
Abstract
Hmo1, a member of the high mobility group B family proteins in Saccharomyces cerevisiae, associates with the promoters of ribosomal protein genes (RPGs) to direct accurate transcriptional initiation. Here, to identify factors involved in the binding of Hmo1 to its targets and the mechanism of Hmo1-dependent transcriptional initiation, we developed a novel reporter system using the promoter of the RPG RPS5. A genetic screen did not identify any factors that influence Hmo1 binding, but did identify a number of mutations in Hmo1 that impair its DNA binding activity in vivo and in vitro. These results suggest that Hmo1 binds to its target promoters autonomously without any aid of additional factors. Furthermore, characterization of Hmo1 mutants showed that the box A domain plays a pivotal role in DNA binding and may be required for the recognition of structural properties of target promoters that occur in native chromatin.
Graphical Abstract
Hmo1 contains three characteristic domains, box A, box B and C-terminal tail. Both HMG boxes play essential role(s) in the DNA binding activity of Hmo1.
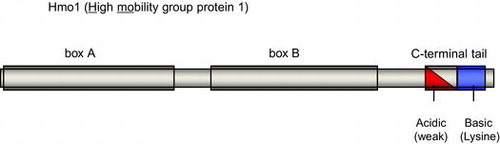
High mobility group B (HMGB) proteins are non-histone architectural proteins that have diverse regulatory roles in various reactions that occur on the eukaryotic chromatin, such as transcription, recombination, and DNA repair.Citation1–4) HMGB proteins usually contain one or more DNA binding motifs known as HMG box, which adopts an L-shaped structure composed of three α-helices and associates preferentially with distorted regions of DNA, such as four-way junctions, minicircles, and cisplatinated DNA, rather than specific primary sequences.Citation5)
Among the seven HMGB proteins in Saccharomyces cerevisiae, four (Hmo1, Nhp10, Nhp6a, and Nhp6b) are predicted to play roles in altering the chromatin architecture, but do not act as sequence-specific transcription factors. Nhp10 is a component of the Ino80 chromatin remodeling complex that is recruited to DNA damage sites through interactions with phosphorylated histone γH2AX and the ends of broken DNA.Citation6–11) Nhp6a is an abundant protein (50,000–70,000 molecules per cell, corresponding to approximately one molecule for every 1–2 nucleosomes) that has specific, but numerous binding sites (present in approximately 23% of promoters for protein-coding genes).Citation12,13) Nhp6b is a less abundant and partially functional redundant paralog of Nhp6a (approximately 87% amino acid sequence identity); however, its binding sites have not yet been investigated. Nhp6a and Nhp6b participate in the transcription of various class II (RNA polymerase II-driven) and class III (RNA polymerase III-driven) genes.Citation14) In contrast to Nhp10, Nhp6a, and Nhp6b, Hmo1 binds specifically to more limited number of target genes. Previous chromatin immunoprecipitation (ChIP)-chip analyses demonstrated that Hmo1 is associated specifically with the promoters of 70% of ribosomal protein genes (RPGs) and the coding region of the 35S ribosomal RNA (rRNA) gene, which are transcribed by RNA polymerases II and I, respectively.Citation15–17) The fact that Hmo1 targets genes encoding different classes of ribosomal components implies that it may play significant roles in coordinating the synthesis of ribosomes in response to certain environmental conditions. Hmo1 regulates the transcription of RPGs by facilitating the binding of Fhl1/Ifhl1 and TFIID to the promoter and by masking a wide nucleosome-free region between the upstream activating sequence (UAS) and core promoter to direct the assembly of the preinitiation complex (PIC) to a biologically relevant site.Citation15,16,18–20) Although recent studies have revealed the roles of Hmo1 at RPG promoters, the mechanisms by which Hmo1 binds specifically to these promoters remain unclear.
Here, to unveil the mechanisms and/or factors that direct specific binding of Hmo1 to RPG promoters, we developed a novel and highly sensitive reporter system to detect binding of Hmo1 to the promoter of the gene encoding ribosomal protein S5 (RPS5). Although an intensive genetic screen using this reporter system identified several genes as possible candidate co-factors that promote binding of Hmo1 to DNA, mutations of these genes did not influence Hmo1 binding itself, but rather affected the transcription start site (TSS) of the reporter gene. Therefore, it is likely that Hmo1 binds to its target promoters without the aid of any additional factors. In the same screen, mutations in Hmo1 that reduce its binding activity both in vitro and in vivo were identified. Similar to the mammalian HMGB1/2 proteins, Hmo1 has two HMG boxes; while box B is highly similar to the classical HMG box, box A has only limited similarities. Previous in vitro studies suggested that box B is responsible for most of the DNA binding activity of Hmo1, whereas the contribution of box A to this activity is very limited.Citation21,22) It was reported recently that the integrity of box A is required for the function of Hmo1 in the RNA polymerase I transcription in vivo, whereas the requirement of box A in the transcription of RPG (such as RPS23A) by RNA polymerase II is unclear.Citation23) In contrast to these previous studies, the box A mutations identified in the genetic screen performed here impaired the ability of Hmo1 to bind to DNA. Furthermore, one of the box A Hmo1 mutants was deficient in DNA binding specifically in vivo. Therefore, we conclude that box A is essential for the DNA binding activity of Hmo1. In addition, box A may also recognize some structural properties that are induced specifically in vivo, thereby directing Hmo1 to specific loci on the genome.
Materials and methods
Yeast strains and plasmids
The yeast strains used in this study are listed in Supplemental Table 1. Standard techniques were used for the growth and transformation of yeast. Detailed descriptions of each yeast strain and the protocols used to construct the plasmids are included in the Supplemental Information. The culture conditions for each experiment are described in the figure legends. The detailed protocols used to construct the plasmids are also described in the Supplemental Information. The oligonucleotides used in this study are listed in Supplemental Table 2.
