
Abstract
Resorcinomycin (1) is composed of a nonproteinogenic amino acid, (S)-2-(3,5-dihydroxy-4-isopropylphenyl)-2-guanidinoacetic acid (2), and glycine. A biosynthetic gene cluster was identified in a genome database of Streptoverticillium roseoverticillatum by searching for orthologs of the genes responsible for biosynthesis of pheganomycin (3), which possesses a (2)-derivative at its N-terminus. The cluster contained a gene encoding an ATP-grasp-ligase (res5), which was suggested to catalyze the peptide bond formation between 2 and glycine. A res5-deletion mutant lost 1 productivity but accumulated 2 in the culture broth. However, recombinant RES5 did not show catalytic activity to form 1 with 2 and glycine as substrates. Moreover, heterologous expression of the cluster resulted in accumulation of only 2 and no production of 1 was observed. These results suggested that a peptide with glycine at its N-terminus may be used as a nucleophile and then maturated by a peptidase encoded by a gene outside of the cluster.
Graphical abstract
Biosynthetic gene cluster of resorcinomycin composed of (S)-2-(3,5-dihydroxy-4-isopropylphenyl)-2-guanidinoacetic acid and glycine was identified and characterized
We recently reported the biosynthetic machinery of pheganomycin (3), which consists of a nonproteinogenic amino acid, (S)-2-(3,5-dihydroxy-4-hydroxymethylphenyl)-2-guanidinoacetic acid (4) at the N-terminus and a proteinogenic core peptide derived from NVKDGPT or NVKDR (Fig. ).Citation1) The biosynthetic gene cluster responsible for its catalysis was identified in Streptomyces cirratus to contain a gene encoding a precursor peptide, which includes both core peptides, and several genes plausibly encoding tailoring enzymes. We also identified a peptide ligase (PGM1) that catalyzed 4-phosphorylation with ATP and the successive nucleophilic attack of the peptides in the cluster. This was the first example of cooperative peptide synthesis achieved by a ribosome and peptide ligase using a peptide as a nucleophile. Unexpectedly, the enzyme accepted a variety of peptides as the nucleophile and its flexibility was elucidated from the crystal structure of the enzyme and mutagenesis analyses.
Fig. 1. Structures of resorcinomycin (1), pheganomycins (3), and their corresponding N-terminal nonproteinogenic amino acids (2 and 4).
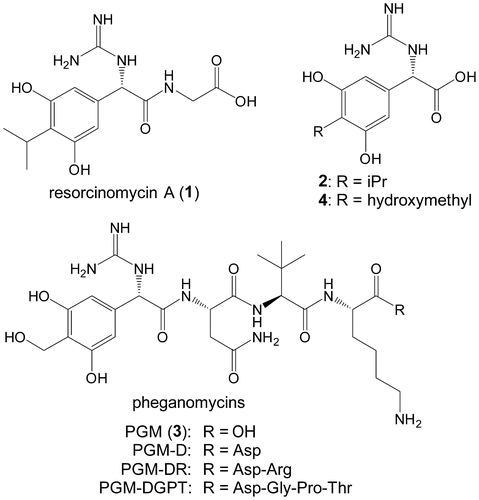
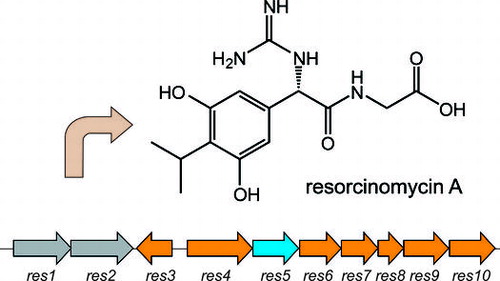
Resorcinomycin A (1) is composed of a nonproteinogenic amino acid, (S)-2-(3,5-dihydroxy-4-isopropylphenyl)-2-guanidinoacetic acid (2), and glycine (Fig. ).Citation2,3) Both 1 and 3 were isolated as anti-Mycobacterium drugs and have the structurally related nonproteinogenic amino acids 2 and 4 at their N-termini. Based on the 3 biosynthetic machinery, the peptide bond formation to yield 1 was suggested to be catalyzed by an ortholog of PGM1 with the ATP-grasp-motif, which perhaps phosphorylates 2 and the successive nucleophilic attack of glycine. If this is the case, we would be interested in the substrate specificities of these enzymes. Both enzymes phosphorylate the structurally related compounds 2 and 4, but utilize quite different compounds, the peptides and glycine, as nucleophiles. In this study, we identified a 1 biosynthetic gene cluster and characterized it by gene-disruption, heterologous expression, and in vitro experiments.
