
Abstract
Homologous recombination is a system for repairing the broken genomes of living organisms by connecting two DNA strands at their homologous sequences. Today, homologous recombination in yeast is used for plasmid construction as a substitute for traditional methods using restriction enzymes and ligases. This method has various advantages over the traditional method, including flexibility in the position of DNA insertion and ease of manipulation. Recently, the author of this review reported the construction of plasmids by homologous recombination in the methanol-utilizing yeast Pichia pastoris, which is known to be an excellent expression host for secretory proteins and membrane proteins. The method enabled high-throughput construction of expression systems of proteins using P. pastoris; the constructed expression systems were used to investigate the expression conditions of membrane proteins and to perform X-ray crystallography of secretory proteins. This review discusses the mechanisms and applications of homologous recombination, including the production of proteins for X-ray crystallography.
Graphical Abstract
High-throughput construction of an expression system using Pichia pastoris: its application to production of a GFP-membrane protein fusion.
To repair their broken genomes, living organisms have a system connecting two DNA strands at their similar (homologous) sequences.Citation1–3) This system is called homologous recombination, and it is employed for genetic recombination during meiosis. In recent years, homologous recombination in yeast has been used for plasmid construction (such as insertion of a target gene into a plasmid) and gene isolation (such as transformation-associated recombination cloning to directly capture genomic loci from Saccharomyces cerevisiae).Citation4,5) Unlike with traditional methods using restriction enzymes and ligases, the method for plasmid construction using homologous recombination can insert a DNA fragment into an appropriate and favorable position of a plasmid. The regions of homology used for recombination need not be at the ends of the linearized plasmid.Citation4) For example, using a plasmid with a green fluorescence protein (GFP) in the region downstream of the promoter, homologous recombination has been applied to high-throughput construction of a yeast S. cerevisiae expression system of GFP membrane protein fusions.Citation6,7) This system has been efficiently used to investigate the expression, purification, and crystallization conditions of membrane proteins. As another example, a method was developed for converting a common Escherichia coli vector to an E. coli yeast shuttle vector to enable the construction of plasmid by homologous recombination in S. cerevisiae,Citation8,9) and this technique has been efficiently used to construct expression plasmids for plants. Recently, two groups demonstrated plasmid constructions using homologous recombination in fission yeast Schizosaccharomyces pombe.Citation10,11) In addition, the author of this review discovered that homologous recombination could be used to construct a plasmid in the methanol-utilizing yeast Pichia pastoris.Citation12,13) I and my collaborators reported high-throughput construction of P. pastoris expression systems of membrane proteins and secretory proteins. For secretory proteins, large-scale preparation and X-ray crystallography of the protein were also achieved. Homologous recombination in S. cerevisiae can connect multiple fragments, and yeast can retain a larger plasmid compared to those in the case of E. coli. A group at the J. Craig Venter Institute (JCVI) reported the preparation of a synthetic bacterial genome by connecting synthesized DNA fragments by homologous recombination in yeast.Citation14) They also reported the “creation” of bacteria by transplantation of the synthetic genome into recipient cells.Citation15) Homologous recombination and other methods connecting multiple DNA fragments, such as overlap extension PCR and Gibson Assembly (a new enzymatic method described later in Chapter IV), also enable the introduction of mutations, preparation of fusion proteins, in vitro splicing, and synthesis of genes.
Today, X-ray crystallography has become one of the most important techniques in bioscience. I and my collaborators have determined and reported the crystal structures of secretory proteins of eukaryote (microorganisms and animals), such as β-1,4-mannnase of a gastropod (the common sea hare, Aplysia kurodai), bilirubin oxidase of a fungus (Myrothecium verrucaria), and lysozyme and transferrin of chicken (Gallus gallus),Citation16–19) and our main target protein was transferrin, which is an iron-binding and iron-transporting protein.Citation20–24) The transferrin molecule is folded into two homologous lobes, and each lobe is further divided into two similarly sized domains.Citation25) An iron-binding site is contained within an inter-domain cleft, and a large-scale domain motion occurs upon the uptake and release of an iron. The apo and holo structures of the chicken transferrin N-lobe revealed the mechanism of the domain motion; the domains were rotated as rigid bodies around a screw axis passing through inter-domain contacts (two β-strand connecting domains and van der Waals contacts around the C-terminal residues).Citation21,24) We also determined an iron-loaded but open-domain crystal structure (iron-binding intermediate) in which two residues in one of the domains bind to iron.Citation23) Transferrin can bind other metal ions, including toxic ones, and is considered to play an important role in their transportation.Citation25) We reported the first structural analysis of aluminum-binding transferrin, which had a domain-closed structure similar to the iron-binding form.Citation20) The extent of domain closure varies slightly according to the size of the bound metal; the aluminum-bound form has a more compact structure than the iron-bound form.Citation20,25) I and my collaborators constructed a P. pastoris expression system of human serum transferrin and chicken transferrin by a traditional method.Citation13,26,27) Recombinant proteins were secreted into media and had N-linked high-mannose carbohydrate chains. Both proteins had tertiary structures similar to native proteins, and bound two irons as native proteins. We determined the crystal structure of the recombinant chicken transferrin N-lobe at atomic resolution, i.e. 0.93 angstrom, at which resolution hydrogen atoms can be observed (Mizutani, unpublished data). Serum transferrin binds iron molecules in the blood serum and delivers them into target cells via the transferrin receptor. Since transferrin binds synergistic anions, such as carbonate with iron, the development of an anion acting as a medical agent would enable drug delivery using transferrin. The high resolution crystal structure clearly revealed the dissociative state () of the bound carbonate anion and hydrogen bond network around the anion. The chicken transferrin N-lobe was crystallized under microgravity at the Japanese Experiment Module (JEM Kibo) of the International Space Station, and X-ray diffraction data were collected. The data is now being analyzed and evaluated. A yeast expression system is appropriate for the production of eukaryotic proteins, especially secretory proteins and membrane proteins. In this review, I describe the mechanism of homologous recombination and its numerous applications, including protein production for X-ray crystallography.
