water at a constant flow rate of 0.3 mL min−1. The oligosaccharide products in the effluent were monitored by UV absorption at 220 nm. Peak area of each GlcNAcn obtained from the elution profile was then converted into molar concentration using the standard calibration curve of the GlcNAcn mixture with known concentrations.
Results and discussion
Time courses of chitooligosaccharide degradation catalyzed by wild-type VhChiA
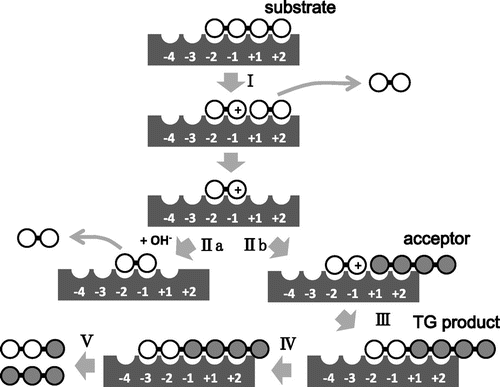
We first evaluated TG activity of the wild-type VhChiA (WT). Incubation of WT with the GlcNAc4 substrate produced GlcNAc2 as the major hydrolytic product after 3 h of reaction (Fig. (A)). A small but detectable amount of GlcNAc3 was also produced after 3 h, but no GlcNAc was detected at all. From the GlcNAc5 substrate, GlcNAc2 and GlcNAc3 were formed as the major hydrolytic products, and a trivial amount of GlcNAc4 was also formed at 2 h (Fig. (B)). The GlcNAc4 formation from GlcNAc5 was not accompanied by GlcNAc formation. The GlcNAc3 product from GlcNAc4 and the GlcNAc4 product from GlcNAc5 were not derived from a simple hydrolysis of the initial substrates. Aronson et al.Citation19) reported a similar hydrolytic profile obtained by S. marcescens chitinase A. Plant class V chitinase from cycad also exhibited a similar reaction profile.Citation35) Both reports explained that GlcNAc3 is produced from initial substrate GlcNAc4 through the TG product GlcNAc6, as shown in Fig. . GlcNAc4 was first hydrolyzed into GlcNAc2 + GlcNAc2 (Step I). After the latter GlcNAc2 is released from the enzyme, the acceptor GlcNAc4 binds to the acceptor-binding site (the positively numbered subsites) (Step IIb), and then attacks the oxazolinium ion intermediate at subsite −1, producing GlcNAc6 as the TG product (Step III). The GlcNAc6 produced is relocated to the more stable binding mode (−3, −2, −1, +1, +2) (Step IV), and hydrolyzed into GlcNAc3 + GlcNAc3 (Step V). In the case of the initial substrate GlcNAc5, WT produced a small amount of GlcNAc4 at 2 h in addition to GlcNAc2 and GlcNAc3 (Fig. (B)). Since GlcNAc5 is assumed to act as an acceptor molecule as well as a substrate in the mechanism shown in Fig. , GlcNAc4 is most likely produced through the TG product GlcNAc7. Thus, we concluded that the WT enzyme has a very low TG activity. From the initial substrate GlcNAc6, WT produced GlcNAc2, GlcNAc3, and GlcNAc4 (Fig. (C)). No evidence for TG reaction was obtained from the reaction toward GlcNAc6. The result suggested that WT not only catalyzes the hydrolysis of the chitooligosaccharide substrates, but also catalyzes TG reaction much less efficiently with the substrates GlcNAc4 and GlcNAc5.
Fig. 2. Reaction time courses of the wild-type and mutated VhChiA toward chitooligosaccharide substrates GlcNAc4–6.