Materials and methods
General
Chemicals and primers were purchased from Sigma–Aldrich Japan (Tokyo, Japan) and Wako Pure Chemical Industry (Osaka, Japan). General genetic manipulations of Escherichia coli and Streptomyces were performed according to standard protocols.Citation4,5) PCR reactions were carried out using a GeneAmp PCR System 9700 thermal cycler (Applied Biosystems) with Tks Gflex DNA polymerase (Takara Bio, Otsu, Shiga, Japan). NMR spectra were obtained using a JEOL ECS-400 spectrometer. Chemical shifts (δ in parts per million) were reported relative to the solvent peak (δ = 3.31 ppm and 49.0 ppm for methanol-d4 in 1H and 13C NMR spectra, respectively). High resolution MS data were acquired using a LTQ-orbitrapXL mass spectrometer (Thermo Scientific).
Draft genome sequences
Draft genome sequences of the S. roseoverticillatum genome were determined by a commercial company (Hokkaido System Science, Hokkaido, Japan) using an Illumina HiSeq (Illumina, San Diego, CA, USA). A genomic DNA library (350 bp insert) was constructed with the TruSeq Nano DNA LT Sample Prep Kit (Illumina). Paired end data (2 × 100 bp) were assembled using VelvetCitation6) (version 1.0.18). Gene prediction and annotation was carried out using MetaGeneAnnotator.Citation7)
Gene disruption
A mutant, in which the res5 gene was in-frame deleted, was constructed by double-crossover homologous recombination. To construct the gene disruption plasmids, two 2-kb DNA fragments, carrying the upstream (EcoRI-XbaI fragment) and downstream (XbaI-HindIII fragment) regions of res5, were amplified by PCR. The primer pair RM-ligase_DEL_up(EcoRI)Fw: 5′-GTCGAATTCACGTTCCACCGGAGTC-3′ and RM-ligase_DEL_up(XbaI)Rv: 5′-GACTCTAGAGAAGACGGCCTTGCTGTTGACGAG-3′ was used to amplify the upstream fragment and the primer pair RM-ligase_DEL_down(XbaI)Fw: 5′-GACTCTAGAGTGATGCTCATCAGCGCCTTCGAC-3′ and RM-ligase_DEL_down(HindIII)Rv: 5′-GCCAAGCTTGACGCGTACGGAGTCGATGAC-3′ was used to amplify the downstream fragment. The introduced restriction sites are underlined. The downstream and upstream PCR products were sequentially inserted into the EcoRI/XbaI and XbaI/HindIII sites of pWHM3,Citation8) respectively. The constructed plasmids were used to transform the 1 producer and thiostrepton-resistant colonies were selected by the same method as for Streptomyces coelicolor A3(2).Citation4) After protoplasting and regeneration of the transformants, thiostrepton-sensitive colonies were screened. Among them, a deletion mutant was selected by PCR analysis and cultivated with X mediumCitation9) (soluble starch 2%, glucose 2%, soy flour 2%, yeast extract 0.5%, NaCl 0.25%, CaCO3 0.35%, MnCl2·4H2O 0.0005%, ZnSO4·7H2O 0.0005%, and CuSO4·5H2O 0.0005%, pH 7.0). Then, production of 1 was analyzed by LC-ESI-MS (Waters Acquity UPLC equipped with a SQD2, Waters). The analytical conditions were as follows: InertSustain C18 column (2.1 × 150 mm, GL sciences, Tokyo, Japan); column temperature, 40 °C; detection, positive mode; mobile phase, 0.05% trifluoroacetic acid (TFA) in water: 0.05% TFA in acetonitrile = 95:5 for 5 min, and a linear gradient to 15:85 in 30 min; flow rate of 0.2 mL/min; cone voltage, 30 V.