I. Significance and mechanism of homologous recombination
When a DNA double strand in a living organism is broken by external factors, such as radiation, it is necessary for the organism to repair the damaged DNA chain. One of the methods for such repair is homologous recombination, which is a reaction for connecting two DNA chains at the same sequence (or very similar sequences).Citation1–3) Living organisms can use another set of DNA chains to repair a broken DNA chain in S and G2 phases of the cell cycle when a sister chromatid is present; homologous recombination can repair the broken DNA more accurately than simple connection of broken chains (non-homologous end joining pathway, as shown below).Citation1–3) It is known that homologous recombination in G1 phase can occur in diploid cells of S. cerevisiae.Citation1,3,28) During meiosis homologous, recombination is used to exchange parts of paired chromosomes for increasing genome diversity of the haploid gamete.Citation1–3) It is known that homologous recombination can occur between the genome and an externally introduced DNA strand. Therefore, homologous recombination is used for the transformation of cultured cells and yeast cells.Citation29,30) Although transformation methods using viruses and autonomous plasmids are often used for cultured cells and yeast, respectively; homologous recombination has now become another valid option for stable integration of external DNA into the genome.Citation31,32)
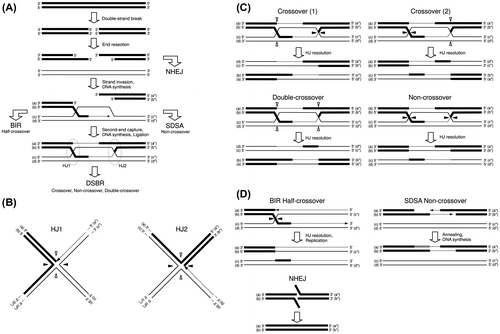
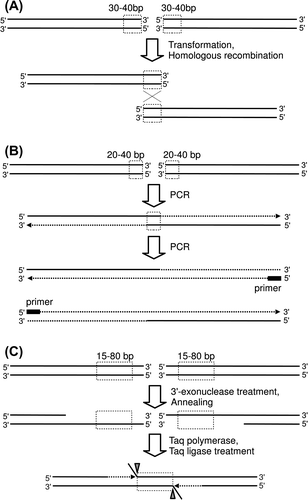
Homologous recombination is carried out in eukaryotic cells as shown in Fig. (A).Citation1–3) From broken DNA chains, 5′ terminal nucleotides are removed (end resection). The generated single-chain DNA binds the homologous part of an intact DNA chain (strand invasion), and DNA synthesis is started. In DSBR (double-strand break repair) pathway, also known as dHJ (double Holliday junction) pathway, binding of the other end of the broken DNA strand to the intact DNA chain (second end capture) and subsequent DNA synthesis yield two Holliday junctions. Two patterns are possible for resolution of the Holliday junctions, as shown in Fig. (B). Combinations of patterns of Holliday junction resolution can generate crossed DNA chains (crossover), restored DNA chains of non-crossover type, or restored DNA chains of double-crossover type (by duplicated crossing), as shown in Fig. (C). Many proteins contribute to the homologous recombination, and investigations of the roles of these proteins are a hot study area today.Citation33) More roles of various protein in homologous recombination are described in other reviews and papers.Citation1–3,34,35) In addition to the DSBR pathway, the BIR (break-induced replication) pathway, SDSA (synthesis-dependent strand annealing) pathway, and NHEJ (non-homologous end joining) pathway are known for their applicability to DNA repair mechanisms, as shown in Fig. (A) and (D).Citation1–3,36,37) In the BIR pathway, DNA replication occurs simultaneously to yield a half-crossover DNA chain. In the SDSA pathway, the synthesized DNA chain is annealed to the original DNA chain, yielding a non-crossover DNA chain. In the NHEJ pathway, no additional DNA chain is used; rather, the ends of broken DNA chains are simply joined. It is known that the activity of the NHEJ pathway is high in mammalian cells, whose genome has large non-protein-coding regions, and homologous recombination is the preferred pathway in yeast.Citation38)
Fig. 1. Mechanism of homologous recombination in eukaryotic cells.
Notes: A schematic of the repair protocol for a DNA double-strand break including homologous recombination and other pathways is shown (A). Structures of two Holliday junctions in the DSBR pathway in (A) are shown (B). The Holliday junction is resolved by cutting and connection at the white arrowheads or black arrowheads. Four possible patterns of homologous recombination are shown (C). Mechanisms of other pathways (BIR, SDSA, and HHEJ) to repair broken DNA chains are shown (D).
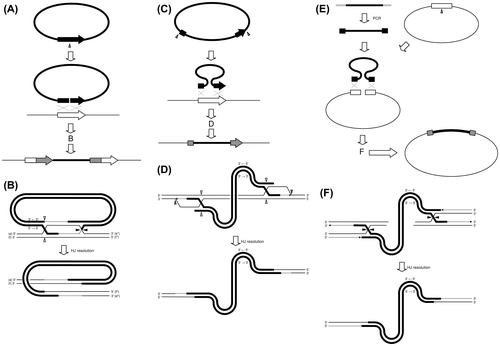
From the 1970s, homologous recombination has been used for protein expression and gene disruption of eukaryotic cells.Citation39–41) Two types of homologous recombination, ends-in (Fig. (A) and (B)) and ends-out (Fig. (C) and (D)) are used for the transformation of cells.Citation29,30,42) For the ends-in type, a plasmid containing a target gene is used; cutting of the plasmid inside the gene creates a linear DNA chain with a homologous sequence (homologous to a target gene) at both ends.Citation29) Introduction of the linear DNA chain starts the DNA repair reaction, as shown in Fig. (A). When the generated two Holliday junctions are resolved as crossover (1) or crossover (2) as shown in Fig. (C), the whole sequence of the linear DNA chain is introduced to the target genome. Detailed mechanism for crossover (1) type was shown in Fig. (B). When homologous sequences are attached to both sides of a linear DNA (with the direction of the two parts being the same as that of the target gene), introduction of the linear DNA chain induces ends-out-type recombination, as shown in Fig. (C).Citation29) In this type, recombination succeeds only when two Holliday junctions are resolved as a “double-crossover”; the recombination fails when other resolution types occur (Fig. (D)). The reported lower efficiency of the ends-out type compared to the ends-in type recombination may be due to the mechanism shown above.Citation29) Homologous recombination occurs between plasmid DNA and a DNA chain with a homologous region (homologous to the plasmid),Citation4) and this technique has been used for the construction of plasmids and the preparation of protein expression system.Citation7,8,43) As seen in Fig. (E), the homologous recombination is of the ends-out type. It is reported that efficiency of this reaction (i.e. the efficiency of DNA fragment insertion into a plasmid) is much higher than that of the ends-out-type DNA introduction into the genome.Citation7,44) The reasons of the high efficiency may be the short sequence of the plasmid compared to the genome DNA and BIR pathway like reaction induced by replication of DNA from a replication origin sequence introduced into an autonomous plasmid.Citation45) As shown in Fig. (F), any type of Holliday junction resolution will yield a DNA fragment-inserted plasmid. This high efficiency reaction has been used for the preparation of protein expression systems, construction of plasmids, and connection of multiple DNA fragments. Unlike with traditional methods by digestion using restriction enzymes and covalent linking of DNA strands (ligation) with ligase, the method by homologous recombination can insert a DNA fragment into an appropriate and favorable position in a plasmid. As seen in Fig. (E) and (F), a DNA fragment can be inserted at any part of the linearized plasmid (and not only the very end of the linearized plasmid) by designing an adequate homologous sequence to be added to the DNA fragment.