Notes: The wild-type VhChiA (5 μM) was incubated with 6.8 mM GlcNAc4 (A), 5.5 mM GlcNAc5 (B), or 4.6 mM GlcNAc6 (C), W570G VhChiA (5 μM) was incubated with 6.8 mM GlcNAc4 (D), 5.5 mM GlcNAc5 (E), or 4.6 mM GlcNAc6 (F). D392N VhChiA (5 μM) was incubated with 6.8 mM GlcNAc4 (G), 5.5 mM GlcNAc5 (H), or 4.6 mM GlcNAc6 (I). Individual reactions were conducted in 20 mM phosphate buffer, pH 7.0 at 40 °C. The products were analyzed by gel-filtration HPLC at various times of incubation. Numbers represent the degree of polymerization. Symbols are □, GlcNAc2; ●, GlcNAc3; ○, GlcNAc4; ▲, GlcNAc5; ∆, GlcNAc6.
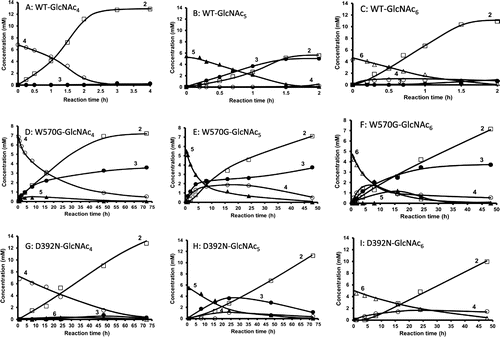
Fig. 3. The reaction scheme for hydrolysis/TG catalyzed by VhChiA with GlcNAc4 substrate.
Notes: Step I: Bond cleavage; GlcNAc4 binds to the −2 to +2 subsites and the glycosidic linkage located between the −1 and +1 subsites is cleaved by the action of Glu315 to form GlcNAc2 with an oxazolinium ion intermediate at subsites −2 and –1 and the intact GlcNAc2 product at subsites +1 and +2, which will diffuse away. Step IIa: Hydration; a water molecule attacks the C1 carbon of the oxazolinium ion intermediate to release the product of GlcNAc2. Steps IIb and III: Acceptor binding and formation of glycosidic linkage; an incoming GlcNAc4 attacks the intermediate instead of a water molecule to form a new glycosidic linkage, producing the TG product of GlcNAc6. Step IV: Shifting the binding mode of GlcNAc6 to subsites −3 to +2. Step V: the newly formed GlcNAc6 is then hydrolyzed to form two molecules of GlcNAc3. GlcNAc residues are represented by open circles, an incoming of GlcNAc4 molecule is represented by grey circles, the oxazolinium ion intermediate is represented by positive signs in open circles and the binding subsites of the enzyme given as integers based on the nomenclature suggested by Davies et al.Citation39) Formation of the TG products by the mutants VhChiA D313A and D313N is represented from Step I to Step III, while the additional steps (Step IV and Step V) should be introduced for the reactions catalyzed by the mutants W570G and D392N.
Time courses of chitooligosaccharide degradation catalyzed by W570G
The hydrolytic activities of mutant W570G toward the substrates GlcNAc4–6 were much less than those of WT (Fig. (D), (E), and (F)), and the results were consistent with the specific activity data reported previously.Citation31) However, a considerable amount of GlcNAc3 was produced in addition to GlcNAc2 from the initial substrate GlcNAc4 (Fig. (D)). The GlcNAc3 produced was clearly derived from the mechanism shown in Fig. , because no GlcNAc was found in the products. GlcNAc5, which may be derived from the TG reaction between the donor GlcNAc2 and the acceptor GlcNAc3, was also detected in the early stage of the reaction. The productions of GlcNAc3 and GlcNAc5 indicate that TG activity was significantly enhanced in W570G. The time-course profiles of mutant W570G with GlcNAc5 substrate (Fig. (E)) showed that GlcNAc2 and GlcNAc3 were the major hydrolytic products. GlcNAc4 was also produced without the formation of GlcNAc, and the maximum level of GlcNAc4 was approximately 2 mM at 16 h of incubation. GlcNAc4 was then gradually degraded to GlcNAc2, and only 0.5 mM remained at 48 h. The GlcNAc4 