To isolate 2, a large scale culture (8 × 100 mL) was centrifuged to remove the cells, and the resulting supernatant was mixed with 100 mL of Diaion HP20 (Diaion). The resin was loaded onto a column and the column was washed with water (1 L). Compound 2 was eluted with 50% aqueous MeOH (400 mL). Fractions with 2 were collected and concentrated by rotary evaporation. The residue was re-dissolved in 1 mL of water and further purified with preparative HPLC, yielding 8.3 mg of a white solid.
Cloning, overexpression, and purification of RES5
Two primers, res5_Fw: 5′-CATATGACGGCGCCCGCCCGCCC-3′ and pColdTF_res5_Rv: 5′-CATATGTCAGGTGTGCCGGGCCGCTGCGGGGGC-3′, were designed to amplify res5 with engineered NdeI restriction sites (underlined). The PCR product was cloned into pGEM-T Easy, sequence confirmed, and then cloned into the NdeI site of the pCold TF vector (Takara Bio) to express RES5 fused with an N-terminal Trigger Factor (TF) chaperone and a His6-tag (TF-RES5). The resulting plasmid pCold-TF-res5 was used to transform E. coli BL21 (DE3) for protein overexpression. An overnight culture of E. coli BL21 (DE3)/pCold-TF-res5 in Luria-Broth (LB) medium was induced by adding 0.5 mM IPTG when the optical density at 600 nm reached about 0.6. The cultivation was continued for an additional 24 h at 15 °C. To express maltose binding protein-fused RES5 (MAL-RES5), the primer pair res5_Fw and pMal_res5_Rv: 5′-GCGGCCGCACTGGTGTGCCGGGCCGCTGCGGGGGC-3′ was used to amplify the res5 gene. The PCR product was first cloned into pGEM-T Easy, sequence confirmed, and then cloned into the NdeI-EcoRI site of pMal-c5X (NEB) to construct the plasmid pMal-res5. The plasmid was introduced into E. coli K12 TB1. A liquid culture of the transformant in LB supplied with 0.2% glucose was induced by adding 0.5 mM IPTG when the optical density at 600 nm reached about 0.6. The cultivation was continued for an additional 16 h at 20 °C. The purification of each recombinant protein was carried out as follows. After the cells were disrupted with an ultrasonic disruptor (TOMY, UD-200, Tokyo, Japan), the TF- and MBP-fused proteins were purified with Ni-NTA agarose column (QIAGEN) and amylose affinity column chromatography (NEB), respectively, and their purity was analyzed by SDS-PAGE on 8% gels (Supplementary Fig. 1). The proteins were visualized by Coomassie brilliant blue staining and the protein concentrations were determined by the Bradford method with bovine serum albumin as a standard.
Preparation of 2
The N-terminal substrate, 2, was prepared by acid hydrolysis from 1, which was purified from the culture broth of the producer by the procedure reported previously.Citation2) The purified 1 (10 mg) was hydrolyzed with 2 mL of 6 N HCl in the presence of 1% hydroquinone at 110 °C under anaerobic conditions for 15 h. The reaction product was fractionated by preparative HPLC to obtain 1 mg of 2. HPLC conditions were as follows: Mightysil RP-18GP Aqua column (10 × 250 mm, KANTO CHEMICAL, Tokyo, Japan); column temperature, 40 °C; detection, UV 210 nm; mobile phase, 0.05% TFA in water: 0.05% TFA in acetonitrile = 95:5 for 5 min, and a linear gradient to 50:50 in 45 min; Table S1 and Figs. S2–S5 show the 1H- and 13C NMR spectra of 2. High resolution (HR)-ESI-MS [M + H]+ found: 268.12915, calcd.: 268.12918 for C12H18N3O4.