TSS shift screening
For random screening of mutations that influence Hmo1 binding to its target gene in vivo, a wild-type S. cerevisiae strain (H2451) was grown at mid-log phase in 50 mL of YPD medium, harvested, and mutagenized by treatment with 2% ethylmethane sulfonate (the ratio of survival was approximately 10–50%). The mutagenized cells were cultivated in 50 mL of YPD medium for 6 h and then transformed with the pKM68 reporter plasmid. The transformants were plated on SD medium lacking histidine (SD −His) and incubated at 30 °C. Growing colonies were isolated after 3–10 d. To exclude false–positive colonies after the primary selection, 3 amino-1,2,4-triazole (0.1–1 mM), an inhibitor of His3 protein, was added to the selection medium. The positive strains were then backcrossed to an isogenic parent strain (YKK16), and subjected to at least three rounds of tetrad dissection analysis to remove unrelated mutations with the His+ phenotype. Segregants that exhibited the His+ phenotype were subjected to library screening or whole genome sequencing to identify the responsible mutations. Sequencing libraries were prepared using the NEBNext DNA Sample Prep Reagent Set 1 (New England Biolabs) following the manufacturer’s protocols. Pooled libraries were sequenced on Illumina Genome Analyzer IIx following the manufacturer’s protocols, generating 100-bp paired-end reads and 6-bp index tags. Screening of mutations in box A that influence the DNA binding activity of Hmo1 was performed by introducing random mutations into the box A region by PCR.
Primer extension analysis
TSSs were analyzed by primer extension analysis as described previously.Citation19) The TK3212 (endogenous RPS5) and KK288 (RPS5*p+HIS3 reporter) primers were used for the analysis. Electrophoretic images were acquired by exposing gels to imaging plates, and were analyzed by image analyzer BAS2500 (Fuji Film).
ChIP assay
ChIP analyses were performed as described previously.Citation15) Immunoprecipitation was performed using Dynabeads Protein G (Invitrogen) and an anti-FLAG monoclonal antibody (Sigma–Aldrich; M2) or anti-Hmo1 polyclonal antibody.Citation19) Real-time quantitative PCR analyses were performed using the KAPA SYBR FAST qPCR Kit (Kapa Biosystems) and the Mx3000P qPCR System (Agilent Technologies). Each experiment was performed in triplicate and the mean and standard deviations of the ratio of immunoprecipitated DNA to input DNA (IP/input) were calculated. The TK9075-TK9076 (35S rDNA), KK255-KK256 (RPS23A), and TK9734-TK11311 (RPS5) primers were used for quantitative PCR.
Preparation of Hmo1 proteins
Detailed descriptions of the methods used to construct the plasmids expressing C-terminal His (x6)-tagged Hmo1 proteins are provided in the supplemental information. Hmo1 proteins were expressed in Escherichia coli BL21 (DE3) Rosetta (pLysS) cells (Novagen) by induction with 0.5 mM IPTG in 50 mL LB medium for 3 h at 30 °C. The harvested cells were suspended in 1.5 mL of lysis buffer (40 mM Tris–HCl, pH 8.0 [at 4 °C], 4 mM EDTA, 100 mM NaCl, 0.1% Triton X-100, 10% glycerol, 1 mM PMSF, and 1 mM DTT) and disrupted by sonication. The lysate was centrifuged and the supernatant was stored at −80 °C. Purification of the Hmo1 protein was performed as described in Supplemental Information.
DNA binding assay
The in vitro DNA binding activity of Hmo1 was investigated using an immobilized template assay. Biotinylated DNA (the promoter sequences of RPS5 and RPS10A, or autonomously replicating sequence on chromosome V (ARS504), which is located between the UAS and core promoter of RPL10) was amplified by PCR and immobilized on Dynabeads M-280 Streptavidin (Life Technologies). A detailed description of the preparation of the biotinylated DNA fragments is provided in the supplemental information. The immobilized DNA was incubated with lysate containing Hmo1 proteins in T (100) buffer (50 mM Tris–HCl, pH 8.0 [at 4 °C], 100 mM NaCl, and 0.1% Triton X-100) at 4 °C from 4 h to overnight, and then washed twice with the same buffer. Proteins that bound to the DNA on the beads were eluted by boiling in SDS sample buffer, and analyzed by immunoblotting with an anti-Hmo1 or anti-FLAG antibody, as described for the ChIP assay.
Results
Construction of a reporter system to investigate binding of Hmo1 to its target genes in vivo
In our previous study, we demonstrated that Hmo1 masks a wide nucleosome-free region between the UAS and core promoter in a subset of target genes. Deletion of HMO1 causes ectopic PIC-assembly in this region and yields upstream TSSs.Citation20) In the endogenous RPS5 promoter, which is targeted by Hmo1, transcription is initiated mainly at the −37 position (relative to the ATG start codon), while upstream TSSs are detected only modestly. However, the use of upstream TSSs was more evident in Δhmo1 cells than wild-type cells (compare lanes 1 and 2 in supplemental Fig. 1). In the reporter system generated here, the RPS5 promoter (Supplemental Fig. 2(A)) was fused to HIS3 and URA3 genes, both of which contained a mutated initiation codon, and the reporter genes were positioned in tandem on a low-copy number plasmid (Fig. (A) and (B)). An artificial ATG start codon was created at position −159, upstream of the RPS5 TSS (−37), and the stop codons at downstream of this ATG were mutated (Fig. (A) and Supplemental Fig. 2(B)). In wild-type cells harboring the reporter plasmid (RPS5*p + HIS3 (URA3)), the initiation of transcription upstream of the artificial ATG was modest; however, it was increased markedly in Δhmo1 cells (compare lanes 1 and 2 in Fig. (A)). Therefore, unlike wild-type cells, Δhmo1 cells can grow on SD media lacking histidine and uracil (SD −His, −Ura), but were unable to grow on SD medium containing 5-fluoroorotic acid due to the expression of functional His3 and Ura3 (Fig. (C)). Thus, this reporter system enables not only positive, but also negative selection that may offer a great advantage to search for any other factors that can assist binding of Hmo1 to its target promoters in vivo.
Fig. 1. Construction of an in vivo reporter system to monitor defects in binding of Hmo1 to the RPS5 promoter.