product may be derived from the mechanism shown in Fig. , where the substrate and the acceptor molecules should be replaced with GlcNAc5. Mutant W570G hydrolyzed GlcNAc6 substrate to GlcNAc2, along with GlcNAc3 and GlcNAc4 (Fig. (F)). GlcNAc5 was also detected, but GlcNAc was not. Thus, the GlcNAc5 product may be produced through the TG product GlcNAc8 as shown in Fig. , where the substrate and the acceptor molecules should be replaced with GlcNAc6. The results obtained from this set of experiments suggested that the mutation of Trp570 located in between subsites −2 and −1 strongly enhanced TG activity, but the TG products obtained from the mutant W570G were only temporarily formed, and then further degraded. Trp570 is responsible for the GlcNAc residue binding at subsites −2 and −1 (Fig. ), so that cleavage of the glycosidic bond between subsites −1 and +1 takes place most efficiently. Substitution of the Trp570 side chain with glycine completely removed the aromatic surface area, thereby causing a dramatic decrease in the hydrolytic activity to about 5% of the WT activity, and decreased the binding affinity (increased Km) that affected sugar–enzyme interaction.Citation31) The reduction of the binding affinity at these two subsites, on the other hand, may relatively enhance the affinity at the acceptor-binding site (positively numbered subsites), resulting in the enhanced TG activity. However, all of the TG products immediately hydrolyzed again into oligosaccharides with shorter chains.
Time courses of chitooligosaccharide degradation catalyzed by D392N
The D392N mutant produced GlcNAc2 as a major product from GlcNAc4 substrate, while a small amount of GlcNAc3 was produced as shown in Fig. (G), probably through the mechanism shown in Fig. . The GlcNAc3 production was slightly enhanced in the D392N mutant, when compared with that in WT (Fig. (A)). The D392N mutant hydrolyzed GlcNAc5 substrate, yielding GlcNAc2 and GlcNAc3 as the major end products (Fig. (H)). The enhanced formation of GlcNAc4 was found in the reaction catalyzed by D392N. Since the GlcNAc4 formation was not accompanied by GlcNAc formation, the tetramer was most likely derived from the mechanism shown in Fig. , where the substrate and the acceptor molecules should be replaced with GlcNAc5. With GlcNAc6 substrate, GlcNAc2 and GlcNAc4 were the major hydrolytic products (Fig. (I)), while no other products were detected. These results suggested that mutation of Asp392, which is involved in sugar residue binding at subsites +1 and +2 (Fig. ), to asparagine enhanced the TG activity of VhChiA with the substrates GlcNAc4 and GlcNAc5, but not with the substrate GlcNAc6. Our previous kinetic data showed that D392N has greater affinity towards pNP-GlcNAc2 and chitooligosaccharide substrates than those of WT.Citation31) The greater affinity of D392N may facilitate the acceptor binding to subsites +1 and +2; hence, the TG reaction for the substrates GlcNAc4 and GlcNAc5 (Fig. (G) and (H)). However, also in this mutant, the TG products were immediately hydrolyzed into oligosaccharides with shorter chains. Mutations of Trp570 and Asp392 are unlikely effective for obtaining chitooligosaccharides with longer chains, even though the mutant enzymes exhibit the enhanced TG activity.