Enzyme assay
The standard assay mixture for the recombinant enzymes contained in a final volume of 100 μL, 1 mM 2 or amidino phenylglycine, 1 mM glycine or pentapeptide (NVKDR), 50 mM Tris–HCl (pH 8.0), 10 mM ATP, 10 mM MgCl2, and a suitable amount of enzymes. The mixture was incubated at 30 °C overnight and the products were analyzed by LC-ESI-MS under the conditions described above.
Heterologous expression of the res biosynthetic gene cluster
A DNA fragment carrying a putative 1 biosynthetic gene cluster (res3–10) was amplified by PCR with two primers, RSM-cluster-11kbp-Fw:5′-ATGAAGCTTGCACACCATCATGGGGCACAAC-3′ and RSM-cluster-11kbp-Rv: 5′-ATGAAGCTTCAAAGGTCAGGTCAAGACGAC-3′. The amplified fragments were digested with HindIII and inserted into the same sites of pWHM3 to give pWHM-RES. The resulting plasmid was introduced in Streptomyces lividans TK23, and the transformants were grown in 200-mL baffled flasks containing 30 mL of X mediumCitation9) supplied with thiostrepton (10 μg/ml). Fermentation was carried out for 5 days at 30 °C with agitation (200 rpm). Metabolites accumulated in the culture broth were analyzed by LC-ESI-MS under the conditions described above.
Accession number
The nucleotide sequences reported here have been submitted to the DDBJ/GenBankTM/EBI Data Bank under accession No. LC035135.
Results and discussion
Identification of the 1 biosynthetic gene cluster
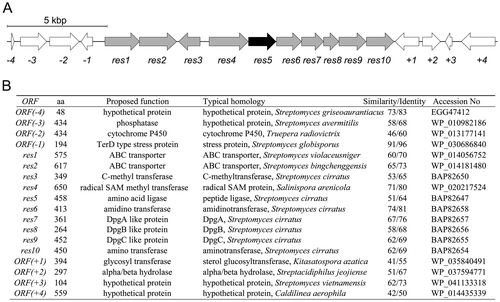
We previously identified the 3 biosynthetic gene cluster including the five genes (pgm8–12) responsible for 4 biosynthesis. Because 1 has the 2 moiety, the structure of which is very similar to 4 (Fig. ), we searched for orthologs of the pgm genes in a draft genome database of the 1 producer. From a BLAST search, we were able to identify five genes (res6–10) corresponding to pgm8–12. A gene encoding an ATP-grasp ligase (res5) also existed in the cluster (Fig. ). Two genes encoding a B12-dependent radical S-adenosyl methionine (SAM)Citation10,11) enzyme (res4) and a methyl transferase (res3), both of which would participate in biosynthesis of the isopropyl moiety of 2, were located next to res5. Two ABC transporterCitation12) genes (res1 and 2) that may participate in efflux of 1 also existed. In the upstream and downstream regions of the cluster, genes encoding a glycosyl transferase and a TerD family hypothetical protein,Citation13) which is found in a stress response operon, were located but these genes were suggested to have no relation to 1 biosynthesis.
Fig. 2. The resorcinomycin biosynthetic gene cluster identified in Streptoverticillium roseoverticillatum.
Disruption of res5
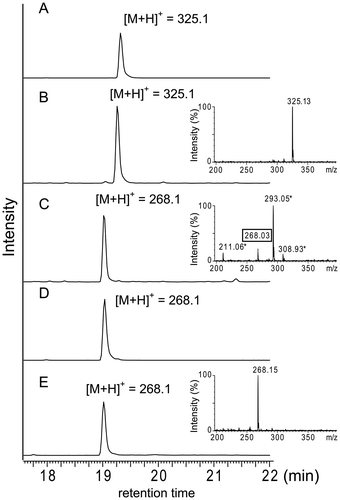
To investigate whether res5 was essential for 1 biosynthesis, we in-frame deleted the res5 gene by a gene replacement technique.Citation14) We successfully obtained res5 gene-deleted mutants (Fig. ). After cultivation of the disruptant in liquid medium, the production of 1 was examined by LC-ESI-MS analysis. As shown in Fig. , we confirmed that 1 was not produced. However, we detected a specific metabolite in the culture broth of the disruptant. The molecular mass was estimated to be 268.1, which corresponds to the [M + H]+ of 2. We purified the compound and its structure was determined by high resolution mass and NMR analyses (Supplemental Table S1 and Figs. S2–S5). These results suggested that the cluster is responsible for 1 biosynthesis and that RES5 catalyzes the amide bond formation.