Notes: (A) A schematic illustration of the endogenous RPS5 gene (upper panel) and the reporter gene (RPS5*p + HIS3 (URA3)) (lower panel). The filled asterisk indicates the major TSS (−37) in the RPS5 promoter and the open asterisk indicates one of the upstream TSSs (−225) that was enhanced in Δhmo1 cells. The positions of the TSSs are numbered relative to the start codon. In the reporter gene, the start codons of the HIS3 and URA3 genes were deleted and an artificial ATG start codon (ATG*), which was in-frame with the HIS3/URA3 gene, was created upstream of the major RPS5 TSS at position −159 (see also Supplemental Fig. 2). (B) A schematic illustration of the reporter plasmid used in the genetic screen. The HIS3 and URA3 genes lacking a start codon were fused to the modified RPS5 promoter (RPS5*p). The arrowheads indicate the artificially created start codons, as shown in Supplemental Fig. 2(B). (C) The growth of wild-type and Δhmo1 cells containing the RPS5*p + HIS3 (URA3) reporter plasmid. Ten-fold dilutions of each strain were spotted in triplicate onto SD (+His,+Ura), SD (−His,−Ura), and SD (+His,+Ura,+5-FOA (5-fluoroorotic acid)) plates and then grown for 3 d (left panel) or 5 d (middle and right panels) at 30 °C.
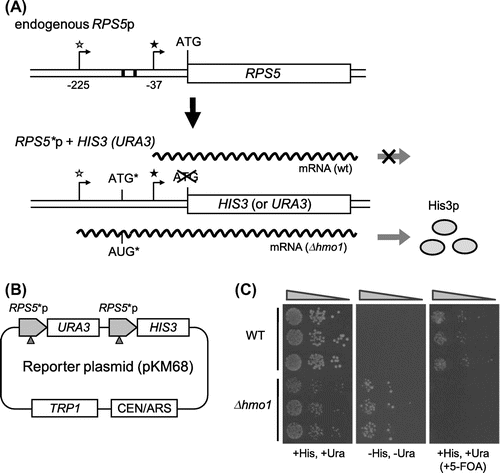
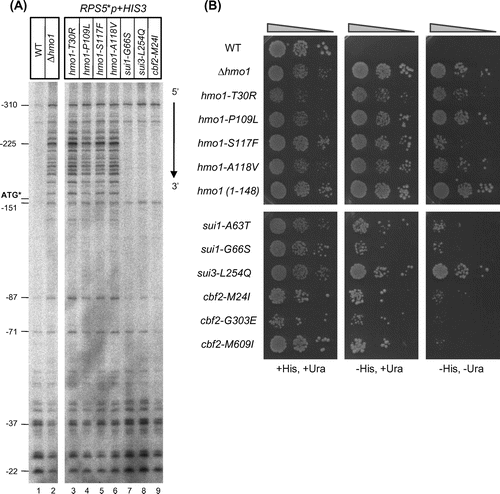
Fig. 2. Characterization of the mutants obtained in the genetic screen.
Notes: (A) The effects of the mutations in the HMO1, SUI1, SUI3, and CBF2 genes on the TSS of the RPS5*p + HIS3 (URA3) reporter gene. The indicated strains were grown at 30 °C and then total RNA (15 µg) was prepared and analyzed by primer extension. The positions of several TSSs, relative to that of the ATG start codon in the endogenous RPS5 promoter, are indicated. The position of the artificially created start codon (ATG*) is also indicated. (B) The effects of mutations in the HMO1, SUI1, SUI3, and CBF2 genes on the growth of yeast cells containing the RPS5*p + HIS3 (URA3) reporter plasmid. The mutant strains obtained in the genetic screen were retransformed with the reporter plasmid, spotted onto the indicated selection plates, and cultivated for 3 d (all strains on the SD + His,+Ura plate), 4 d (hmo1 mutants on the SD −His,+Ura or SD −His, −Ura plate) or 6 d (sui1, sui3, and cbf2 mutants on the SD −His,+Ura or SD −His, −Ura plate) at 30 °C.
The genetic screen to search for mutants that are deficient in binding of Hmo1 to its target promoter in vivo
To understand the mechanisms by which Hmo1 binds to its target promoters, the RPS5*p + HIS3 (URA3) reporter system was used to perform a genetic screen for mutants that are deficient in Hmo1 binding to the RPS5 promoter in vivo. To avoid the selection of colonies that could grow on medium lacking histidine due to a mutation in the reporter gene (for example, reversion of the mutated ATG at the 5′ region of HIS3), the reporter plasmid was transformed into yeast cells that had previously been treated with ethylmethane sulfonate to introduce random genetic mutations. The colonies that grew on SD −His medium (His+ phenotype) were isolated and backcrossed to an isogenic parent strain several times to reduce the number of unrelated mutations. Finally, 12 mutants that showed a 2:2 segregation of the His+ phenotype in the tetrad analysis, indicating that the phenotype was caused by monogenic mutation, were selected as possible candidates. Among these candidates, four strains exhibited temperature-sensitive (ts) growth phenotypes at 37 °C that co-segregated with the His+ phenotype. A yeast genomic library was transformed into these strains and the plasmids that suppressed the ts phenotype were isolated. Sequencing of the isolated plasmids revealed that the ts phenotype of three of the four strains was suppressed by plasmids containing the SUI1 gene, which encodes eukaryotic translation initiation factor 1 (eIF1).Citation24) As expected, the His+ phenotype of these strains was also suppressed by the same plasmid. Furthermore, the endogenous SUI1 gene of these three strains contained a mutation within the open reading frame (ORF) (A63T, A63V, or G66S; Table ), indicating that their His+ phenotype was caused by these mutations. Similarly, a mutation in the SUI3 gene (L254Q; Table ), which encodes the β-subunit of eIF2,Citation25,26) was found to underlie the ts and His+ phenotypes of the remaining ts strain.
Table 1. Mutations causing the His+ phenotype.
