
Abstract
An abnormal form of prion protein (PrP) is considered to be the pathogen in prion diseases. However, the structural details of this abnormal form are not known. To characterize the non-native structure of PrP, we synthesized position-specific double-fluorescent labeled PrP for a fluorescence resonance energy transfer (FRET) experiment. Using FRET, we observed a conformational change in the labeled PrP associated with amyloid fibril formation. The FRET analysis indicated that the distance between fluorescent labeled N- and C-terminal sites of PrP increased upon the formation of amyloid fibrils compared with that of the native state. This approach using FRET analysis is useful for elucidating the structure of abnormal PrP.
Graphical abstract
The double-fluorescent labeled PrP is a useful for analyses of the structure and the formation of amyloid fibrils or PrPSc by using FRET.
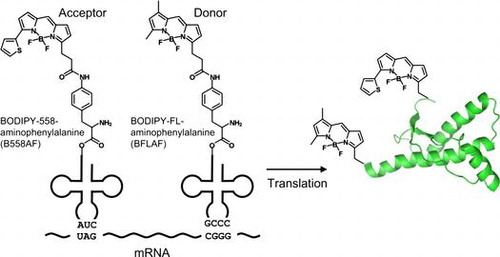
Prion diseases, such as Creutzfeldt–Jakob disease in humans and scrapie and bovine spongiform encephalopathy in animals, are fatal neurodegenerative diseases.Citation1−3) It is considered that the accumulation of the scrapie form of prion protein (PrPSc), an abnormal form derived from cellular PrP (PrPC), is associated with pathogenesis.Citation1,2) Determining the structure of PrPSc is essential for understanding the mechanism of prion propagation, the existence of different prion strains, and species barriers.Citation4) However, the structural details of PrPSc are not known because of difficulties faced in its solubilization, purification, and crystallization processes. Although many studies using various techniques, such as electron microscopy,Citation5) fiber X-ray diffraction,Citation6) small-angle X-ray scattering,Citation7) limited proteolysis,Citation2,8) FTIR,Citation9) or hydrogen/deuterium exchange mass spectrometry (HXMS),Citation10,11) have been performed to elucidate the structure of PrPSc, they have been insufficient. For example, HXMS is a powerful method for measuring the solvent accessibility of peptide backbone amide hydrogens, but gives no information about the three-dimensional packing. In this study, we propose fluorescence resonance energy transfer (FRET) analysis using the double-fluorescent labeled PrP, which is incorporated position-specifically, to estimate the distance between two specific positions in amyloid fibril as a model of the infectious PrPSc.
The incorporation of fluorescent groups into proteins is useful for analyzing structures, functions, localization, and conformational changes of proteins.Citation12) However, it is not easy to introduce fluorescent groups into proteins in a position-specific and reliable manner. For example, fluorescent molecules have been introduced into proteins by chemical modifications of specific amino acid side-chains.Citation13) In thiol-specific chemical modification, the replacement of the amino acid at the labeling position by cysteine is required. This chemical modification does not always result in complete reactions, and cysteine replacement may affect the structure and function of the protein. Fusion expression with green fluorescent protein is often restricted to N- or C-terminal labeling and may be unsuitable for position-specific labeling due to its large size.Citation14)
To avoid these difficulties, the position-specific incorporation of fluorescent non-natural amino acids into proteins using an amber codonCitation15−17) or a four-base codonCitation18−21) has been developed. Using this fluorescent-based approach, intermolecular distances are determined by measuring the FRET between the fluorescent amino acid incorporated into the neurokinin-2 receptor and the fluorescently labeled antagonist.Citation17) Further, a method for the incorporation of two different fluorescent non-natural amino acids into single proteins using a pair of amber and four-base codons,Citation14,22,23) or two four-base codons,Citation24) has been developed. This double-incorporation technology was used for double-fluorescent labeling of proteins and to analyze the protein structure by FRET.
In this study, we incorporated two fluorescent non-natural amino acids BODIPY558- and BODIPYFL-linked p-amino-L-phenylalanine (B558AF and BFLAF, respectively; Fig. ) into PrP using amber and four-base codonsCitation14) and synthesized six proteins double-labeled at different positions. We then investigated the structure of amyloid fibril of the double-labeled PrP using FRET and found that the distance between the N- and C-terminal sites of PrP increased upon the formation of amyloid fibrils. Thus, the double-fluorescent labeled PrP is useful for the analyses of the structure and the formation of amyloid fibrils or PrPSc using FRET.