Fig. 3. Disruption of the res5 gene.
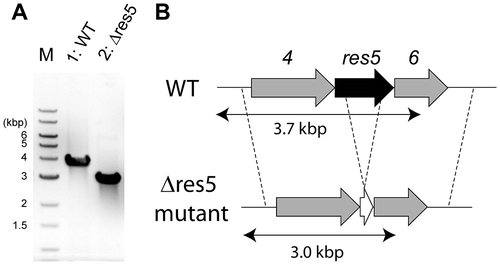
Fig. 4. LC-ESI-MS analysis of products accumulated in the culture broth of the res5 disruptant and S. lividans carrying the res gene cluster.
In vitro assay with recombinant RES5
To examine whether RES5 catalyzes the peptide bond formation between 2 and glycine, we tried to express RES5 recombinant enzymes. We obtained soluble forms of recombinant RES5 with pMAL c5X and pCold TF as expression vectors, and used them for in vitro experiments. We previously demonstrated that PGM1 could utilize amidino-phenylglycine as an N-terminal substrate instead of the intrinsic substrate, 4. Therefore, we used amidino-phenylglycine and glycine as substrates and the reaction was performed in the presence of ATP and Mg2+. The reaction product was analyzed by LC-ESI-MS. However, we detected no specific product with either recombinant enzyme. Next, we prepared the intrinsic substrate, 2, by acid hydrolysis of 1. After purification of 2, we used it for PGM1 assay to examine its quality. As shown in Fig. S6, PGM1 could accept 2 with pentapeptide (NVKDR) as a counterpart substrate, confirming that 2 was of sufficient quality for the enzyme assay. We then performed the RES5 enzyme reaction with 2 and glycine as the substrates, but no formation of 1 was observed with either recombinant enzyme.
To examine the possibility that a peptide with glycine at its N-terminus is utilized as the nucleophilic substrate instead of glycine, we carefully searched for a specific compound in the culture broth of the res5-disruptant but only 2 could be detected. We also used several di- (GF, GG, GI and GR) and tripeptides (GGG, and GGH) with glycine at the N-terminus as the nucleophile. However, none of the peptides examined were accepted.
Heterologous expression of the res gene cluster
Next, we performed a heterologous expression experiment to examine whether the cluster identified in this study contained all of the genes responsible for 1 biosynthesis. An 11 kb DNA fragment carrying the eight genes (res3–10) was amplified by PCR and inserted into the appropriate restriction sites of pWHM3. The plasmid was introduced into S. lividans TK23. After cultivation of the transformants, the products were analyzed by LC-ESI-MS. Unexpectedly, none of the transformants we examined produced 1 but all accumulated 2, suggesting that all res genes except res5 could be functionally expressed in the heterologous host (Fig. (E)). To examine the possibility of a mutation in the res5 gene, we determined the nucleotide sequence of the DNA fragment carrying the res5 gene in the plasmid. However, no errors were detected. We also tested plasmids containing an additional 9 kb upstream region (i.e. orf-4–res2) and 5 kb downstream region (orf+1–+4) for heterologous expression. However, only 2 was detected in the culture broth.
Conclusion
In this study, we tried to identify the 1 biosynthetic gene cluster as a first step to studying the enzyme that catalyzes the peptide bond formation between 2 and glycine. We successfully identified the 1 biosynthetic gene cluster, which consisted of ten genes. Of these, res5 was confirmed to be essential for 1 biosynthesis by a gene disruption experiment. Seven other genes (res3, 4, 6–10) were also suggested to participate in 2 biosynthesis by a heterologous expression experiment. However, we could not clarify the exact function of RES5 by either an in vitro experiment with recombinant enzyme or a heterologous expression experiment. However, considering the results obtained in this study and the high similarity between PGM1 and RES5 (51%), the following biosynthetic machinery might be involved in 1 biosynthesis: a peptide with glycine at its N-terminus may be used as a nucleophile and then maturated by a peptidase, encoded by a gene outside of the cluster, to yield 1.