Fig. 2. DNA fragment insertion by homologous recombination.
Notes: For insertion of DNA fragment into a eukaryotic genome by homologous recombination, a schematic view (A) and the mechanism (B) of Ends-in type insertion are shown, and a schematic view (C) and current model (D) of Ends-out type insertion are shown. For DNA fragment insertion into an autonomous plasmid in yeast cells, a schematic view (E) and possible mechanism (F) are shown. Homologous regions in the DNA fragments and the genome were shown as black and white boxes, respectively (A) and (C). Homologous regions in the DNA fragment and the plasmid were shown in the same way (E). Regions used for homologous recombination were shown as gray boxes (A), (C), and (E). Holliday junctions are resolved by cutting and connection at the white and black arrowheads (B), (D), and (F).
II. Construction of expression plasmid using homologous recombination in yeast S. cerevisiae
As described above, autonomous plasmids are widely used for the transformation and preparation of protein expression systems in S. cerevisiae, and the plasmid construction can be done by homologous recombination in S. cerevisiae itself. Drew and Newstead et al. developed a plasmid (pDDGFP-2) with a GFP in the region downstream of the S. cerevisiae GAL1 promoter that is induced by galactose, and established a method to for the preparation of a membrane protein expression system by inserting a membrane protein gene between the promoter and GFP via homologous recombination in yeast S. cerevisiae.Citation6,7) This is a high-throughput expression system construction method using homologous recombination; the common transformation treatment (PEG-Li method)Citation46,47) using a linearized plasmid by cutting at the SmaI restriction enzyme site (between the promoter and GFP) and target gene with a homologous sequence (homologous to the ends of the linearized plasmid) at both sides can enable construction of an expression plasmid, and thus can be used to prepare a S. cerevisiae expression system. For the transformed yeast cells, expression of the membrane protein-GFP fusion can be induced by galactose, and the expression level of the fusion can be estimated by fluorescence from GFP. Observation of the yeast cells by fluorescence microscope also enables investigation of the localization of the fusion protein in the yeast cells.Citation6,7) Fig. (A) shows a fluorescence microscopic photograph of the rhodopsin of medaka fish-GFP fusion prepared using the above-described method in our laboratory.Citation12) Optimization of the conditions for expression, purification, and solubilization by detergent can also be done using GFP fluorescence.Citation6,7,48) Because the goal of the study is crystallization and structure determination of the membrane proteins, Drew and Newstead et al. tested possibility of preparing mono-disperse samples using gel filtration. Because a fluorescence detector enables the employment of crude solubilized samples, the method (preparing the membrane protein-GFP fusion) is robust and effective.Citation6,7) Afterwards, Shimamura and Shiroishi et al. succeeded in the crystallization and structure determination of membrane proteins investigated by the homologous recombination method.Citation49–51) In addition to X-ray crystallography, the plasmid pDDGFP-2 was also successfully employed for further analyses, such as receptor–ligand binding assays.Citation52)
Fig. 3. Construction of an expression plasmid using S. cerevisiae.
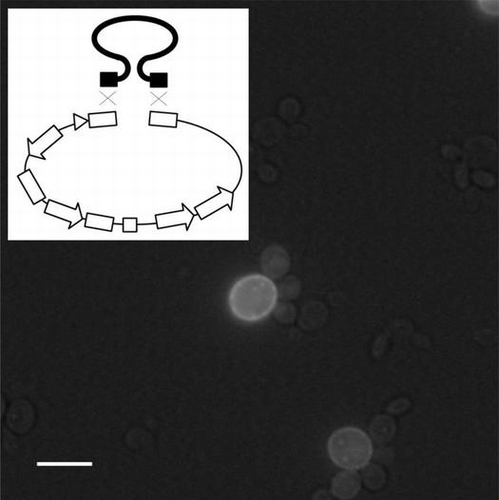
Notes: A photograph of S. cerevisiae cells expressing a GFP-membrane protein fusion (medaka rhodopsin) taken using a fluorescence microscope is shown (A). A low level of bright-field light was included to detect yeast cells. A scale bar in the photograph is 5 μm. Schematic view of introduction of mutation by joining of multiple fragments is shown (B). The mutation sites are indicated with stars.
By exploiting the ability of homologous recombination to connect multiple (more than two) fragments, Shimamura and Shiroishi et al. reported a simple method for the preparation of mutants and fusion proteins (in addition to membrane protein and GFP).Citation49,50) As shown in Fig. (B) and Chapter IV, mutants can be prepared using severed PCR amplified fragments of the gene with mutation at the ends of the fragments.Citation44,51) They reported a change in the expression level by mutations and also reported a crystallization study of the fusion proteins.Citation44,49–51) The details of this method and its application to the joining of multiple fragments are described in Chapter IV.