Mutation of Asp313 is the most effective for obtaining chitooligosaccharides with longer chains
Asp313 is an essential residue located at the middle of the catalytic DxDxE motif (Asp311-x-Asp313-x-Glu315), and plays multiple roles in the catalytic cycle of chitin degradation by VhChiACitation32). Mutation of Asp313 to alanine (D313A) abolished the hydrolytic activity of the enzyme almost completely, while mutation of Asp313 to asparagine (D313N) retained slight hydrolytic activity. HPLC profiles of the products from incubation of the mutant D313A or D313N with GlcNAc4 substrate indicated that a significant amount of GlcNAc6 as the TG product was generated in addition to the major hydrolytic product GlcNAc2 after 120 h of incubation, as shown in Fig. (B) and (C). In contrast, no GlcNAc6 was found in the chromatogram for WT (Fig. (A)). In the reactions catalyzed by D313A and D313N, the TG product GlcNAc6 was not hydrolyzed into GlcNAc3. Similarly, when GlcNAc6 was incubated with the Asp313 mutants, a significant amount of GlcNAc8, which was produced by the TG reaction between the donor GlcNAc2 and the acceptor GlcNAc6, was detected by HPLC (Fig. (B) and (C)). WT did not produce GlcNAc8 at all (Fig. (A)). The chain length of the TG product, GlcNAc8, was confirmed based on the theoretical retention time obtained by the simulation of the gel-filtration profile.Citation29) The donor for the TG reaction appears to be GlcNAc2, because VhChiA hydrolyzes most frequently the second β-1,4-glycosidic linkage from the nonreducing end of chitooligosaccharide substrates.Citation36) Thus, from the substrate GlcNAc5, the Asp313 mutants may produce GlcNAc7 by the TG reaction between the donor GlcNAc2 and the acceptor GlcNAc5.
Fig. 4. HPLC profiles showing the reaction of the wild type (A) and the mutants D313A (B) and D313N (C) VhChiA.
Notes: A reaction mixture containing 6.8 mM GlcNAc4 and the enzyme (5 μM wild type, 16 μM D313A, or 8 μM D313N) in 20 mM phosphate buffer, pH 7.0, was incubated at various times at 40 °C. The reaction products were analyzed by gel-filtration HPLC. The TG product GlcNAc6 is designated by arrow.
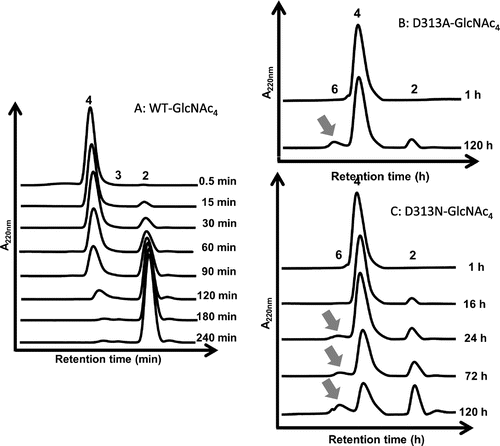
Fig. 5. HPLC profiles showing the reaction of the wild type (A) and the mutants D313A (B) and D313N (C) VhChiA.
Notes: A reaction mixture containing 4.6 mM GlcNAc6 and the enzyme (5 μM wild type, 16 μM D313A, or 8 μM D313N) in 20 mM phosphate buffer, pH 7.0, was incubated at various times at 40 °C. The reaction products were analyzed by gel-filtration HPLC. The TG product GlcNAc8 is indicated by arrow.