Author Contribution
Study concept and design: Tohru Dairi
Acquisition of data: Koichi Ooya and Motoyoshi Noike
Analysis and interpretation of data: Koichi Ooya, Yasushi Ogasawara, Motoyoshi Noike, and Tohru Dairi
Drafting of the manuscript: Yasushi Ogasawara and Tohru Dairi
All authors reviewed and approved the final manuscript.
Supplemental material
The supplemental material for this paper is available at http://dx.doi.org/10.1080/09168451.2015.1050992.
Supplemental Material
Download PDF (455.4 KB)Acknowledgments
This study was supported by Grants-in-Aid for Scientific Research (23108101 and 25560397 to T. Dairi) from the Japan Society for the Promotion of Science (JSPS). We also thank Shinogi & Co. Ltd., Osaka, Japan, for providing Streptoverticillium roseoverticillatum and the resorcinomycin A standard.
Related Research Data
References
- Noike M, Matsui T, Ooya K, et al. A peptide ligase and the ribosome cooperate to synthesize the peptide pheganomycin. Nat. Chem. Biol. 2015;11:71–76.
- Kondo E, Katayama T, Kawamura Y, et al. Isolation and characterization of new antibiotics resorcinomycins A and B. J. Antibiot. 1989;42:1–6.10.7164/antibiotics.42.1
- Masaki S, Konishi T, Tsuji N, et al. New antibiotics, resorcinomycins A and B: antibacterial activity of resorcinomycin A against mycobacteria in vitro. J. Antibiot. 1989;42:463–466.10.7164/antibiotics.42.463
- Kieser T, Bibb MJ, Buttner MJ, et al. Practical streptomyces genetics. The John Innes Foundation: Norwich; 2000.
- Sambrook J, Russel W. Molecular cloning: a laboratory manual. 3rd ed. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory; 2001.
- Zerbino DR, Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008;18:821–829.10.1101/gr.074492.107
- Noguchi H, Taniguchi T, Itoh T. MetaGeneAnnotator: detecting species-specific patterns of ribosomal binding site for precise gene prediction in anonymous prokaryotic and phage genomes. DNA Res. 2008;15:387–396.10.1093/dnares/dsn027
- Vara J, Lewandowska-Skarbek M, Wang YG, et al. Cloning of genes governing the deoxysugar portion of the erythromycin biosynthesis pathway in Saccharopolyspora erythraea (Streptomyces erythreus). J. Bacteriol. 1989;171:5872–5881.
- Kondo E, Kawamura Y, Konishi T, et al.; Shionogi & Co., Ltd. Antibiotics DO-248-A and B and process for preparing the same. United States Patent US4,647, 693A. 1987 Mar 3.
- Kim HJ, McCarty RM, Ogasawara Y, et al. GenK-catalyzed C-6′ methylation in the biosynthesis of gentamicin: isolation and characterization of a cobalamin-dependent radical SAM enzyme. J. Am. Chem. Soc. 2013;135:8093–8096.10.1021/ja312641f
- Werner WJ, Allen KD, Hu K, et al. In vitro phosphinate methylation by PhpK from Kitasatospora phosalacinea. Biochemistry. 2011;50:8986–8988.10.1021/bi201220r
- Walsh C. Molecular mechanisms that confer antibacterial drug resistance. Nature. 2000;406:775–781.10.1038/35021219
- Anantharaman V, Iyer LM, Aravind L. Ter-dependent stress response systems: novel pathways related to metal sensing, production of a nucleoside-like metabolite, and DNA-processing. Mol. Biosyst. 2012;8:3142–3165.10.1039/c2mb25239b
- Anzai H, Kumada Y, Hara O, et al. Replacement of Streptomyces hygroscopicus genomic segments with in vitro altered DNA sequences. J. Antibiot. 1988;41:226–233.10.7164/antibiotics.41.226