Iizasa and Nagano applied homologous recombination to the manipulation of general plasmid (originally not for yeast) using a method which they subsequently patented.Citation8) Many E. coli plasmids have a common Amp-resistant gene (Amp) and a common origin of replication (pBR322 origin). They have developed “helper plasmids” for insertion of the 2μ origin (replication origin of yeast S. cerevisiae) and URA3 gene (selection marker for S. cerevisiae) between Amp and pBR322 replication origin. They reported construction of an expression plasmid for plants using this method and introduction of the purified constructed plasmid into plants to prepare an expression system.Citation8,9) Transformation of S. cerevisiae using three DNA fragments, an E. coli plasmid (cut at multiple cloning sites), a target gene (with homologous sequence of the multiple cloning site at both ends), and a helper plasmid (cut by two restriction enzymes), enables construction of an expression plasmid by homologous recombination in yeast cells.Citation8,9) The constructed plasmid can be prepared from S. cerevisiae cells and can be amplified in E. coli cells.Citation9) Since the helper plasmid has replication origins for both E. coli and S. cerevisiae, construction of a plasmid (assembly of the three DNA fragments) may fail if an uncut helper plasmid remains or NHEJ pathway occurs, as shown in Chapter I. Recently, Goto and Nagano developed a new plasmid without E. coli replication origin, and reported a construction method with much higher efficiency than the original method.Citation53)
III. Construction of an expression system using homologous recombination in the methylotrophic yeast P. pastoris
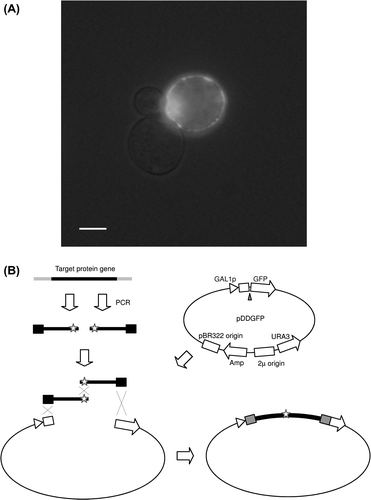
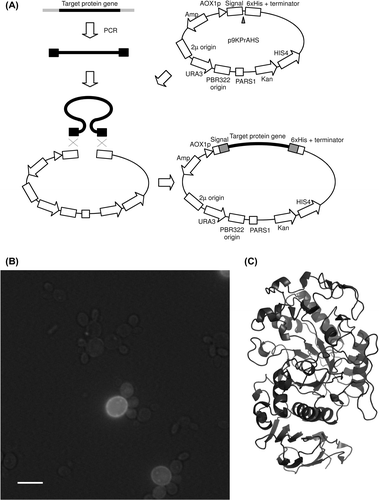
Because the methanol-utilizing yeast P. pastoris can be used for high cell density cultivation and has the strong promoter AOX1 (that is induced by methanol), it is known to be an excellent expression host for eukaryotic secretory proteins and membrane proteins.Citation26,27,49,50,54,55) In general, the construction of an expression system using P. pastoris is time consuming; the expression plasmid is usually constructed using the traditional method (manipulation using enzymes and amplification in E. coli), and then the constructed plasmid is inserted into the genome of P. pastoris. Cregg et al. developed the first P. pastoris expression system using autonomous plasmids with PARS1 or PARS2 (replication sequence derived from the genome of P. pastoris).Citation56) The autonomous plasmids had not been widely used because of their relatively low stability in non-selective medium, while non-autonomous plasmids for insertion into the genome by homologous recombination, such as pPIC3.5, pPIC3.5K, pPIC9 and pPIC9K, all available from Life Technologies, had been widely used.Citation26,54,55) Recently, I and my collaborators introduced the PARS1 sequence into the Life Technologies plasmids and developed new autonomous plasmids (such as p9KPrAHS) compatible with the Life Technologies plasmids (Fig. (A)).Citation12,13) We found that construction of an expression system by homologous recombination between the autonomous plasmid and DNA fragment in P. pastoris cells was possible as in S. cerevisiae cells.Citation12,13) We developed autonomous plasmids (pPrAGS-DDGFP) containing the GFP gene and inserted membrane protein genes between the promoter and GFP using homologous recombination in P. pastoris, as Drew and Newstead et al. did in S. cerevisiae. Using the high-throughput P. pastoris expression system construction method, we constructed an expression system of membrane proteins or micro-organisms and animals, such as ammonium transporter and melatonin receptor of medaka fish, and CTR1 and HSP30 of S. cerevisiae. For the expressed membrane proteins, we reported localization in cells observed using fluorescence microscopy (Fig. (B)), investigation of solubilization conditions, and determination of approximate molecular weight by in-gel fluorescence measurement, followed by SDS-PAGE.Citation12) We also developed autonomous plasmids containing a secretion signal that enables high-throughput construction of secretory proteins using homologous recombination in P. pastoris (Fig. (A)).Citation13) In general, the conventional method using restriction enzymes results in the insertion of undesired residues; however, our system can connect the secretion signal and protein gene precisely. This method can also connect the His-tag at the C-terminal precisely and easily, which also enables high-throughput purification of recombinant proteins.Citation13) Using this method, we constructed an expression system of secretory proteins of medaka fish, such as α-amylase, lysozyme, and transferrin. We already reported on the purification, enzymatic properties, and crystal structure of the recombinant α-amylase (Fig. (C)).Citation13) The raw starch degradation activity of medaka α-amylase was lower than that of mammalian α-amylase, and the crystal structure revealed that one of the Trp residues that were considered a raw starch binding site apart from catalytic residues was not conserved in medaka α-amylase. I am currently trying to develop a high-throughput method for construction of cell surface protein production system of P. pastoris, which will be applicable for biotechnological applications, such as collection of metals by recombinant transferrins,Citation13,25,26) using a plasmid with the gene for cell wall protein PIR3.Citation57) It is our hope that the high-throughput expression system construction method using P. pastoris, which is proving to be an excellent host for eukaryotic membrane protein and secretory proteins, will be used for further biochemical studies.
Fig. 4. High-throughput construction of an expression system using P. pastoris.
Notes: Structure of one of the developed plasmids and a diagram of expression plasmid construction by homologous recombination in P. pastoris are shown (A). A photograph of P. pastoris cells expressing a GFP-membrane protein fusion (S. cerevisiae HSP30) taken using a fluorescence microscope is shown (B). A low level of bright-field light was included to detect yeast cells. A scale bar in the photograph is 5 μm. Determined crystal structure of recombinant medaka α-amylase, an application of the P. pastoris expression system construction method, is shown (C).
Since our plasmids also contain 2μ replication origin and URA3 gene of S. cerevisiae introduced using the helper plasmid of Iizasa et al. (Fig. (A)), management of our plasmids, such as insertion of a target gene into the plasmid, using homologous recombination in S. cerevisiae is also possible.Citation12) We tried to use a CUP1 promoter, which has been reported to be an effective promoter for both S. cerevisiae and P. pastoris, to make a plasmid which can be used for S. cerevisiae and P. pastoris expression system.Citation12,58) However, the plasmid was not an effective expression plasmid; the CUP1 promoter was weakly induced by the copper included in the medium, but not strongly induced by added copper ions, and higher concentration copper had toxicity to yeast.Citation12) Therefore, we made plasmids with the P. pastoris AOX1 promoter that is strongly induced by methanol, and plasmids with the S. cerevisiae GAL1 promoter that is induced by galactose, to enable high-throughput construction of an expression system for respective yeast.Citation12,13) The expression level of proteins may depend on the expression host; our two types of plasmids have enabled high-throughput investigation of expression efficiency using two yeasts.Citation12,13) Since the backbone of the plasmids is the same as that of the plasmid with a CUP1 promoter, plasmids with an AOX1 promoter or a GAL1 promoter are also kept autonomously in both yeasts.Citation12,13) Therefore, our plasmids having advantages of both yeasts can be used in a range of applications, such as connection of multiple DNA fragments by homologous recombination in S. cerevisiae.