Fig. 1. Schema of double-fluorescent labeled PrP synthesis.
Materials and methods
Materials
The pIVEX2.3d vector and RTS 100 E. coli Disulfide Kit were from Roche Diagnostics (Basel, Switzerland) or 5-Prime GmbH (Hamburg, Germany). BODIPYFL and BODIPY558 succinimide esters were from Invitrogen (Carlsbad, CA, USA).
Construction of mouse PrP expression plasmid
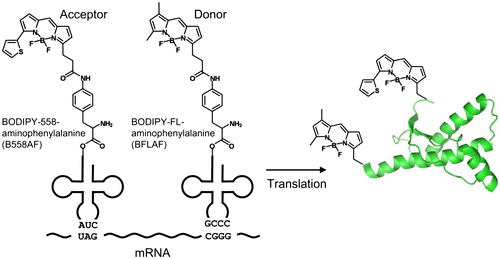
The DNA of mouse PrP, consisting of amino acid residues of 121–231 (mPrP(121–231)) with C-terminal His6 tag, was amplified by PCR using 5′-ATACCATGGTGGGGGGCCTTGGTGG-3′ and 5′-GCCGGATCCTTACCTTATTAATGATGATGATGATGATGAGAACCACCGCTGGATCTTCTCCCGTCG-3′ as primers and was cloned into NcoI and BamHI sites of the expression vector pIVEX2.3d. We first inserted a TAG (amber) and a CGGG four-base codons into the amino acid residues of 120 and 232 at the mPrP(121–231) gene, respectively (Fig. ). We also substituted TAG (amber) codons for the amino acid residues of Tyr127, Phe140, Phe174, Phe197, and Tyr217 and inserted a CGGG four-base codon into the amino acid residue of 232 at the mPrP(121–231) gene, respectively.
Fig. 2. The introduction of mutations into expression vector.

Preparation of aminoacyl tRNAs
B558AF-tRNA containing the CUA anticodon, and BFLAF-tRNA containing the four-base anticodon CCCG were obtained as commercially available reagents (CoverDirectTM tRNA reagents for site-directed protein labeling, Protein Express, Chiba, Japan). Briefly, a yeast phenylalanine tRNA that contained the four-base anticodon CCCG and lacked two nucleotides at its 3′-end was prepared as described previously.Citation14) An amber suppressor tRNA derived from Mycoplasma capricolum Trp1 tRNACitation25) was prepared in a similar manner. Ligation of the truncated tRNA and pdCpA, aminoacylated with B558AF or BFLAF,Citation24) was performed as described previously.Citation14)
Cell-free translation and purification of labeled PrP
Incorporation of B558AF and BFLAF into mPrP(121–231) was performed using an RTS 100 E. coli Disulfide kit (Roche Diagnostics, Basel, Switzerland) as described previously.Citation26) The reaction mixture, containing 500 ng plasmid DNA, 400 pmol B558AF-tRNA, and 400 pmol BFLAF-tRNA, was incubated at 20 °C by shaking on a RTS ProteoMaster (Roche Diagnostics, Basel, Switzerland) at 600 rpm for 2 h, and then at 4 °C without shaking for 16 h. To purify the labeled protein, the total reaction mixture (50 μL × 6 = 300 μL) was centrifuged at 16,000 × g for 15 min, and 90 μL of the supernatant was diluted in Wash-A buffer (20 mM phosphate, 500 mM NaCl, 30 mM imidazole, 0.1% polyoxyethylene(23)lauryl ether, pH 7.4) to a final volume of 180 μL and was applied to a His SpinTrap column (GE Healthcare, Piscataway, NJ). After incubation at room temperature for 15 min, the column was washed eight times with Wash-A buffer and four times with Wash-B buffer (20 mM phosphate, 500 mM NaCl, 0.1% polyoxyethylene(23)lauryl ether, pH 7.4). The labeled proteins were eluted with three 90 μL volumes of Elute-B buffer (30 mM acetate, 500 mM NaCl, 600 mM imidazole, 0.1% polyoxyethylene(23)lauryl ether, pH 8.0). Part of the eluate was subjected to 15% SDS-PAGE using a Real Gel Plate (Bio craft, Tokyo, Japan). A fluorescence scanner FMBIO-III (Hitachi, Tokyo, Japan) was used to visualize the gel. The gel was analyzed by Western blotting using penta-His antibody (Qiagen, Venlo, Netherlands), alkaline phosphatase-conjugated anti-mouse IgG (Promega, Madison, WI), anti-PrP antibody M-20 (Santa cruz, Dallas, TX), and alkaline phosphatase-conjugated anti-goat IgG (Promega). The signals were visualized with Western Blue (Promega).