In contrast to the sui1 and sui3 ts mutants, five of the isolated strains exhibited a slow-growth phenotype, especially at a low temperature (25 °C), that co-segregated with the His+ phenotype. Library screening revealed that the slow growth of these strains was restored by plasmids containing the HMO1 gene. Sequencing of the endogenous HMO1 gene of these strains identified four missense mutations in the ORF (T30R, P109L, S117F, and A118V) and a nonsense mutation (W149 stop) (Table ). These hmo1 alleles co-segregated with the His+ phenotype during multiple backcross generations (data not shown), excluding the possibility that these His+ phenotypes were caused by as-yet-unidentified secondary mutations. The remaining three His+ phenotype mutants did not exhibit any significant growth defects; therefore, a whole genome sequencing analysis of these strains was performed after five rounds of backcrossing. A comparison of these mutant strains with the wild-type strain revealed that all mutant strains contained a mutation in the CBF2 gene (Table ), which encodes a kinetochore protein that binds to the centromere and plays an essential role in chromosomal segregation.Citation27–29) Furthermore, a plasmid expressing CBF2 suppressed the His+ phenotype of these three mutant strains, confirming that it was caused by the cbf2 mutations (M24I, G303E, and M609I).
Next, a primer extension analysis was performed to determine whether the TSS of the RPS5*p + HIS3 (URA3) reporter gene was altered in the mutant strains. The locations of the TSSs of the reporter gene and endogenous RPS5 were similar in wild-type cells (Fig. (A) and Supplemental Fig. 1). Notably, numerous ectopic TSSs at the region upstream of the artificially created ATG sequence (ATG*) were identified in the hmo1 mutants (Fig. (A), lanes 2–6). This TSS change was more prominent for the reporter gene than the endogenous RPS5 gene (compare Fig. (A) and Supplemental Fig. 1). A slight but reproducible enhancement of specific TSSs was also observed for the sui1, sui3, and cbf2 mutants. Retransformation of the RPS5*p + HIS3 (URA3) reporter plasmid into the mutant cells indicated that the sui1, sui3, and cbf2 mutants grew slower than the hmo1 mutants on SD −His or SD −His, −Ura medium (Fig. (B)), although they exhibited similar growth to the hmo1 mutants on non-selectable medium (data not shown). It is unclear whether the enhancement of specific upstream TSSs in the reporter gene was responsible for the His+ phenotype of the sui1, sui3, and cbf2 mutants; however, the strength of the His+ phenotype did appear to depend on the amount of transcripts generated at the region upstream of ATG*.
Mutations in HMO1, but not SUI1, SUI3, and CBF2, affect the binding of Hmo1 to its target genes in vivo
As described above, the His+ phenotype and changes in the TSS profile were less pronounced in the sui1, sui3, and cbf2 mutants than the hmo1 mutants. Therefore, we hypothesized that the mutations in sui1, sui3, and cbf2 caused these phenotypes by reducing Hmo1 protein levels and/or inhibiting its ability to bind to the RPS5 gene.
However, immunoblot analyses revealed that the expression levels of the Hmo1 protein were similar among the hmo1, sui1, sui3, cbf2 mutants and the wild-type strain (Fig. (A)). Hmo1 protein expression was not detected in a hmo1mutant with nonsense mutation (W149 stop), presumably due to degradation of mRNA by the NMD (nonsense-mediated mRNA decay) pathway (hmo1 (1–148); Fig. (A), lane 7).
Fig. 3. The effects of the hmo1, sui1, sui3, and cbf2 mutations on the expression level of the Hmo1 protein.
Notes: (A) Immunoblot analyses of Hmo1 protein levels in wild-type and hmo1/sui1/sui3/cbf2 mutant cells. Whole cell extracts prepared from mid-log phase cells grown at 30 °C in YPD media were electrophoresed through an SDS-polyacrylamide gel and then transferred to a nitrocellulose membrane. Hmo1 proteins were detected using an anti-Hmo1 antibody. As the loading control, α tubulin was detected using an anti-α tubulin antibody (abcam). (B) ChIP analyses of the ability of Hmo1 to bind to its target promoters (RPS5 and 35s rDNA) in vivo. The strains used in A were cultivated to mid-log phase at 30 °C in SD medium lacking Trp. Cross-linked chromatin was then prepared and immunoprecipitated with an anti-Hmo1 antibody (0.1 μg) and Dynabeads Protein G (lanes 2–10). As a negative control, precipitation of whole cell extracts of wild-type cells was performed without antibody (lane 1). a.u., arbitrary unit. (C) Binding of wild-type and mutant Hmo1 proteins to DNA in vitro, as determined by an immobilized template assay. Extracts of E. coli cells expressing each the indicated hmo1 mutants were mixed with immobilized DNA fragments derived from the RPS5 or RPS10B promoter, or from ARS504 (U10-ARS504-C10), on Dynabeads M-280 Streptavidin at 4 °C. After elution by heating, the bound proteins were analyzed by immunoblotting as described in A. A portion of each diluted cell extract was analyzed as the input control. (D) Binding of purified Hmo1 proteins to DNA in vitro, as determined by an immobilized template assay. Purified Hmo1 protein (0.5 μg) was subjected to in vitro DNA binding assay as described in C.
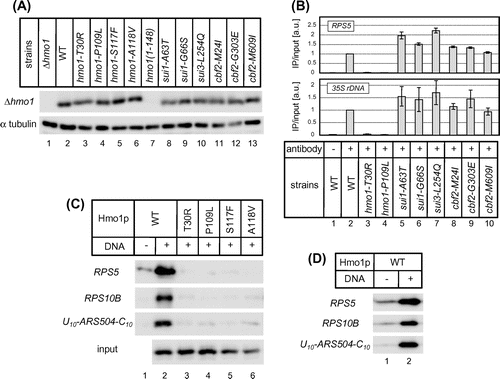
Next, the effects of the hmo1, sui1, sui3, and cbf2 mutations on binding of Hmo1 to its target promoters were investigated by ChIP analysis. Binding of Hmo1 to the promoters of the endogenous RPS5 and 35S rRNA genes, as well as the RPS5*p + HIS3 (URA3) reporter gene, was almost completely abolished in all of the hmo1 mutants (Fig. (B) and data not shown). By contrast, binding of Hmo1 to these genes was not decreased by the sui1, sui3, and cbf2 mutations (Fig. (B) and data not shown), suggesting that they exert their phenotypic effects via some Hmo1-independent mechanism(s).