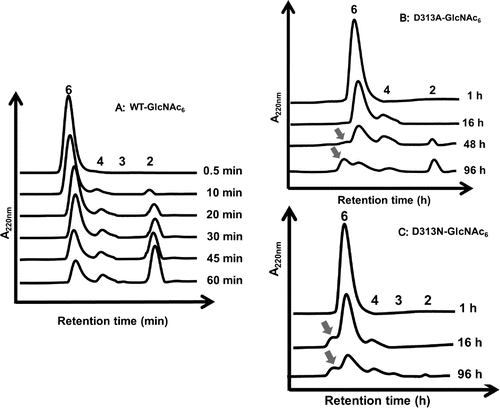
VhChiA is a bacterial GH18 chitinase that cleaves a chitin chain into various chitooligosaccharide fragments.Citation37) Based on our previous studies,Citation30,36) VhChiA has structure and function similar to those of S. marcescens chitinase A, and it degrades GlcNAc4 substrate mostly to GlcNAc2, GlcNAc5 substrate to GlcNAc, GlcNAc2, and GlcNAc3, while GlcNAc6 is degraded to GlcNAc2, GlcNAc3, and GlcNAc4. GH18 chitinases have a catalytic motif specified by a sequence DxDxE, which correspond to Asp311-x-Asp313-x-Glu315 in VhChiA. Glu315 is a catalytic acid, which donates a proton to the β-1,4-glycosidic oxygen to cleave the linkage. Asp313 is located at the bottom of the substrate binding cleft (Fig. ). This aspartic acid plays multiple roles in the catalytic cycle of chitin hydrolysis.Citation32,38) It interacts with the 2-acetamido group of the sugar residue at subsite −1 (the cleavage site) and helps to lower the pKa value of the catalytic residue Glu315, so that bond cleavage can be achieved more easily. Moreover, it helps to orient the 2-acetamido group in the correct position to stabilize the oxazolinium ion intermediate in the substrate-assisted mechanism. Mutations of Asp313 to Ala and Asn abolished the hydrolytic activity almost completely by disrupting hydrogen-bond interactions with the sugar residue. Instead, the mutations enhanced the TG activity. We tried to compare the efficiencies of TG reaction obtained by our D313A/N mutants with those obtained by the corresponding mutants of the two Serratia enzymes, SmChiA and SmChiB.Citation20) In the Serratia enzymes, the mutations of the middle Asp of the DxDxE motif to Asn were reported to enhance the TG reaction more strongly than the mutations to Ala. In our VhChiA mutants, however, no significant difference was found in the highest yields of the TG products (GlcNAc6 from the initial substrate GlcNAc4, Fig. ; or GlcNAc8 from the initial substrate GlcNAc6, Fig. ) between D313A and D313N. The TG efficiencies in the mutants from Serratia enzymes were evaluated from the GlcNAc3 production from the initial substrate GlcNAc4, indicating that the TG product GlcNAc6 was decomposed into GlcNAc3 as shown in Fig. .Citation20) The evaluation of TG efficiency based on the yield of GlcNAcn with longer chains (TG products) may be more informative for practical use of the transglycosylating chitinases. Thus, the mutants of the middle Asp of the DxDxE motif from VhChiA are likely more effective for obtaining GlcNAcn with longer chains than the corresponding mutants from the Serratia enzymes, SmChiA and SmChiB.Citation20)
In the Asp313 mutants from VhChiA, the Km values toward GlcNAc6 were four- (D313N) or six-fold (D313A) higher than that of the wild type.Citation32) The lower affinity may result in the spontaneous release of the TG product from the enzyme without relocation to the productive binding mode (process IV in Fig. ). This situation may bring about the accumulation of the TG products in the Asp313 mutants. In the other mutants W570G and D392N, however, the TG products may be immediately relocated to the productive binding mode spanning the catalytic center, due to the affinity with Asp313, and subsequently broken down by the hydrolytic action of the enzyme. Zakariassen et al.Citation20) who reported a hypertransglycosylating mutants obtained from the Serratia enzymes, explained that the mutation of Asp313 changes the electrostatics around the catalytic center, decreasing the probability of nucleophilic attack of a water molecule to the oxazolinium ion intermediate. Similar situation may possibly take place in the VhChiA mutants, D313A and D313N. Aronson et al. reported that the mutation of Trp167 of SmChiA to alanine (W167A) significantly enhances the TG reaction.Citation19) In W167A, the side chain of Asp313 is oriented only toward Glu315, whereas in the wild type, the Asp313 side chain is equally distributed between two orientations, toward Asp311 or toward Glu315. They explained that the orientation of Asp313 toward Glu315 may interfere with the attack of a water molecule to the oxazolinium ion intermediate. Thus, the state of the side chain of Asp313 appears to be related to the efficiency of TG reaction. Crystal structure analysis of VhChiA D313N or D313A will afford valuable information on the structural factor for enhancing the TG reaction in VhChiA.