Optimization of the mPrP(121–231) gene
To enhance the synthetic efficiency, the mPrP(121–231) gene was optimized as follows: (i) codon usage was brought close to that of E. coli K-12 strain expressed in a cell-free translation system, (ii) repeat sequences such as CGCCGCCGCCGC were decreased, (iii) GC content was adjusted to 0.4–0.65, and (iv) the codons were arranged to avoid a rigid RNA secondary structure.
Electron microscope (EM) observation
A 5-μL aliquot of the sample solution was placed for 1 min on a 400-mesh copper grid covered with a carbon film, and the excess solution was removed by blotting with filter paper. The grid was negatively stained for 1 min with a 5-μL droplet of 2% phosphotungstic acid. The liquid on the grid was again removed by blotting, and the grid was dried. Electron micrographs were acquired with a JEM-2100F transmission electron microscope (JEOL, Tokyo, Japan), operating at 200 kV acceleration with a magnification of 15,000×.
Circular dichroism (CD) and fluorescence spectral measurements
CD spectra were measured at 25 °C using an AVIV model 215s spectropolarimeter (AVIV Biomedical, Lakewood, NJ, USA) with a step size of 0.2 nm. The CD spectra of monomer and amyloid fibril were measured in 50 mM 2-(N-morpholino)ethanesulfonic acid (MES)-NaOH buffer (pH 5.5) containing 2 M GdnHCl, using a quartz cell with a light path of 1 mm and a protein concentration of 0.1 mg/ml. The results are expressed as the mean residue ellipticity [θ] (deg cm2 dmol−1).
Fluorescence spectra were measured from 500 to 620 nm, with excitation at 488 nm using a F-7000 fluorescence spectrophotometer (Hitachi High-Technologies, Tokyo, Japan) at 25 °C. Excitation and emission slit widths were set to 5.0 nm.
Reaction of labeled PrP with the amyloid fibril
To make seeds, an ELESTEIN 070-GOT (Elekon, Chiba, Japan) water bath-type ultrasonic transmitter with a temperature controller was used as described previously.Citation27) Non-labeled mPrP(23–231) was dissolved in 50 mM MES-NaOH buffer (pH 5.5) containing 2 M GdnHCl to yield a final concentration of 20 μM mPrP(23–231). The frequency of the instrument was 17–20 kHz, and the power output was set to a maximum of approximately 550 W. Reaction mixtures were ultrasonicated from three directions (both sides and the bottom) for 30 s and then incubated for 9 min without sonication. This process was repeated during incubation at 37 °C.
Amyloid fibril of the double-labeled PrP was made by adding the non-labeled mPrP(23–231) seeds and starting ultrasonication in 50 mM MES-NaOH buffer (pH 5.5) containing 2 M GdnHCl.
Results
Synthesis of double-labeled PrP
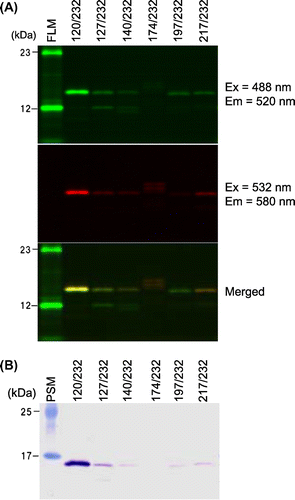
We expressed six proteins double-labeled at different positions by adding fluorescently labeled tRNAs to an E. coli cell-free translation system together with the mutated expression vector (Table ). Henceforth, we refer to the double-fluorescent labeled mPrP(121–231) containing B558AF at positions 120, 127, 140, 174, 197, and 217 and BFLAF at position 232 as 120/232, 127/232, 140/232, 174/232, 197/232, and 217/232, respectively. To confirm that these proteins were double-labeled, we performed SDS-PAGE, followed by fluorescence imaging and Western blotting, using part of the elute of a His SpinTrap column. Fluorescent imaging showed that bands of five proteins, excluding 174/232, were located at around 15 kDa, corresponding to a molecular weight of mPrP(121–231) and that these five proteins emitted fluorescence derived from both BODIPY558 and BODIPYFL (Fig. (A)). Western blot analysis using the M-20 antibody of PrP revealed that the amino acid sequences of these five proteins, except for 174/232, corresponded with that of mPrP(121–231) (Fig. (B)). To evaluate the concentration of fluorescence-labeled PrP, 50, 25, 12.5, 6.25, and 3.125 fmol of BODIPY558 succinimide ester were loaded onto an SDS-PAGE gel as a standard. Using a standard curve, the concentration of synthesized protein was determined (Table ). Except for 120/232, the concentration of each synthesized protein did not exceed 5 nM.