As mentioned above, Cbf2 binds to the centromere and plays an essential role in chromosomal segregation. One cbf2 mutant has been reported to generate polyploid and aploid cells as a result of abnormal chromosomal segregation; therefore, we hypothesized that the His+ phenotype and/or changes in the TSS profiles of the cbf2 mutant strains were due to an increased copy number of the reporter plasmid caused by unequal distributions in the cbf2 mutants. To address this possibility, the copy numbers of the RPS5*p + HIS3 (URA3) reporter plasmid in Δhmo1, sui1, sui3, and cbf2 mutant cells cultivated in SD −His, in which wild-type cells could not grow, were compared. A quantitative PCR analysis showed that the copy number of the reporter plasmid was significantly higher in the cbf2 mutants than the Δhmo1, sui1, and sui3 mutants (approximately 10-fold higher than that in Δhmo1 cells; data not shown). This result suggests that the cbf2 mutations induced the His+ phenotype and/or changes in the TSS profiles by increasing the copy number of the reporter plasmid, although it is unclear why only some of the specific upstream TSSs were enhanced in the cbf2 mutants. To confirm that the His+ phenotype caused by the Δhmo1 mutation does not involve a similar copy number issue, we examined the phenotype of a strain carrying the RPS5*p+HIS3 reporter gene at the URA3 locus. The results showed that the same His+ phenotype could be caused by the Δhmo1 mutation but not by the cbf2 mutation, in this copy-number fixed strain (data not shown). Therefore, we assume that the Δhmo1 mutation caused the His+ phenotype via the TSS shift alone, even though the Δhmo1 mutation is known to affect plasmid maintenance.Citation30)
As mentioned above, SUI1 and SUI3 encode the translation initiation factors eIF1 and eIF2β, respectively, which are involved in the recognition of initiation codons on mRNAs.Citation24–26) Despite the current lack of direct evidence, we speculate that the mutation of these factors might cause the His+ phenotype by promoting translation of the His3 protein from codons, other than the AUG located within the region near the promoter. Given that mutations in the specific genes mentioned above were repeatedly isolated (note that more sui1 and sui3 mutants were isolated, but excluded from further analyses by testing whether plasmids containing the SUI1 or SUI3 gene suppressed the ts and His+ phenotypes of the mutant strains), the genetic screen appeared to be saturated. Therefore, the fact that we did not identify any factors that affect the binding of Hmo1 to its target promoters suggests that Hmo1 binds to its target genes in vivo without the aid of any additional proteins.
Box A is essential for binding of Hmo1 to DNA both in vitro and in vivo
The results described above suggested that Hmo1 can recognize and bind to its target genes autonomously in vivo. Because some other transcription factors containing HMG boxes (such as Sry, Lef-1, etc.) bind to DNA at specific sequences,Citation5) the sequence specificity of Hmo1 was determined by performing an in vitro DNA binding assay using immobilized DNA template. Biotinylated DNA fragments containing the RPS5 promoter, the RPS10B promoter, or ARS504, which are located between the UAS and core promoter of RPL10, were used as DNA templates. Among these sequences, only the RPS5 promoter is associated with Hmo1 significantly in vivo.Citation15,20) Recombinant His-tagged Hmo1 protein in the bacterial cell extract bound to all three of these DNA fragments at similar levels (Fig. (C)). The same results were obtained when Hmo1 protein, which was purified by Ni-affinity chromatography, was used (Fig. (D)). These results suggest that Hmo1 can bind to DNA autonomously, and that Hmo1 has either a lack of or weak DNA sequence specificity, or that it may recognize chromosomal structures rather than primary DNA sequences. In this case, there are two possible explanations for the loss of Hmo1 binding to its target genes in hmo1 mutants: (i) mutant Hmo1 proteins are deficient in the recognition of particular chromosomal structures or (ii) mutant Hmo1 proteins are simply deficient in DNA binding itself. To differentiate between these possibilities, a DNA binding assay using recombinant mutant Hmo1 proteins was performed. The results indicated that the Hmo1 proteins obtained in the genetic screen were all deficient in DNA binding in vitro (Fig. (C), lanes 3–6). These results suggest that Hmo1 mutants are also unable to bind to DNA in vivo, possibly due to defects in domains that are important for binding to naked DNA.
Box A is essential for DNA binding
Grove et al. reported that, while box B is responsible for most of the DNA binding activity of Hmo1, box A contributes to the bending of duplex DNA and has only a slight, if any, influence on DNA binding.Citation21,22) In contrast to these findings, a mutation in box A of Hmo1 (T30R) identified in the genetic screen abolished the DNA binding activity of Hmo1 both in vivo and in vitro (Fig. (B) and (C)). To confirm the importance of box A to the DNA binding activity of Hmo1, another genetic screen based on the RPS5*p + HIS3 (URA3) reporter system described earlier was performed to identify additional box A mutations that reduce this activity of Hmo1 (Supplemental Fig. 3). After introducing random mutations into the box A region by error-prone PCR, several missense mutations that conferred the His+ phenotype to yeast cells containing the reporter plasmid were identified (Fig. (A)), namely K13E, V17A, S18P, E22K, K25E, A31V, S33P, V35A, F37S, and N39D/K93L. The expression levels of these mutant Hmo1 proteins were similar to that of the wild-type protein (Fig. (B)). A ChIP analysis demonstrated that all of these mutations reduced the ability of Hmo1 to bind to its target genes (RPS5, RPS23A, and 35S rDNA) in vivo, albeit to different extents (Fig. (C)). Furthermore, an immobilized template assay revealed that, with the exception of the F37S mutation, the mutations also reduced the DNA binding activity of Hmo1 in vitro (Fig. (D)). Intriguingly, the DNA binding activity of the hmo1-F37S mutant was not affected in vitro (Fig. (D)), but affected significantly in vivo (Fig. (C)). It is likely that the mutant F37S may be deficient specifically in recognition of some specific chromosomal structures that occur in vivo.
Fig. 4. The effects of mutations in box A on the ability of Hmo1 to bind its target promoters in vivo.