IV. Connection of multiple DNA fragments and their applications
Since homologous recombination in S. cerevisiae can connect multiple fragments, as briefly shown in Chapters II and III, construction of plasmids using S. cerevisiae enables joining of multiple protein genes (assembly of complicated fusion protein) (Fig. (A)). The partitioning of protein genes and introduction of mutations on the homologous region between the fragments enables the high-throughput construction of an expression system of mutant protein (Fig. (B)).Citation44,51) Because of the lower transformation efficiency of P. pastoris, it is currently quite difficult to connect multiple fragments by homologous recombination in P. pastoris.Citation13) However the combination of another connection method, such as oePCR and homologous recombination in P. pastoris, enables the high-throughput construction of mutant proteins and/or fusion protein expression systems using P. pastoris (Fig. (B)). We have made a mutant of the medaka lysozyme using the homologous recombination method and have investigated the enzymatic properties and crystal structure (Mizutani, unpublished data). One of the residues that were not conserved between medaka lysozyme and chicken egg lysozyme near the catalytic site, Leu 31, was mutated to the corresponding residue, Ala. The mutant revealed that the residue was not related to the observed four times higher activity of medaka lysozyme relative to chicken lysozyme at optimal pH and this residue was related to the optimal pH of the lysozymes. We, very recently, found that a mutation in the region connecting the secretion signal and protein gene largely affected the expression level of a secretory protein (Mizutani, unpublished data); I am now trying to establish an optimal method for the production of secretory proteins by introducing a mutation using degenerate primers.
Fig. 5. Methods used to connect multiple DNA fragments.
Notes: Schematic view and models for the connection of two DNA fragments by homologous recombination (A), by oePCR (overlap extension PCR) (B), and by the techniques of Gibson et al. (C) are shown. Homologous regions are shown as boxes with broken lines.
In addition to the ability to connect multiple DNA fragments by homologous recombination, S. cerevisiae has the ability to maintain extremely large plasmids.Citation14) Gibson et al. (JCVI) succeeded in creating an artificial bacterial genome by connecting synthesized DNA fragments.Citation14) J. Craig Venter (founder and president of JCVI) is known for inventing the expressed sequence tag,Citation59) and leading contributors to human genome project.Citation60) Gibson et al. also reported success in creating “artificial bacteria” by introducing the artificial genome to the cells of closely related bacteria,Citation15) For synthesis of the artificial genome, they developed and used the enzymatic process shown in Fig. (C) to connect 5–7 kb DNA fragments; the prepared BAC plasmids were amplified in E. coli and then were connected by the same process to make larger DNA chains. However, a larger (than 144 kb) plasmid could not be kept in E. coli cells; the whole artificial genome (580 kb) was assembled by using homologous recombination in yeast S. cerevisiae and was kept in yeast cells.Citation15) These remarkable and elegant experiments used the ability of yeast to maintain a huge plasmid and to connect DNA fragments by homologous recombination. A kit for the enzymatic method of Gibson et al. has been released by New England BioLabs; the product is named Gibson Assembly Master Mix after the inventor of the method. Another kit, the In-fusion kit provided by Clontech, uses vaccinia virus DNA polymerase with 3′-5′ exonuclease activity; after two initial steps similar to those of Gibson et al., the combined DNA is introduced to E. coli and the gap in the DNA may be repaired in the cells. The 5′-3′ exonuclease and ligation treatments included in the Gibson Assembly master Mix are not included in the In-fusion kit, and thus in the latter system (In-fusion kit) the very ends of the DNA fragments to be combined by the kit should be complementary. Another kit provided by Life Technology, GeneArt Seamless Cloning and Assembly Enzyme Mix, looks similar to the In-fusion kit, but the details of the process of this kit have not been disclosed. (Life Technology also provides a cloning kit using homologous recombination of S. cerevisiae, the GeneArt High-Order Genetic Assembly System, but this kit is expensive. As shown in this review, special enzymes, chemicals, and strains are not necessary for the cloning method using homologous recombination in yeast.)
Commercially available synthesized DNA, such as GeneArt Strings DNA Fragment of Life Technologies, has become less and less expensive, such that it has become reasonable to make a genome by joining together relatively cheap synthesized DNA chains (<1000 bp costing 18,000 yen per DNA chain), while synthesis of a whole genome is still difficult and highly expensive. Eukaryotic genes are often divided by introns; the three methods shown in Fig. enable “in vitro splicing” by connecting the exon regions amplified by PCR from the genome. Construction of the P. pastoris expression system by in vitro splicing is also possible; high-throughput construction of a plasmid and expression system can be done by combining oePCR (or Gibson Assembly) and homologous recombination in P. pastoris. Through the contribution of scientists around the world, including Venter, genome sequences and cDNA sequences of many species have been reported. Our group is currently making various expression systems using the sequence information of various species, especially fish, and trying to determine the crystal structures of proteins. Through the use of such novel applications of “high-throughput structural biology,” I hope to contribute to the progress of bioscience.
Acknowledgments
I would like to express my sincere gratitude to Dr Masaaki Hirose, Professor Emeritus of Kyoto University, for his gentle guidance and courteous encouragement. I would also like to express my thanks to Dr Bunzo Mikami, Professor of Kyoto University, for his kind guidance and valuable discussion. Dr Shigeo Aibara, Dr Nobuyuki Takahashi, and Dr Honami Yamashita provided encouragement and essential input. I would also like to acknowledge Dr So Iwata, Professor of Imperial College London and Kyoto University, Dr Kenji Kano, Professor of Kyoto University, and Dr Yukio Nagano, Associate Professor of Saga University, for their encouragement and support, and the late Dr Youji Sakagami, Professor of Nagoya University, for his guidance and inspiration. This work would not have been possible without my dedicated collaborators, and the advice and technical support of the members of the Laboratory of Applied Structural Biology, Graduate School of Agriculture, Kyoto University. Finally, I gratefully acknowledge the support of the National BioResource Project (NBRP medaka and yeast), Japan Synchrotron Radiation Research Institute (JASRI), and Japan Aerospace Exploration Agency (JAXA), a fellowship from the Canon Foundation, and Grants-in-Aid from the Ministry of Education, Culture, Sports, Science, and Technology of Japan.
Notes
This review was written in response to the author’s receipt of the JSBBA Award for Young Scientists in 2013.