Table 1. Concentrations of double-fluorescent labeled PrP synthesized in this study.
Fig. 3. Analysis of purified, double-labeled protein containing BFLAF and B558AF.
Improvement of protein synthesis
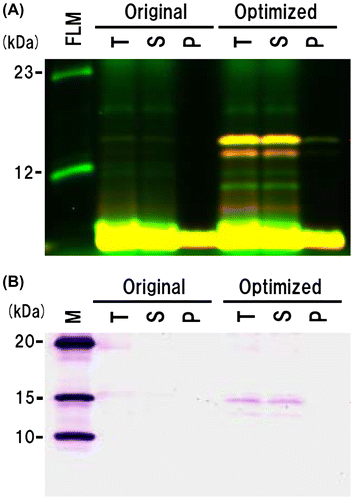
To improve synthetic efficiency, we optimized a gene sequence of the protein 140/232 as a representative of the constructs. After the optimization of the 140/232 plasmid, the yield of the protein increased about 25-fold compared with that of the original plasmid (Fig. ), indicating that optimization of the gene sequence enhanced the double-labeled PrP synthesis. Thus, gene optimization may improve the synthesis efficiency of the other proteins. Although we tried the expression and purification of the protein 140/232, the purification was not successful. So in this study, we used only the protein 120/232 for the FRET experiment.
Fig. 4. Each double-labeled protein 140/232 from the original and optimized sequences was separated into total fraction (pre-separation, T), supernatant fraction (S), and pellet fraction (P).
Amyloid fibril formation of double-fluorescent labeled PrP under ultrasonication
In the FRET experiment, we firstly examined the interaction between the double-labeled protein 120/232 and the seeds made by unlabeled mPrP(121–231) or mPrP(23–231) without the ultrasonication. The FRET spectrum of the protein 120/232 shifted only by the interaction with the mPrP(23–231) seeds at the concentration of 20 μM (Supplemental Fig. S1). It was reported that proteinase K-resistant core of full-length PrP corresponded to the C-terminal region of PrP,Citation28) which could work as seeds of amyloid formation of mPrP(121–231). In addition, EM observation showed that the mPrP(23–231) seeds were 50–200 nm in length and approximately 20 nm in diameter (Fig. (A)). CD spectra showed that mPrP(23–231) monomer formed a partially unfolded conformation in the amyloid buffer containing 2 M GdnHCl (Fig. (B), black) and converted into amyloid fibrils with a β-sheet-rich conformation after an incubation under the ultrasonication (Fig. (B), red). These data indicate that the mPrP(23–231) have typical fibril structure and can be used for the seeding experiment. Hence, we performed the amyloid formation of double-labeled protein 120/232 using the mPrP(23–231) seeds.
Fig. 5. Amyloid fibril formation of the double-labeled protein 120/232.
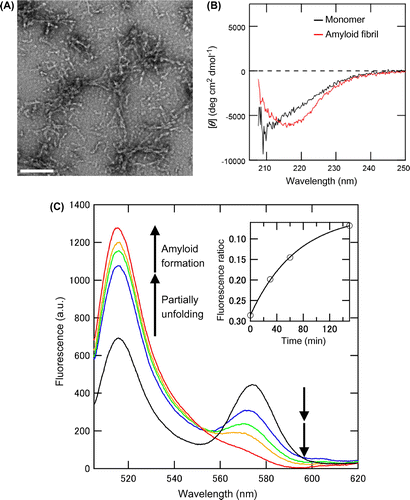
Amyloid formation of the double-labeled protein 120/232, seeded with the unlabeled mPrP(23–231) seeds, was performed at 37 °C under the ultrasonication. The mPrP(23–231) seeds were mixed with the protein 120/232 at a final concentration of 0.2 μM (monomer concentration of mPrP(23–231)), and the fluorescence spectra of the protein 120/232 were recorded time dependently (Fig. (C)). Under the native conditions, the protein 120/232 showed fluorescence of both the BFLAF donor and the B558AF acceptor at 515 and 573 nm, respectively (Fig. (C), black). In the amyloid-forming buffer containing 2 M GdnHCl, an increase and a decrease of the fluorescence at 515 and 573 nm, respectively, were observed (Fig. (C), blue), since approximately half of the PrP was unfolded in these conditions.Citation27) Upon the formation of amyloid fibrils, further changes in fluorescence were observed, depending on incubation time (Fig. (C), blue–red). These results indicated that the distance between the donor and acceptor increased upon the formation of amyloid fibril, compared with the native state (see Discussion). After incubation for 150 min, the fluorescence ratio at 573 and 515 nm (acceptor and donor, respectively) was almost saturated (Fig. (C), inset).