Notes: (A) Growth of Δhmo1 yeast cells containing the RPS5*p + HIS3 (URA3) reporter plasmid that were transformed with a plasmid expressing the indicated hmo1 mutants. The strains were spotted onto SD + His or SD −His plates and cultivated for 5 d at 25 °C. For the N39D/K93L double mutant, we confirmed that the N39D mutation was mainly responsible for the His+ phenotype of this strain, while the K93L mutation had only a modest effect (data not shown). On the other hand, the S33P mutation was solely responsible for the His+ phenotype of the F21L/S33P double mutant (data not shown). (B) Immunoblot analyses of Hmo1 protein levels in the indicated hmo1 mutant cells. Whole cell extracts were prepared from mid-log phase cells grown in SD −Leu, −Trp medium at 30 °C. Hmo1 proteins were detected using an anti-FLAG antibody. α tubulin was also detected as described in Fig. (A). (C) ChIP analyses of the ability of Hmo1 to bind to its target promoters (RPS5, RPS23A, and 35s rDNA) in vivo. The ChIP assay was performed as described in Fig. (B). (D) Binding of wild-type and mutant Hmo1 proteins to DNA in vitro, as determined by an immobilized template assay, which was performed as described in Fig. (C).
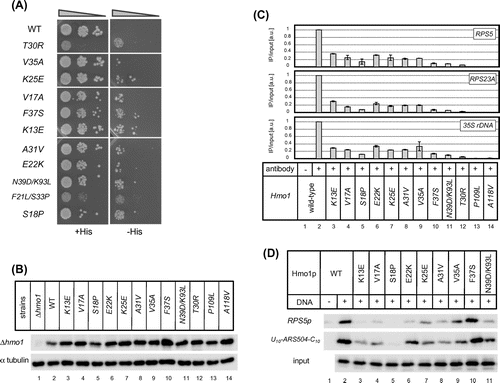
Given that some mutations in box A were drastic replacement of the amino acid residues, e.g. K13E, E22K, and K25E (positively charged to negatively charged and vice versa) or F37S (a bulky side chain for a small side chain), it is possible that such mutations might reduce the DNA binding activity of Hmo1 by affecting the configuration of box B, rather than by affecting the function of box A (which is required for DNA binding). Therefore, to exclude this possibility, we replaced K13, E22, K25, and F37 with alanine residues. In addition, we created an S18A mutant and various K13 mutants (i.e. K13E, K13P, K13G, K13L, K13S, and K13Δ). We tested the effects of these mutations on DNA binding both in vitro and in vivo. As indicated in Supplemental Fig. 4, the E22A, K25A, and K13A mutants were deficient in DNA binding in vitro and in vivo (showing binding activity similar to that of the original mutants, E22K, K25E, and K13E, respectively). Furthermore, like the original F37S mutant, F37A affected DNA binding activity only in vivo. Taken together, the replacing six different amino acid residues (i.e. K13, E22, K25, F37, V17, and V35) with alanine reduced the DNA binding activity of Hmo1 significantly. In addition, replacing of K13 with any type of amino acid residue (e.g. K13E, K13A, K13W, K13P, K13G, K13L, and K13S) affected the DNA binding activity of Hmo1 both in vitro and in vivo. In addition, we isolated K13Q and K13I mutations during the original screen (data not shown). These results strongly suggest that at least several amino acid residues (i.e. K13, E22, K25, F37, V17, and V35) within the box A are directly involved in the DNA binding activity of Hmo1. In contrast to the mutations described above, and unlike the original mutant (S18P), the S18A mutation did not reduce the binding activity of Hmo1 in vitro and in vivo. As described below, we assume that, unlike the S18A mutation, the S18P mutation could break the first α-helix within box A, which is essential for the function of box A in the DNA binding activity of Hmo1.
Prediction of the secondary structure of Hmo1 using the PSIPRED sequence analysis package (http://bioinf.cs.ucl.ac.uk/psipred/) revealed that box B contains three α-helices that may form the L-shaped structure common to HMG boxes (Supplemental Fig. 5, amino acids 111–130 (4th), 138–149 (5th), and 154–180 (6th)). The fact that mutations affecting the DNA binding ability of Hmo1 were located at or close to the first α-helix of box B is consistent with the previously held notion that box B is responsible for most of the DNA binding activity of Hmo1.Citation21,22) On the other hand, two large α-helices were also predicted within box A of Hmo1 (Supplemental Fig. 5, amino acids 7–41 (1st) and 45–63 (2nd)). Notably, all of the box A mutations identified in the genetic screen mapped to the first α-helix (K13E, V17A, S18P, E22K, K25E, A31V, S33P, V35A, F37S, and N39D), although mutations were introduced across the entire box A region (amino acids 2–88). Furthermore, substitution of various amino acids in the first α-helix of box A with helix-breaking proline resulted in the His+ phenotype and abolished the DNA binding activity of Hmo1 both in vivo and in vitro (S18P and S33P, Fig. (A); K13P, Supplemental Fig. 4; and T30P, data not shown). These results suggest that box A, especially the first α-helix, may play a critical role in the DNA binding activity of Hmo1.
Discussion
In this study, a novel and highly sensitive in vivo reporter system was developed and used to perform a genetic screen for mutations that affect binding of Hmo1 to RPG promoters. In addition to mutations in HMO1, several other mutations in SUI1, SUI3, and CBF2 were identified in the screen. As mentioned above, the mechanism by which the TSS profiles of the RPS5 promoter were altered in the sui1, sui3, and cbf2 mutant strains, and whether changes in the TSS profiles were responsible for the His+ phenotype of these mutants, are still unclear. However, ChIP analyses did suggest that these mutations caused the His+ phenotype by some Hmo1-independent mechanism(s); hence, we were unable to identify factors that affect the binding of Hmo1 to its target genes in vivo, indicating that Hmo1 may recognize and bind to its targets autonomously. In a previous study, Berger et al. demonstrated that treatment of cells with rapamycin, an inhibitor of the kinase activity of the TORC1 complex, induces prompt dissociation of Hmo1 from its target genes.Citation18) Therefore, while Hmo1 may bind to its target genes without the aid of additional proteins, the dissociation process may be regulated by other factors, possibly via post-translational modifications.