In conclusion, mutations of Trp570 and Asp392 of VhChiA significantly enhanced the TG reaction, but the TG products were immediately hydrolyzed into chitooligosaccharides with shorter chains. In contrast, mutations of Asp313 strongly enhanced the TG reaction, and the products, chitooligosaccharides with longer chains, were not hydrolyzed but accumulated in the reaction mixture. The results obtained from this study may suggest a convenient, strategic design for new chitinase molecules with suitable property for producing the biologically active chitooligosaccharides required for pharmaceutical and industrial uses.
Acknowledgments
We would like to acknowledge Biochemistry Laboratory, the Center for Scientific and Technological Equipment, SUT for providing all the facilities for this research.
Additional information
Funding
Notes
Abbreviations: GlcNAcn, β-1,4 linked oligomers of N-acetyl-D-glucosamine units where n is a chain length, 1–6; IPTG, isopropyl thio-β-D-galactoside; TG, transglycosylation; VhChiA, Vibrio harveyi chitinase A; WT, wild-type VhChiA; HPLC, high performance liquid chromatography.
References
- Arbia W, Arbia L, Adour L, Amrane A. Chitin recovery from crustacean shells by biological methods—A review. Food Technol. Biotechnol. 2013;51:12–25.
- Merzendorfer H, Zimoch L. Chitin metabolism in insects: Structure, function and regulation of chitin synthases and chitinases. J. Exp. Biol. 2003;206:4393–4412.10.1242/jeb.00709
- Free SJ. Fungal cell wall organization and biosynthesis. Adv. Genet. 2013;81:33–82.10.1016/B978-0-12-407677-8.00002-6
- Funkhouser JD, Aronson NN. Chitinase family GH18: Evolutionary insights from the genomic history of a diverse protein family. BMC Evol. Biol. 2007;7:96.10.1186/1471-2148-7-96
- Kawase T, Saito A, Sato T, Kanai R, Fujii T, Nikaidou N, Miyashita K, Watanabe T. Distribution and phylogenetic analysis of family 19 chitinases in actinobacteria. Appl. Environ. Microbiol. 2004;70:1135–1144.10.1128/AEM.70.2.1135-1144.2004
- van Aalten D, Komander D, Synstad B, Gaseidnes S, Peter M, Eijsink V. Structural insights into the catalytic mechanism of a family 18 exo-chitinase. Proc. Nat. Acad. Sci. 2001;98:8979–8984.10.1073/pnas.151103798
- Terwisscha van Scheltinga AC, Hennig M, Dijkstra BW. The 1.8 Å resolution structure of hevamine, a plant chitinase/lysozyme, and analysis of the conserved sequence and structure motifs of glycosyl hydrolase family 18. J. Mol. Biol. 1996;262:243–257.10.1006/jmbi.1996.0510
- Brameld KA, Goddard WA III. The role of enzyme distortion in the single displacement mechanism of family 19 chitinases. Proc. Nat. Acad. Sci. 1998;95:4276–4281.10.1073/pnas.95.8.4276
- Hoell IA, Dalhus B, Heggset EB, Aspmo SI, Eijsink VGH. Crystal structure and enzymatic properties of a bacterial family 19 chitinase reveal differences from plant enzymes. FEBS J. 2006;273:4889–4900.10.1111/ejb.2006.273.issue-21
- Rinaudo M. Chitin and chitosan: Properties and applications. Prog. Polym. Sci. 2006;31:603–632.10.1016/j.progpolymsci.2006.06.001
- Kaku H, Nishizawa Y, Ishii-Minami N, Akimoto-Tomiyama C, Dohmae N, Takio K, Minami E, Shibuya N. Plant cells recognize chitin fragments for defense signaling through a plasma membrane receptor. Proc. Nat. Acad. Sci. 2006;103:11086–11091.10.1073/pnas.0508882103
- Miya A, Albert P, Shinya T, Desaki Y, Ichimura K, Shirasu K, Narusaka Y, Kawakami N, Kaku H, Shibuya N. CERK1, a LysM receptor kinase, is essential for chitin elicitor signaling in Arabidopsis. Proc. Nat. Acad. Sci. 2007;104:19613–19618.10.1073/pnas.0705147104
- Yamaguchi K, Yamada K, Ishikawa K, Yoshimura S, Hayashi N, Uchihashi K, Ishihama N, Kishi-Kaboshi M, Takahashi A, Tsuge S, Ochiai H, Tada Y, Shimamoto K, Yoshioka H, Kawasaki T. A receptor-like cytoplasmic kinase targeted by a plant pathogen effector is directly phosphorylated by the chitin receptor and mediates rice immunity. Cell Host Microbe. 2013;13:347–357.10.1016/j.chom.2013.02.007
- VishuKumar AB, Varadaraj MC, Gowda LR, Tharanathan RN. Characterization of chito-oligosaccharides prepared by chitosanolysis with the aid of papain and Pronase, and their bactericidal action against Bacillus cereus and Escherichia coli. Biochem. J. 2005;391:167–175.