Discussion
The accumulation of PrPSc is known to be associated with the pathogenesis of prion disease, but the structural details of PrPSc are still unknown. Elucidation of the structure of PrPSc and the conversion mechanism from PrPC to PrPSc are urgent for understanding the mechanism of pathogenesis and for the development of pharmaceuticals. In general, NMR and X-ray diffraction are used for analysis of protein structure. However, these methods require large amounts of purified protein or crystallization, so PrPSc structural analysis has not yet been fully exploited. Given a FRET experiment, a relatively small amount of double-labeled protein will allow conformational analysis of non-native PrPs, including infectious PrPSc and the intermediate prepared from PMCA conversion,Citation10,29,30) amyloid fibril,Citation27,28,31) β-oligomer,Citation28) and molten globule state.Citation32) Thus far, the incorporation of two different fluorescent non-natural amino acids has been reported for calmodulinCitation24) and maltose-binding protein.Citation14) We applied this position-specific incorporation technique to PrP and estimated the distance between two specific positions in PrP amyloid fibril.
In this study, we used a C-terminal His6 containing tag for the purification. The His6 tag is connected to the protein 120/232 by linker consisting of several amino acid residues and is much smaller compared with the protein. So, the tag can be considered to have small effect on the amyloid formation. Actually, we conformed that the unlabeled mPrP(121–231) with C-terminal His6 tag formed amyloid fibrils under the ultrasonication, as shown by thioflavin T fluorescence assay (Supplemental Fig. S2). So, we used the double-labeled mPrP(121–231) with C-terminal His6 tag for the experiment of amyloid fibril formation.
The incorporation efficiency of non-natural amino acids into a protein depends strongly on the structures of their side-chains.Citation20) It has been reported that p-substituted phenylalanine derivatives are best substrates for translational machinery.Citation19) On the basis of this finding, BODIPYFL-labeled amino acids and BODIPY-labeled aminophenylalanines were synthesized, and the incorporation efficiencies of BFLAF and B558AF were 46 and 15% in previous experiment,Citation24) respectively. We used the same incorporation strategy, and the efficiency of B558AF was similarly low, owing to the small amounts of double-labeled PrPs synthesized (Fig. ). We showed that the yield of protein could be improved by optimizing the gene sequence of the expression plasmid (Fig. ). In contrast, the incorporation efficiency of B558AF changes depending on the incorporation sites, especially it tends to increase inversely proportional to the distance from the N-terminus in the translated amino acid residues.Citation33) Therefore, it is considered that the incorporation efficiency at 120TAG is higher than that at 140TAG even in the same gene sequence. So, it is considered that the gene optimization of the protein 140/232 mainly increased its expression level to approximately 25-fold. Consequently, gene optimization may be a useful approach for improving the synthesis efficiency of other mammalian proteins.
In the FRET experiment, the protein 120/232 formed amyloid fibrils without a lag time by the seeding (Fig. (C), inset), and it took a few hours before the saturation of amyloid formation, which coincides with the time scale as shown by the previous studies.Citation27,28) It is also known that the unfolding of mPrP is very fastCitation34,35) and typically occurs within few seconds. So, this slow reaction cannot be explained by the unfolding of the PrP. Therefore, we considered that the FRET spectra showed the formation of amyloid fibrils of the double-labeled PrP.