In addition to trans-acting factors, it is unclear, whether any cis-acting elements are recognized by Hmo1. Our previous in vivo study using chimeric promoters between Hmo1-enriched (RPS5, RPL27B) and Hmo1-limited (RPL10) RPG promoters demonstrated that Hmo1 binds to the intervening region (IVR) between the UAS and the core promoter; this region also determines the level of Hmo1 binding in vivo, whereas the UAS and core promoter are dispensable.Citation20) However, we were unable to identify any specific cis-elements that are responsible for Hmo1 binding within the IVR. In previous bioinformatics approaches, cis-elements, such as the IFHL motif and GGY(n) repeat were proposed as binding sites for Hmo1; however, deletion of the IFHL motif reduces Hmo1 binding only modestly.Citation16,31) Here, we demonstrated that Hmo1 does not have sequence specificity for DNA binding, at least in vitro. The HMG box tends to interact with specific DNA structures, such as four-way junctions, minicircles, and cisplatinated DNA, rather than specific sequences.Citation5) Therefore, the DNA binding specificity of Hmo1 observed only in vivo may reflect the characteristic recognition of some structural properties that occur specifically in the context of native chromatin.
Using the genetic screen, a number of mutations within box A and box B of Hmo1 were found to reduce the DNA binding activity of the protein in vitro and in vivo. In previous in vitro studies, several functions including DNA binding activity were proposed for box A. For example, while box A has only a slight contribution to the DNA binding activity of Hmo1 compared with that of box B, it is responsible for structure-specific DNA binding of the protein, albeit less significantly.Citation21) Furthermore, box A may interact with the carboxy-terminal tail of Hmo1 to bend DNA,Citation22) or with box A itself to form dimer or oligomer.Citation32) However, the significance of these properties of box A in vivo has not been verified.
In our system, the importance of box A for DNA binding was observed similarly both in vitro and in vivo. Remarkably, the characterization of hmo1 mutants (especially hmo1-F37S) suggests that Hmo1 has two independent activities for DNA binding, i.e. recognition of the naked DNA or the structured DNA, the latter of which may play a significant role in the chromatin environment. A previous study suggests that the specific localization of Nhp6a on the genome is determined by the chromatin environments rather than DNA sequences,Citation12) as proposed here for Hmo1. However, the specific properties of chromatin that are recognized by Nhp6a or Hmo1 are still unknown. Therefore, isolation and characterization of additional Hmo1 mutants that are defective in various aspects of DNA binding will provide novel insights into the mechanism of how particular HMGB proteins can recognize specific sites in vivo.
It was recently reported that box A is essential for stimulating rRNA transcription, but it is not fully required for the expression of RPS23A, an Hmo1-targeted RPG.Citation23) By contrast, we confirmed that the box A mutations described here also impaired binding of Hmo1 to the RPS23A promoter (Fig. (C)). Notably, although Hmo1 binding was largely impaired, growth of the box A mutants identified in the genetic screen was similar to or slightly worse than that of wild-type cells (Fig. (A) and (C)). Given that the growth properties of these mutants on the selective media (SD −His) are more consistent with their DNA binding properties (Fig. (A) and (C)), the effect of each box A mutant on DNA binding to other target genes may differ significantly. For instance, hmo1-T30R and hmo1-S33P showing severe growth phenotype on both selective and non-selective media may affect DNA binding to much broader set of target genes, when compared with other hmo1-box A mutants. Additional studies are required to define the contributions of the HMG boxes of Hmo1 to its DNA binding and other functions required for its diverse biological roles.
Supplemental material
The supplemental material for this paper is available at http://dx.doi.org/10.1080/09168451.2014.978258.
978258.zip
Download Zip (351.7 KB)Acknowledgments
We thank Drs T. Chibazakura and S. Watanabe for their advice and helpful discussions.
Additional information
Funding
Notes
Abbreviations: TSS, transcription start site; RPG, ribosomal protein gene; rRNA, ribosomal RNA.
Related Research Data
References
- Bianchi ME, Agresti A. HMG proteins: dynamic players in gene regulation and differentiation. Curr. Opin. Genet. Dev. 2005;15:496–506.10.1016/j.gde.2005.08.007
- Agresti A, Bianchi ME. HMGB proteins and gene expression. Curr. Opin. Genet. Dev. 2003;13:170–178.10.1016/S0959-437X(03)00023-6
- Reeves R, Adair JE. Role of high mobility group (HMG) chromatin proteins in DNA repair. DNA Repair (Amst). DNA Repair. 2005;4:926–938.10.1016/j.dnarep.2005.04.010
- Grasser KD. Chromatin-associated HMGA and HMGB proteins: versatile co-regulators of DNA-dependent processes. Plant Mol. Biol. 2003;53:281–295.10.1023/B:PLAN.0000007002.99408.ba
- Thomas JO, Travers AA. HMG1 and 2, and related ‘architectural’ DNA-binding proteins. Trends Biochem. Sci. 2001;26:167–174.10.1016/S0968-0004(01)01801-1
- Ray S, Grove A. Interaction of Saccharomyces cerevisiae HMO2 domains with distorted DNA. Biochemistry. 2012;51:1825–1835.10.1021/bi201700h