- Petutschnig EK, Jones AM, Serazetdinova L, Lipka U, Lipka V. The lysin motif receptor-like kinase (LysM-RLK) CERK1 is a major chitin-binding protein in Arabidopsis thaliana and subject to chitin-induced phosphorylation. J. Biol. Chem. 2010;285:28902–28911.10.1074/jbc.M110.116657
- Kanie O, Ito Y, Ogawa T. Orthogonal glycosylation strategy in oligosaccharide synthesis. J. Am. Chem. Soc. 1994;116:12073–12074.10.1021/ja00105a066
- Aly MRE, Ibrahim E-SI, Ashry ESHE, Schmidt RR. Synthesis of chitotetraose and chitohexaose based on dimethylmaleoyl protection. Carbohydr. Res. 2001;331:129–142.10.1016/S0008-6215(01)00024-6
- Fukamizo T, Sasaki C, Schelp E, Bortone K, Robertus JD. Kinetic properties of chitinase-1 from the fungal pathogen Coccidioides immitis. Biochemistry. 2001;40:2448–2454.10.1021/bi001537s
- Aronson NN, Halloran BA, Alexeyev MF, Zhou XE, Wang Y, Meehan EJ, Chen L. Mutation of a conserved tryptophan in the chitin-binding cleft of Serratia marcescens chitinase a enhances transglycosylation. Biosci., Biotechnol. Biochem. 2006;70:243–251.
- Zakariassen H, Hansen MC, Jøranli M, Eijsink VG, Sørlie M. Mutational effects on transglycosylating activity of family 18 chitinases and construction of a hypertransglycosylating mutant. Biochemistry. 2011;50:5693–5703.10.1021/bi2002532
- Nanjo F, Sakai K, Ishikawa M, Isobe K, Usui T. Properties and transglycosylation reaction of a chitinase from Nocardia orientalis. Agric. Biol. Chem. 1989;53:2189–2195.10.1271/bbb1961.53.2189
- Purushotham P, Podile AR. Synthesis of long-chain chitooligosaccharides by a hypertransglycosylating processive endochitinase of Serratia proteamaculans 568. J. Bacteriol. 2012;194:4260–4271.10.1128/JB.06473-11
- Martinez EA, Boer H, Koivula A, Samain E, Driguez H, Armand S, Cottaz S. Engineering chitinases for the synthesis of chitin oligosaccharides: Catalytic amino acid mutations convert the GH-18 family glycoside hydrolases into transglycosylases. J. Mol. Catal. B: Enzym. 2012;74:89–96.10.1016/j.molcatb.2011.09.003
- Madhuprakash J, Tanneeru K, Purushotham P, Guruprasad L, Podile AR. Transglycosylation by chitinase D from Serratia proteamaculans improved through altered substrate interactions. J. Biol. Chem. 2012;287:44619–44627.10.1074/jbc.M112.400879
- Rupley JA. The hydrolysis of chitin by concentrated hydrochloric acid, and the preparation of low-molecular-weight substrate for lysozyme. Biochim. Biophys. Acta. 1964;83:245–255.