We investigated the structure of amyloid fibrils using the double-labeled protein 120/232. The FRET efficiency (E) between the donor and the acceptor is calculated from the Equation. (Equation1(1) ):
(1)
where IDA is the donor fluorescence of the double-labeled protein and ID is that of the singly labeled protein without the acceptor. To obtain the E value without the singly labeled protein, we used the Förster Equation. (Equation2(2) ):
(2)
where R0 is the Förster distance, at which the transfer efficiency between a specific donor–acceptor pair is 50%, and R is the distance between the donor and acceptor. We used a R0 of 59.6 Å,Citation24) calculated from the overlap of the donor fluorescence and the acceptor absorption, with the assumption that the orientation factor κ2 = 2/3.Citation17,36,37) Firstly, we presumed the R distance at the native state. Although a distance between Cα atoms of amino acid residues of 120 and 232 is 41.3 Å from the PDB entry of 2L1H, the distance between two fluorophores of B558AF and BFLAF should be used for calculation of the E value in the native state. Both of these fluorophores are connected with the protein by the linker, and the distances from Cα atoms to the B558AF and BFLAF fluorophores are estimated to be 10–15 Å, respectively, though these lengths change depending on the rotation angles of the linkers. Hence, we assumed a distance between these fluorophores, which move and rotate flexibly, to be 60 Å at the native state. Then, using Equation. (Equation2(2) ), we calculated the E value as 0.49 at the native state (Fig. (C), black) and estimated ID as 1,350 using intensity IDA from Equation. (Equation1
(1) ).
Subsequently, in the amyloid-forming buffer containing 2 M GdnHCl at pH 5.5 with no incubation (Fig. (C), blue), the distance between the two labeled sites was calculated as 73 Å by Equations. (Equation1(1) ) and (Equation2
(2) ). This result indicated that protein 120/232 was partially unfolded in this buffer. With the increase incubation time, the donor fluorescence at 515 nm gradually increased. After incubation for 150 min, the fluorescence ratio (acceptor/donor) at 573 and 515 nm was almost saturated (Fig. (C), inset), showing that most of protein 120/232 formed amyloid fibrils. Using Equations. (Equation1
(1) ) and (Equation2
(2) ), we calculated the R distance to be 95 Å in the amyloid state, demonstrating that the distance between N- and C-terminal amino acid residues increased by 1.6-fold compared with that of the native state. The E values at the native and amyloid states are 0.49 and 0.06, respective, roughly corresponding to the fluorescence intensities of B558AF in Fig. (C) (black and red). Thus, it would be allowable for rough estimation of the distance, though this assumption is not complete and is affected by the fluorescence fluctuation of B558AF and BFLAF depending on the solution conditions, such as concentration of GdnHCl, pH, and other factors.
In the PrPSc model constructed from electron crystallography,Citation5) the distance between N- and C-terminal amino acid residues was roughly estimated to be 29 Å by presuming the helix C of PrP elongated to amino acid residue of 232, while structure between 227 and 232 was missing in the model, and slightly decreased compared with that in the native state. Our result differs from this model structure of PrPSc. In contrast to the results of the electron crystallography experiment, it has been reported that the NH proton near the helices B and C in the amyloid fibril of PrP is highly protected from hydrogen exchange with bulk water, as revealed by HXMS.Citation11,36,38) This result suggests that the main-chain of PrP is extended by formation of an amyloid core near the helices B and C and that the distance between N- and C-terminus increases in amyloid fibril compared to that in native structure.
In this study, we have proposed a useful strategy for characterizing the amyloid fibril of PrP using the double-fluorescent labeling method and FRET analysis. This method enables us to calculate the distance between any two given positions in non-native PrPs. In this study, we determined only one distance in amyloid structure. In future, we propose to measure the FRET efficiency between various sites in the PrP and to determine the structure of amyloid fibril as well as PrPSc.
Author contribution
Junji Hosokawa-Muto has designed the experiment and prepared double-fluorescent labeled mPrP. Kei-ichi Yamaguchi performed the formation of amyloid fibril. Junji Hosokawa-Muto, Kei-ichi Yamaguchi, and Yuji O. Kamatari prepared the manuscript with the useful suggestions by Kazuo Kuwata. All authors reviewed and approved the final manuscript.
Supplemental material
The supplemental material for this paper is available at http://dx.doi.10.1080/09168451.2015.1050991
Supplemental Materials
Download PDF (212.2 KB)Acknowledgments
The coordinates of the model PrPSc structure were kindly provided by Cédric Govaerts. We thank Ryoji Abe and Kazuhide Imai of ProteinExpress Co., Ltd., for their assistance with the preparation of and experiments with double-labeled proteins.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Notes
Abbreviations: CD, circular dichroism; EM, electron microscope; FRET, fluorescence resonance energy transfer; HXMS, hydrogen/deuterium exchange mass spectrometry; PrP, prion protein; PrPSc, scrapie form of prion protein; PrPC, cellular form of prion protein; B558AF, BODIPY558-linked p-amino-L-phenylalanine; BFLAF, BODIPYFL-linked p-amino-L-phenylalanine; mPrP(121–231), mouse prion protein of amino acid residues 121–231; mPrP(23–231), mouse prion protein of amino acid residues 23–231.