Related Research Data
References
- Aylon Y, Kupiec M. New insights into the mechanism of homologous recombination in yeast. Mutation Res. 2004;566:231–248.10.1016/j.mrrev.2003.10.001
- San Filippo J, Sung P, Klein H. Mechanism of eukaryotic homologous recombination. Annu. Rev. Biochem. 2008;77:229–257.10.1146/annurev.biochem.77.061306.125255
- Heyer W-D, Ehmsen KT, Liu J. Regulation of homologous recombination in eukaryotes. Annu. Rev. Genet. 2010;44:113–139.10.1146/annurev-genet-051710-150955
- Ma H, Kunes S, Schatz PJ, Botstein D. Plasmid construction by homologous recombination in yeast. Gene. 1987;58:201–216.10.1016/0378-1119(87)90376-3
- Kouprina N, Larionov V. Innovation – TAR cloning: insights into gene function, long-range haplotypes and genome structure and evolution. Nat. Rev. Genet. 2006;7:805–812.10.1038/nrg1943
- Drew D, Newstead S, Sonoda Y, Kim H, von Heijne G, Iwata S. GFP-based optimization scheme for the overexpression and purification of eukaryotic membrane proteins in Saccharomyces cerevisiae. Nat. Protoc. 2008;3:784–798.10.1038/nprot.2008.44
- Newstead S, Kim H, von Heijne G, Iwata S, Drew D. High-throughput fluorescent-based optimization of eukaryotic membrane protein overexpression and purification in Saccharomyces cerevisiae. Proc. Nat. Acad. Sci. U.S.A. 2007;104:13936–13941.10.1073/pnas.0704546104
- Iizasa E, Nagano Y. Highly efficient yeast-based in vivo DNA cloning of multiple DNA fragments and the simultaneous construction of yeast/Escherichia coli shuttle vectors. Biotechniques. 2006;40:79–83.10.2144/000112041
- Nagano Y, Takao S, Kudo T, Iizasa E, Anai T. Yeast-based recombineering of DNA fragments into plant transformation vectors by one-step transformation. Plant Cell Rep. 2007;26:2111–2117.10.1007/s00299-007-0428-2
- Matsuo Y, Kishimoto H, Horiuchi T, Tanae K, Kawamukai M. Simple and effective gap-repair cloning using short tracts of flanking homology in fission yeast. Biosci. Biotechnol. Biochem. 2010;74:685–689.10.1271/bbb.90967
- Chino A, Watanabe K, Moriya H. Plasmid construction using recombination activity in the fission yeast Schizosaccharomyces pombe. PloS one. 2010;5: e9652.
- Mizutani K, Yoshioka S, Mizutani Y, Iwata S, Mikami B. High-throughput construction of expression system using yeast Pichia pastoris, and its application to membrane proteins. Protein Expression Purif. 2011;77:1–8.10.1016/j.pep.2010.12.009
- Mizutani K, Toyoda M, Otake Y, Yoshioka S, Takahashi N, Mikami B. Structural and functional characterization of recombinant medaka fish alpha-amylase expressed in yeast Pichia pastoris. Biochim. Biophys. Acta. 2012;1824:954–962.
- Gibson DG, Benders GA, Andrews-Pfannkoch C, Denisova EA, Baden-Tillson H, Zaveri J, Stockwell TB, Brownley A, Thomas DW, Algire MA, Merryman C, Young L, Noskov VN, Glass JI, Venter JC, Hutchison CA, Smith HO. Complete chemical synthesis, assembly, and cloning of a Mycoplasma genitalium genome. Science. 2008;319:1215–1220.10.1126/science.1151721
- Gibson DG, Glass JI, Lartigue C, Noskov VN, Chuang RY, Algire Ma, Benders Ga, Montague MG, Ma L, Moodie MM, Merryman C, Vashee S, Krishnakumar R, Assad-Garcia N, Andrews-Pfannkoch C, Denisova Ea, Young L, Qi ZQ, Segall-Shapiro TH, Calvey CH, Parmar PP, Hutchison Ca, Smith HO, Venter JC. Creation of a bacterial cell controlled by a chemically synthesized genome. Science. 2010;329:52–56.10.1126/science.1190719
- Kimura F, Mizutani K, Mikami B, Kimura T. Single-crystal X-ray diffraction study of a magnetically oriented microcrystal array of lysozyme. Cryst. Growth Des. 2011;11:12–15.
- Mizutani K, Toyoda M, Sagara K Takahashi N, Sato A, Kamitaka Y, Tsujimura S, Nakanishi Y, Sugiura T, Yamaguchi S, Kano K, Mikami B. X-ray analysis of bilirubin oxidase from Myrothecium verrucaria at 2.3 angstrom resolution using a twinned crystal. Acta Crystallogr. Sect. F-Structural Biol. Cryst. Commun. 2010;66:765–770.
- Mizutani K, Tsuchiya S, Toyoda M, Nanbu Y, Tominaga K, Yuasa, K, Takahashi N, Tsuji A and Mikami B, Structure of beta-1,4-mannanase from the common sea hare Aplysia kurodai at 1.05 angstrom resolution. Acta Crystallogr. Sect. F-Structural Biol. Cryst. Commun. 2012;68:1164–1168.
- Aibara S, Mizutani K, Suzuki A, Horiuchi H, Hashimoto K, Yamane T. Comparison of molecular packing between two kinds of hen egg-white lysozyme orthorhombic crystals. J. Jpn. Soc. Microgravity Appl. 2008;25:82–88.