- Hsu SC, Lockwood JL. Powdered chitin agar as a selective medium for enumeration of actinomycetes from water and soil. Appl. Microbiol. 1975;29:422–426.
- Umemoto N, Ohnuma T, Mizuhara M, Sato H, Skriver K, Fukamizo T. Introduction of a tryptophan side chain into subsite +1 enhances transglycosylation activity of a GH-18 chitinase from Arabidopsis thaliana, AtChiC. Glycobiology. 2013;23:81–90.10.1093/glycob/cws125
- Hurtado-Guerrero R, Schuttelkopf AW, Mouyna I, Ibrahim AF, Shepherd S, Fontaine T, Latge JP, van Aalten DM. Molecular mechanisms of yeast cell wall glucan remodeling. J. Biol. Chem. 2009;284:8461–8469.10.1074/jbc.M807990200
- Fukamizo T, Goto S, Torikata T, Araki T. Enhancement of transglycosylation activity of lysozyme by chemical modification. Agric. Biol. Chem. 1989;53:2641–2651.10.1271/bbb1961.53.2641
- Songsiriritthigul C, Pantoom S, Aguda AH, Robinson RC, Suginta W. Crystal structures of Vibrio harveyi chitinase a complexed with chitooligosaccharides: Implications for the catalytic mechanism. J. Struct. Biol. 2008;162:491–499.10.1016/j.jsb.2008.03.008
- Suginta W, Songsiriritthigul C, Kobdaj A, Opassiri R, Svasti J. Mutations of Trp275 and Trp397 altered the binding selectivity of Vibrio carchariae chitinase A. Biochim. Biophys. Acta. 2007;1770:1151–1160.10.1016/j.bbagen.2007.03.012
- Suginta W, Sritho N. Multiple roles of Asp313 in the refined catalytic cycle of chitin degradation by Vibrio harveyi chitinase A. Biosci., Biotechnol. Biochem. 2012;76:2275–2281.10.1271/bbb.120559
- Pantoom S, Songsiriritthigul C, Suginta W. The effects of the surface-exposed residues on the binding and hydrolytic activities of Vibrio carchariae chitinase A. BMC Biochem. 2008;9:2.10.1186/1471-2091-9-2
- Pace CN, Vajdos F, Fee L, Grimsley G, Gray T. How to measure and predict the molar absorption coefficient of a protein. Protein Sci. 1995;4:2411–2423.10.1002/pro.v4:11
- Taira T, Fujiwara M, Dennhart N, Hayashi H, Onaga S, Ohnuma T, Letzel T, Sakuda S, Fukamizo T. Transglycosylation reaction catalyzed by a class V chitinase from cycad, Cycas revoluta: A study involving site-directed mutagenesis, HPLC, and real-time ESI-MS. Biochim. Biophys. Acta. 2010;1804:668–675.10.1016/j.bbapap.2009.10.015
- Suginta W, Pantoom S, Prinz H. Substrate binding modes and anomer selectivity of chitinase A from Vibrio harveyi. J. Chem. Biol. 2009;2:191–202.10.1007/s12154-009-0021-y
- Suginta W, Vongsuwan A, Songsiriritthigul C, Prinz H, Estibeiro P, Duncan RR, Svasti J, Fothergill-Gilmore LA. An endochitinase A from Vibrio carchariae: Cloning, expression, mass and sequence analyses, and chitin hydrolysis. Arch. Biochem. Biophys. 2004;424:171–180.10.1016/j.abb.2004.01.017
- Synstad B, Gaseidnes S, van Aalten DM, Vriend G, Nielsen JE, Eijsink VG. Mutational and computational analysis of the role of conserved residues in the active site of a family 18 chitinase. Eur. J. Biochem. 2004;271:253–262.10.1046/j.1432-1033.2003.03923.x
- Davies GJ, Wilson KS, Henrissat B. Nomenclature for sugar-binding subsites in glycosyl hydrolases. Biochem. J. 1997;321:557–559.