Related Research Data
References
- Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science. 1982;216:136–144.10.1126/science.6801762
- Prusiner SB. Prions. Proc. Nat. Acad. Sci. U.S.A. 1998;95:13363–13383.10.1073/pnas.95.23.13363
- Riek R, Hornemann S, Wider G, et al. NMR structure of the mouse prion protein domain PrP(121-231). Nature. 1996;382:180–182.10.1038/382180a0
- Diaz-Espinoza R, Soto C. High-resolution structure of infectious prion protein: the final frontier. Nat. Struct. Mol. Biol. 2012;19:370–377.10.1038/nsmb.2266
- Govaerts C, Wille H, Prusiner SB, et al. Evidence for assembly of prions with left-handed beta-helices into trimers. Proc. Nat. Acad. Sci. U.S.A. 2004;101:8342–8347.10.1073/pnas.0402254101
- Wille H, Bian W, McDonald M, et al. Natural and synthetic prion structure from X-ray fiber diffraction. Proc. Nat. Acad. Sci. U.S.A. 2009;106:16990–16995.10.1073/pnas.0909006106
- Amenitsch H, Benetti F, Ramos A, et al. SAXS structural study of PrP(Sc) reveals ~11 nm diameter of basic double intertwined fibers. Prion. 2013;7:496–500.10.4161/pri.27190
- Sajnani G, Pastrana MA, Dynin I, et al. Scrapie prion protein structural constraints obtained by limited proteolysis and mass spectrometry. J. Mol. Biol. 2008;382:88–98.10.1016/j.jmb.2008.06.070
- Caughey B, Raymond GJ, Bessen RA. Strain-dependent differences in beta-sheet conformations of abnormal prion protein. J. Biol. Chem. 1998;273:32230–32235.10.1074/jbc.273.48.32230
- Miller MB, Wang DW, Wang F, et al. Cofactor molecules induce structural transformation during infectious prion formation. Structure. 2013;21:2061–2068.10.1016/j.str.2013.08.025
- Smirnovas V, Baron GS, Offerdahl DK, et al. Structural organization of brain-derived mammalian prions examined by hydrogen-deuterium exchange. Nat. Struct. Mol. Biol. 2011;18:504–506.10.1038/nsmb.2035
- Zhang J, Campbell RE, Ting AY, et al. Creating new fluorescent probes for cell biology. Nat. Struct. Mol. Biol. 2002;3:906–918.10.1038/nrm976
- Eftink MR. Fluorescence quenching reactions: probing biological macromolecular structures. In: Dewey TG, editor. Biophysical and biochemical aspects of fluorescence spectroscopy. New york (NY): Springer; 1991. p. 1–41.
- Iijima I, Hohsaka T. Position-specific incorporation of fluorescent non-natural amino acids into maltose-binding protein for detection of ligand binding by fret and fluorescence quenching. Chem. Biol. Chem. 2009;10:999–1006.10.1002/cbic.v10:6
- Cohen BE, McAnaney TB, Park ES, et al. Probing protein electrostatics with a synthetic fluorescent amino acid. Science. 2002;296:1700–1703.10.1126/science.1069346
- Cornish VW, Benson DR, Altenbach CA, et al. Site-specific incorporation of biophysical probes into proteins. Proc. Nat. Acad. Sci. U.S.A. 1994;91:2910–2914.10.1073/pnas.91.8.2910
- Turcatti G, Nemeth K, Edgerton MD, et al. Probing the structure and function of the tachykinin neurokinin-2 receptor through biosynthetic incorporation of fluorescent amino acids at specific sites. J. Biol. Chem. 1996;271:19991–19998.10.1074/jbc.271.33.19991
- Hamada H, Kameshima N, Szymanska A, et al. Position-specific incorporation of a highly photodurable and blue-laser excitable fluorescent amino acid into proteins for fluorescence sensing. Bioorg. Med. Chem. 2005;13:3379–3384.10.1016/j.bmc.2005.03.014
- Hohsaka T, Kajihara D, Ashizuka Y, et al. Efficient incorporation of non natural amino acids with large aromatic groups into streptavidin in in vitro protein synthesizing systems. J. Am. Chem. Soc. 1999;121:34–40.10.1021/ja9813109
- Hohsaka T, Muranaka N, Komiyama C, et al. Position-specific incorporation of dansylated non-natural amino acids into streptavidin by using a four-base codon. FEBS Lett. 2004;560:173–177.10.1016/S0014-5793(04)00099-7