- Mizutani K, Mikami B, Aibara S, Hirose M. Structure of aluminium-bound ovotransferrin at 2.15 Å resolution. Acta Crystallogr., Sect D: Biol. Crystallogr. 2005;61:1636–1642.10.1107/S090744490503266X
- Mizutani K, Mikami B, Hirose M. Domain closure mechanism in transferrins: new viewpoints about the hinge structure and motion as deduced from high resolution crystal structures of ovotransferrin N-lobe. J. Mol. Biol. 2001;309:937–947.10.1006/jmbi.2001.4719
- Mizutani K, Muralidhara BK, Yamashita H, Tabata S, Mikami B, Hirose M. Anion-mediated Fe3+ release mechanism in ovotransferrin C-lobe – a structurally identified SO42− binding site and its implications for the kinetic pathway. J. Biol. Chem. 2001;276:35940–35946.10.1074/jbc.M102590200
- Mizutani K, Yamashita H, Kurokawa H, Mikami B, Hirose M. Alternative structural state of transferrin – the crystallographic analysis of iron-loaded but domain-opened ovotransferrin N-lobe. J. Biol. Chem. 1999;274:10190–10194.10.1074/jbc.274.15.10190
- Mizutani K, Yamashita H, Mikami B, Hirose M. Crystal structure at 1.9 angstrom resolution of the apoovotransferrin N-lobe bound by sulfate anions: implications for the domain opening and iron release mechanism. Biochemistry. 2000;39:3258–3265.10.1021/bi992574q
- Mizutani K, Toyoda M, Mikami B. X-ray structures of transferrins and related proteins. Biochim. Biophys. Acta, Gen. Subj. 2012;1820:203–211.10.1016/j.bbagen.2011.08.003
- Mizutani K, Okamoto I, Fujita K, Yamamoto K, Hirose M. Structural and functional characterization of ovotransferrin produced by Pichia pastoris. Biosci. Biotechnol. Biochem. 2004;68:376–383.10.1271/bbb.68.376
- Okamoto I, Mizutani K, Hirose M. Iron-binding process in the amino- and carboxyl-terminal lobes of ovotransferrin: quantitative studies utilizing single Fe3+-binding mutants. Biochemistry. 2004;43:11118–11125.10.1021/bi049147j
- Fabre F. Induced intragenic recombination in yeast can occur during G1 mitotic phase. Nature. 1978;272:795–798.10.1038/272795a0
- Hastings PJ, McGill C, Shafer B, Strathern JN. Ends-in vs. ends-out recombination in yeast. Genetics. 1993;558:973–980.
- Rong YS. Gene targeting by homologous recombination in Drosophila. Science. 2000;288:2013–2018.10.1126/science.288.5473.2013
- Kato H, Matsuda F, Yamada R, Nagata K, Shirai T, Hasunuma T, Kondo A. Cocktail delta-integration of xylose assimilation genes for efficient ethanol production from xylose in Saccharomyces cerevisiae. J. Biosci. Bioeng. 2013;116:333–336.10.1016/j.jbiosc.2013.03.020
- Sakai A, Shimizu Y, Hishinuma F. Integration of heterologous genes into the chromosome of Saccharomyces cerevisiae using a delta-sequence of yeast retrotransposon Ty. Appl. Microbiol. Biotechnol. 1990;33:302–306.10.1007/BF00164526
- Murayama Y, Kurokawa Y, Mayanagi K, Iwasaki H. Formation and branch migration of Holliday junctions mediated by eukaryotic recombinases. Nature. 2008;451:1018–1021.10.1038/nature06609
- Krejci L, Altmannova V, Spirek M, Zhao X. Homologous recombination and its regulation. Nucleic Acids Res. 2012;40:5795–5818.10.1093/nar/gks270
- Mazón G, Lam AF, Ho CK, Kupiec M, Symington LS. The Rad1-Rad10 nuclease promotes chromosome translocations between dispersed repeats. Nat. Struct. Mol. Biol. 2012;19:964–971.10.1038/nsmb.2359
- McEachern MJ, Haber JE. Break-induced replication and recombinational telomere elongation in yeast. Annu. Rev. Biochem. 2006;75:111–135.10.1146/annurev.biochem.74.082803.133234
- McMahill MS, Sham CW, Bishop DK. Synthesis-dependent strand annealing in meiosis. PLoS Biol. 2007;5:e299–e299.10.1371/journal.pbio.0050299
- Orrweaver TL, Szostak JW, Rothstein RJ. Yeast transformation – a model system for the study of recombination. Proc. Natl. Acad. Sci. U. S. A. 1981;78:6354–6358.10.1073/pnas.78.10.6354
- Hinnen A, Hicks JB, Fink GR. Transformation of yeast. Proc. Natl. Acad. Sci. U. S. A. 1978;75:1929–1933.10.1073/pnas.75.4.1929
- Thomas KR, Capecchi MR. Site-directed mutagenesis by gene targeting in mouse embryo-eerived stem-cells. Cell. 1987;51:503–512.10.1016/0092-8674(87)90646-5
- Zimmer A, Gruss P. Production of chimaeric mice containing embryonic stem (ES) cells carrying a homeobox Hox-1.1 allele mutated by homologous recombination. Nature. 1989;338:150–153.10.1038/338150a0
- Astromskas E, Cohn M. Ends-in vs. ends-out targeted insertion mutagenesis in Saccharomyces castellii. Curr. Genet. 2009;55:339–347.10.1007/s00294-009-0248-8
- Oldenburg KR, Vo KT, Michaelis S, Paddon C. Recombination-mediated PCR-directed plasmid construction in vivo in yeast. Nucleic Acids Res. 1997;25:451–452.10.1093/nar/25.2.451
- Ito K, Sugawara T, Shiroishi M, Tokuda N, Kurokawa A, Misaka T, Makyio H, Yurugi-Kobayashi T, Shimamura T, Nomura N, Murata T, Abe K, Iwata S, Kobayashi T. Advanced method for high-throughput expression of mutated eukaryotic membrane proteins in Saccharomyces cerevisiae. Biochem. Biophys. Res. Commun. 2008;371:841–845.10.1016/j.bbrc.2008.04.182
- Morrow DM, Connelly C, Hieter P. “Break copy” duplication: a model for chromosome fragment formation in Saccharomyces cerevisiae. Genetics. 1997;382:371–382.
- Gietz RD, Woods RA. Transformation of yeast by lithium acetate/single-stranded carrier DNA/polyethylene glycol method. Methods Enzym. 2002;350:87–96.10.1016/S0076-6879(02)50957-5
- Ito H, Fukuda Y, Murata K, Kimura A. Transformation of intact yeast-cells treated with alkali cations. J. Bacteriol. 1983;153:163–168.
- Sugawara T, Ito K, Shiroishi M, Tokuda N, Asada H, Yurugi-Kobayashi T, Shimamura T, Misaka T, Nomura N, Murata T, Abe K, Iwata S, Kobayashi T. Fluorescence-based optimization of human bitter taste receptor expression in Saccharomyces cerevisiae. Biochem. Biophys. Res. Commun. 2009;382:704–710.10.1016/j.bbrc.2009.03.089
- Shiroishi M, Kobayashi T, Ogasawara S, Tsujimoto H, Ikeda-Suno C, Iwata S, Shimamura T. Production of the stable human histamine H-1 receptor in Pichia pastoris for structural determination. Methods. 2011;55:281–286.10.1016/j.ymeth.2011.08.015
- Shimamura T, Shiroishi M, Weyand S. Structure of the human histamine H1 receptor complex with doxepin. Nature. 2011;475:65–70.10.1038/nature10236
- Shiroishi M, Tsujimoto H, Makyio H, Asada H, Yurugi-Kobayashi T, Shimamura T, Murata T, Nomura N, Haga T, Iwata S, Kobayashi T. Platform for the rapid construction and evaluation of GPCRs for crystallography in Saccharomyces cerevisiae. Microb. Cell Fact. 2012;11:78–78.10.1186/1475-2859-11-78
- Iizasa E, Mitsutomi M, Nagano Y. Direct binding of a plant LysM receptor-like kinase, LysM RLK1/CERK1, to chitin in vitro. J. Biol. Chem. 2010;285:2996–3004.