- Ray S, Grove A. The yeast high mobility group protein HMO2, a subunit of the chromatin-remodeling complex INO80, binds DNA ends. Nucleic Acids Res. 2009;37:6389–6399.10.1093/nar/gkp695
- Morrison AJ, Highland J, Krogan NJ, Arbel-Eden A, Greenblatt JF, Haber JE, Shen X. INO80 and γ-H2AX interaction links ATP-dependent chromatin remodeling to DNA damage repair. Cell. 2004;119:767–775.10.1016/j.cell.2004.11.037
- van Attikum H, Fritsch O, Hohn B, Gasser SM. Recruitment of the INO80 complex by H2A phosphorylation links ATP-dependent chromatin remodeling with DNA double-strand break repair. Cell. 2004;119:777–788.10.1016/j.cell.2004.11.033
- Shen X, Mizuguchi G, Hamiche A, Wu C. A chromatin remodelling complex involved in transcription and DNA processing. Nature. 2000;406:541–544.10.1038/35020123
- Au TJ, Rodriguez J, Vincent JA, Tsukiyama T. ATP-dependent chromatin remodeling factors tune S phase checkpoint activity. Mol. Cell. Biol. 2011;31:4454–4463.10.1128/MCB.05931-11
- Dowell NL, Sperling AS, Mason MJ, Johnson RC. Chromatin-dependent binding of the S. cerevisiae HMGB protein Nhp6A affects nucleosome dynamics and transcription. Genes Dev. 2010;24:2031–2042.10.1101/gad.1948910
- Paull TT, Carey M, Johnson RC. Yeast HMG proteins NHP6A/B potentiate promoter-specific transcriptional activation in vivo and assembly of preinitiation complexes in vitro. Genes Dev. 1996;10:2769–2781.10.1101/gad.10.21.2769
- Stillman DJ. Nhp6: A small but powerful effector of chromatin structure in Saccharomyces cerevisiae. Biochim. Biophys. Acta. 2010;1799:175–180.10.1016/j.bbagrm.2009.11.010
- Kasahara K, Ohtsuki K, Ki S, Aoyama K, Takahashi H, Kobayashi T, Shirahige K, Kokubo T. Assembly of regulatory factors on rRNA and ribosomal protein genes in Saccharomyces cerevisiae. Mol. Cell. Biol. 2007;27:6686–6705.10.1128/MCB.00876-07
- Hall DB, Wade JT, Struhl K. An HMG protein, Hmo1, associates with promoters of many ribosomal protein genes and throughout the rRNA gene locus in Saccharomyces cerevisiae. Mol. Cell. Biol. 2006;26:3672–3679.10.1128/MCB.26.9.3672-3679.2006
- Bermejo R, Capra T, Gonzalez-Huici V, Fachinetti D, Cocito A, Natoli G, Katou Y, Mori H, Kurokawa K, Shirahige K, Foiani M. Genome-organizing factors Top2 and Hmo1 prevent chromosome fragility at sites of S phase transcription. Cell. 2009;138:870–884.10.1016/j.cell.2009.06.022
- Berger AB, Decourty L, Badis G, Nehrbass U, Jacquier A, Gadal O. Hmo1 is required for TOR-dependent regulation of ribosomal protein gene transcription. Mol. Cell. Biol. 2007;27:8015–8026.10.1128/MCB.01102-07
- Kasahara K, Ki S, Aoyama K, Takahashi H, Kokubo T. Saccharomyces cerevisiae HMO1 interacts with TFIID and participates in start site selection by RNA polymerase II. Nucleic Acids Res. 2008;36:1343–1357.
- Kasahara K, Ohyama Y, Kokubo T. Hmo1 directs pre-initiation complex assembly to an appropriate site on its target gene promoters by masking a nucleosome-free region. Nucleic Acids Res. 2011;39:4136–4150.10.1093/nar/gkq1334
- Kamau E, Bauerle KT, Grove A. The Saccharomyces cerevisiae high mobility group box protein HMO1 contains two functional DNA binding domains. J. Biol. Chem. 2004;279:55234–55240.10.1074/jbc.M409459200
- Bauerle KT, Kamau E, Grove A. Interactions between N- and C-terminal domains of the Saccharomyces cerevisiae high-mobility group protein HMO1 are required for DNA bending. Biochemistry. 2006;45:3635–3645.10.1021/bi0522798
- Albert B, Colleran C, Leger-Silvestre I, Berger AB, Dez C, Normand C, Perez-Fernandez J, McStay B, Gadal O. Structure-function analysis of Hmo1 unveils an ancestral organization of HMG-Box factors involved in ribosomal DNA transcription from yeast to human. Nucleic Acids Res. 2013;41:10135–10149.10.1093/nar/gkt770
- Naranda T, MacMillan SE, Donahue TF, Hershey JW. SUI1/p16 is required for the activity of eukaryotic translation initiation factor 3 in Saccharomyces cerevisiae. Mol. Cell. Biol. 1996;16:2307–2313.
- Donahue TF, Cigan AM, Pabich EK, Valavicius BC. Mutations at a Zn(II) finger motif in the yeast elF-2β gene alter ribosomal start-site selection during the scanning process. Cell. 1988;54:621–632.10.1016/S0092-8674(88)80006-0
- Laurino JP, Thompson GM, Pacheco E, Castilho BA. The beta subunit of eukaryotic translation initiation factor 2 binds mRNA through the lysine repeats and a region comprising the C2–C2 motif. Mol. Cell. Biol. 1999;19:173–181.
- Jiang W, Lechner J, Carbon J. Isolation and characterization of a gene (CBF2) specifying a protein component of the budding yeast kinetochore. J. Cell Biol. 1993;121:513–519.10.1083/jcb.121.3.513
- Goh PY, Kilmartin JV. NDC10: a gene involved in chromosome segregation in Saccharomyces cerevisiae. J. Cell Biol. 1993;121:503–512.10.1083/jcb.121.3.503
- Espelin CW, Simons KT, Harrison SC, Sorger PK. Binding of the essential Saccharomyces cerevisiae kinetochore protein Ndc10p to CDEII. Mol. Biol. Cell. 2003;14:4557–4568.10.1091/mbc.E02-08-0533
- Lu J, Kobayashi R, Brill SJ. Characterization of a high mobility group 1/2 homolog in yeast. J. Biol. Chem. 1996;271:33678–33685.10.1074/jbc.271.52.33678
- Lavoie H, Hogues H, Mallick J, Sellam A, Nantel A, Whiteway M. Evolutionary tinkering with conserved components of a transcriptional regulatory network. PLoS Biol. 2010;8:e1000329.10.1371/journal.pbio.1000329
- Xiao L, Williams AM, Grove A. The C-terminal domain of yeast high mobility group protein HMO1 mediates lateral protein accretion and in-phase DNA bending. Biochemistry. 2010;49:4051–4059.10.1021/bi1003603