- Murakami H, Hohsaka T, Ashizuka Y, et al. Site-directed incorporation of fluorescent nonnatural amino acids into streptavidin for highly sensitive detection of biotin. Biomacromolecules. 2000;1:118–125.10.1021/bm990012g
- Anderson RD 3rd, Zhou J, Hecht SM. Fluorescence resonance energy transfer between unnatural amino acids in a structurally modified dihydrofolate reductase. J. Am. Chem. Soc. 2002;124:9674–9675.10.1021/ja0205939
- Tokuda Y, Watanabe T, Horiike K, et al. Biosynthesis of proteins containing modified lysines and fluorescent labels using non-natural amino acid mutagenesis. J. Biosci. Bioeng. 2011;111:402–407.10.1016/j.jbiosc.2010.12.012
- Kajihara D, Abe R, Iijima I, et al. FRET analysis of protein conformational change through position-specific incorporation of fluorescent amino acids. Nat. Methods. 2006;3:923–929.10.1038/nmeth945
- Taira H, Matsushita Y, Kojima K, et al. Comprehensive screening of amber suppressor tRNAs suitable for incorporation of non-natural amino acids in a cell-free translation system. Biochem. Biophys. Res. Commun. 2008;374:304–308.10.1016/j.bbrc.2008.07.020
- Abe R, Ohashi H, Iijima I, et al. “Quenchbodies”: quench-based antibody probes that show antigen-dependent fluorescence. J. Am. Chem. Soc. 2011;133:17386–17394.10.1021/ja205925j
- Yamaguchi K, Matsumoto T, Kuwata K. Proper calibration of ultrasonic power enabled the quantitative analysis of the ultrasonication-induced amyloid formation process. Protein Sci. 2012;21:38–49.10.1002/pro.755
- Bocharova OV, Breydo L, Parfenov AS, et al. In vitro conversion of full-length mammalian prion protein produces amyloid form with physical properties of PrP(Sc). J. Mol. Biol. 2005;346:645–659.10.1016/j.jmb.2004.11.068
- Saborio GP, Permanne B, Soto C. Sensitive detection of pathological prion protein by cyclic amplification of protein misfolding. Nature. 2001;411:810–813.10.1038/35081095
- Wang F, Wang X, Yuan CGet al. Generating a prion with bacterially expressed recombinant prion protein. Science. 2010;327:1132–1135.10.1126/science.1183748
- Legname G, Baskakov IV, Nguyen HO, et al. Synthetic mammalian prions. Science. 2004;305:673–676.10.1126/science.1100195
- Honda RP, Yamaguchi K, Kuwata K. Acid-induced molten globule state of a prion protein: crucial role of Strand 1-Helix 1-Strand 2 segment. J. Biol. Chem. 2014;289:30355–30363.10.1074/jbc.M114.559450
- Abe R, Shiraga K, Ebisu S, et al. Incorporation of fluorescent non-natural amino acids into N-terminal tag of proteins in cell-free translation and its dependence on position and neighboring codons. J. Biosci. Bioeng. 2010;110:32–38.10.1016/j.jbiosc.2010.01.003
- Apetri AC, Maki K, Roder H, et al. Early intermediate in human prion protein folding as evidenced by ultrarapid mixing experiments. J. Am. Chem. Soc. 2006;128:11673–11678.10.1021/ja063880b
- Apetri AC, Surewicz K, Surewicz WK. The effect of disease-associated mutations on the folding pathway of human prion protein. J. Biol. Chem. 2004;279:18008–18014.10.1074/jbc.M313581200
- dos Remedios CG, Moens PD. Fluorescence resonance energy transfer spectroscopy is a reliable “ruler” for measuring structural changes in proteins. Dispelling the problem of the unknown orientation factor. J. Struct. Biol. 1995;115:175–185.10.1006/jsbi.1995.1042
- Gustiananda M, Liggins JR, Cummins PL, et al. Conformation of prion protein repeat peptides probed by fret measurements and molecular dynamics simulations. Biophys. J. 2004;86:2467–2483.10.1016/S0006-3495(04)74303-9
- Lu X, Wintrode PL, Surewicz WK. Beta-Sheet core of human prion protein amyloid fibrils as determined by hydrogen/deuterium exchange. Proc. Nat. Acad. Sci. U.S.A. 2007;104:1510–1515.10.1073/pnas.0608447104