- Goto K, Nagano Y. Ultra-low background DNA cloning system. PloS one. 2013;8:e56530–e56530.10.1371/journal.pone.0056530
- Mizutani K, Hashimoto K, Takahashi N, Hirose M, Aibara S, Mikami B. Structural and functional characterization of recombinant human serum transferrin secreted from Pichia pastoris. Biosci. Biotechnol. Biochem. 2010;74:309–315.10.1271/bbb.90635
- Cregg JM, Vedvick TS, Raschke WC. Recent advances in the expression of foreign genes in Pichia pastoris. Bio/Technology. 1993;11:905–910.10.1038/nbt0893-905
- Cregg JM, Barringer KJ, Hessler AY, Madden KR. Pichia pastoris as a host system for transformations. Mol. Cell. Biol. 1985;5:3376–3385.
- Toh-E A, Oguchi T, Matsui Y, Yasunaga S, Nisogi H, Tanaka K. Three yeast genes, Pir1, Pir2 and Pir3, containing internal tandem repeats, are related to each other, and Pir1 and Pir2 are required for tolerance to heat-shock. Yeast. 1993;9:481–494.10.1002/(ISSN)1097-0061
- Koller A, Valesco J, Subramani S. The CUP1 promoter of Saccharomyces cerevisiae is inducible by copper in Pichia pastoris. Yeast. 2000;16:651–656.10.1002/(ISSN)1097-0061
- Adams MD, Kelley JM, Gocayne JD, Dubnick M, Polymeropoulos MH, Xiao H, Merril CR, Wu A, Olde B, Moreno RF, Kerlavage AR, Mccombie WR, Venter JC. Complementary-DNA sequencing – expressed sequence tags and human genome project. Science. 1991;252:1651–1656.10.1126/science.2047873
- Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, Sutton GG, Smith HO, Yandell M, Evans CA, Holt RA, Gocayne JD, Amanatides P, Ballew RM, Huson DH, Wortman JR, Zhang Q, Kodira CD, Zheng XQH, Chen L, Skupski M, Subramanian G, Thomas PD, Zhang JH, Miklos GLG, Nelson C, Broder S, Clark AG, Nadeau C, McKusick VA, Zinder N, Levine AJ, Roberts RJ, Simon M, Slayman C, Hunkapiller M, Bolanos R, Delcher A, Dew I, Fasulo D, Flanigan M, Florea L, Halpern A, Hannenhalli S, Kravitz S, Levy S, Mobarry C, Reinert K, Remington K, Abu-Threideh J, Beasley E, Biddick K, Bonazzi V, Brandon R, Cargill M, Chandramouliswaran I, Charlab R, Chaturvedi K, Deng ZM, Di Francesco V, Dunn P, Eilbeck K, Evangelista C, Gabrielian AE, Gan W, Ge WM, Gong FC, Gu ZP, Guan P, Heiman TJ, Higgins ME, Ji RR, Ke ZX, Ketchum KA, Lai ZW, Lei YD, Li ZY, Li JY, Liang Y, Lin XY, Lu F, Merkulov GV, Milshina N, Moore HM, Naik AK, Narayan VA, Neelam B, Nusskern D, Rusch DB, Salzberg S, Shao W, Shue BX, Sun JT, Wang ZY, Wang AH, Wang X, Wang J, Wei MH, Wides R, Xiao CL, Yan CH, Yao A, Ye J, Zhan M, Zhang WQ, Zhang HY, Zhao Q, Zheng LS, Zhong F, Zhong WY, Zhu SPC, Zhao SY, Gilbert D, Baumhueter S, Spier G, Carter C, Cravchik A, Woodage T, Ali F, An HJ, Awe A, Baldwin D, Baden H, Barnstead M, Barrow I, Beeson K, Busam D, Carver A, Center A, Cheng ML, Curry L, Danaher S, Davenport L, Desilets R, Dietz S, Dodson K, Doup L, Ferriera S, Garg N, Gluecksmann A, Hart B, Haynes J, Haynes C, Heiner C, Hladun S, Hostin D, Houck J, Howland T, Ibegwam C, Johnson J, Kalush F, Kline L, Koduru S, Love A, Mann F, May D, McCawley S, McIntosh T, McMullen I, Moy M, Moy L, Murphy B, Nelson K, Pfannkoch C, Pratts E, Puri V, Qureshi H, Reardon M, Rodriguez R, Rogers YH, Romblad D, Ruhfel B, Scott R, Sitter C, Smallwood M, Stewart E, Strong R, Suh E, Thomas R, Tint NN, Tse S, Vech C, Wang G, Wetter J, Williams S, Williams M, Windsor S, Winn-Deen E, Wolfe K, Zaveri J, Zaveri K, Abril JF, Guigo R, Campbell MJ, Sjolander KV, Karlak B, Kejariwal A, Mi HY, Lazareva B, Hatton T, Narechania A, Diemer K, Muruganujan A, Guo N, Sato S, Bafna V, Istrail S, Lippert R, Schwartz R, Walenz B, Yooseph S, Allen D, Basu A, Baxendale J, Blick L, Caminha M, Carnes-Stine J, Caulk P, Chiang YH, Coyne M, Dahlke C, Mays AD, Dombroski M, Donnelly M, Ely D, Esparham S, Fosler C, Gire H, Glanowski S, Glasser K, Glodek A, Gorokhov M, Graham K, Gropman B, Harris M, Heil J, Henderson S, Hoover J, Jennings D, Jordan C, Jordan J, Kasha J, Kagan L, Kraft C, Levitsky A, Lewis M, Liu XJ, Lopez J, Ma D, Majoros W, McDaniel J, Murphy S, Newman M, Nguyen T, Nguyen N, Nodell M, Pan S, Peck J, Peterson M, Rowe W, Sanders R, Scott J, Simpson M, Smith T, Sprague A, Stockwell T, Turner R, Venter E, Wang M, Wen MY, Wu D, Wu M, Xia A, Zandieh A, and Zhu XH. The sequence of the human genome. Science. 2001;291:1304–1351.10.1126/science.1058040