
Abstract
The synthesis of compounds with the structures proposed for the oxyneolignan apteniols B, C, and G is described. The diphenyl ether skeletons of the proposed apteniols were formed via Ullmann ether synthesis. In particular, the spectral data for the synthesized apteniols B, C, and G did not agree with those previously reported for the isolated compounds. Furthermore, the synthesized proposed apteniol B did not show degranulation-inhibiting activity, while the prepared proposed apteniols C and G exhibited activities considerably weaker than that of the methyl ester of proposed apteniol A.
Graphical Abstract
The synthesis of proposed apteniols B, C, and G is described. The spectral data of the synthesized apteniols B, C, and G did not agree with those of isolated compounds.
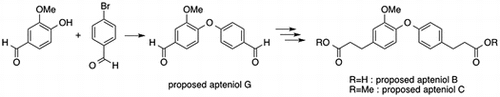
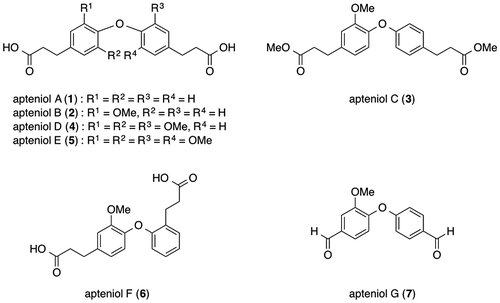
Apteniols A–G (1–7, Fig. ) were first isolated by DellaGreca et al. as phytotoxic metabolites from Aptenia cordifolia (Aizoaceae), a perennial herb native to South Africa that has now spread throughout Europe and become a well-known groundcover or creeping plant; their structures were also determined.Citation1,2) Furthermore, Devi et al. reported the isolation of apteniol A (1) using the marine bacterium Bacillus licheniformis SAB1 as an antimicrobial metabolite.Citation3) In 2014, we synthesized the compound reported as apteniol A (1),Citation4) and Jung and Bräse synthesized the compound reported as apteniol C (3) in 2009.Citation5) These two studies revealed that the spectral data for the isolated apteniols A (1) and C (3) slightly differed from those of the synthesized compounds. Moreover, DellaGreca et al. described the conversion of apteniol B (2) to apteniol C (3) via esterification. On the other hand, the synthesis of apteniols B and G is not reported previously. Therefore, it is necessary to evaluate the structure of apteniol B.
Fig. 1. Proposed structures of apteniols A–G.
If the characterization data for a compound synthesized using a method different from that employed by Jung and Bräse agrees with the data reported for natural apteniol C, then it could be concluded that the proposed structure of apteniol C (3) is correct. However, if the analytical data for two compounds prepared using different synthetic methods do not agree with the data for isolated 3, then it could be concluded that the proposed structure for the natural product is incorrect. Here we describe the synthesis of compounds with the structures proposed for apteniols B (2), C (3), and G (7), using a method different from that employed by Jung and Bräse. Furthermore, we describe the degranulation-inhibiting activities of selected compounds synthesized during the present study, because the methyl ester of the compound proposed to be apteniol A showed strong degranulation-inhibiting activity.Citation4)
Experimental
General experimental procedures
The melting points were measured using an MP-J3 (Yanaco, Kyoto, Japan) and are uncorrected. The IR spectra were obtained using a Nicolet iS10 FT-IR spectrometer (Thermo Fisher Scientific, Waltham, MA, USA) with a diamond horizontal attenuated total reflectance (ATR) accessory and co-addition of 16 interferograms. The calibration models were generated using OMNIC 9.2.98 software. The 1H and 13C nuclear magnetic resonance (NMR) spectra were recorded using ECA-500 (JEOL, Tokyo, Japan) and Agilent 400-MR DD2 (Agilent, Santa Clara CA, USA) spectrometers, respectively, with tetramethylsilane as the internal standard. The mass spectra were recorded using a JMS-700 (JEOL) mass spectrometer. Column chromatography was performed on silica gel 60 N (100–210 mesh, Kanto Chemical Co., Tokyo, Japan).
Chemicals
Both p-nitrophenyl-2-acetamido-2-deoxy-β-d-glucopyranoside and a penicillin–streptomycin mixed solution were obtained from Nacalai Tesque, Kyoto, Japan. The antibody monoclonal anti-dinitrophenyl and DNP-labeled human serum albumin were purchased from Sigma-Aldrich Co., St. Louis, MO, USA. Dulbecco’s modified Eagle’s medium was purchased from Corning, NY, USA. Fetal bovine serum was acquired from HyClone, Logan, UT, USA. Wortmannin was purchased from Wako Pure Chemical Industries, Osaka, Japan. All other chemicals were of at least reagent grade and used as received without further purification.
Evaluation of degranulation-inhibiting activity
The inhibitory activity against the release of β-hexosaminidase from RBL-2H3 cells was evaluated using a modified version of the method reported by Watanabe et al.Citation6) RBL-2H3 cells were purchased from the JCRB Cell Bank (Osaka, Japan). Dulbecco’s modified Eagle’s medium containing 10% heat-inactivated fetal bovine serum, 100 U/mL penicillin, and 100 μg/mL streptomycin was used as the growth medium. The cells were cultured in a 96-well plate (5.0 × 104 cells/well) for 24 h at 37 °C under a humidified 5% CO2 atmosphere and incubated in a growth medium containing 50 ng/mL mouse monoclonal anti-dinitrophenyl (DNP) IgE for 2 h. The cells were then washed with modified Tyrode’s (MT) buffer before a test compound or wortmannin (2.5 μM) was added. The test compounds and wortmannin were first dissolved in dimethyl sulfoxide (DMSO) and diluted with MT buffer to obtain a final DMSO concentration of 0.25%. After 20 min of incubation, DNP-labeled human serum albumin (50 ng/mL final concentration) was added to the cells, and the culture was incubated for 1 h. The supernatant was subsequently collected, and the cells were lysed with MT buffer containing 0.1% Triton X-100. The β-hexosaminidase activities of the supernatant and cell lysate were determined using the method reported by Demo et al.Citation7) The supernatant or cell lysate (20 μL) was mixed with 3.3 mM p-nitrophenyl-2-acetamide-2-deoxy-β-d-glucopyranoside (40 μL) in a 100 mM citrate buffer (pH 4.5), and the mixture was incubated in a 96-well plate at 37 °C for 90 min. The reaction was terminated by adding a 2 M glycine buffer (pH 10.4, 40 μL), and the absorbance at 405 nm was measured using a microplate reader.
Data analysis of the degranulation-inhibiting assays
The results of the degranulation-inhibiting assays are expressed as means and standard deviations (SDs) of three independent cultures. Multiple data comparisons were performed by analyzing the variance using Dunnett’s test. P-values of less than 5% were regarded as significant. One asterisk (*) indicates a p-value <0.05, and two asterisks (**) indicate a p-value <0.01.
Synthesis
Coupling of bromobenzene with phenols (general coupling procedure)
A mixture of bromobenzene (6.60 mmol), phenol (7.40 mmol), Cs2CO3 (3.92 g, 12.0 mmol), copper iodide (140 mg, 0.72 mmol), N,N-dimethylglycine HCl salt (250 mg, 1.81 mmol), and DMF (10 mL) was heated to 100 °C in a sealed tube under a nitrogen atmosphere for 4 days with stirring. The cooled mixture was then poured into water and extracted three times with CH2Cl2. The combined organic layers were washed with brine and dried over Na2SO4. Evaporation of the solvent and purification of the residue by column chromatography (n-hexane/ethyl acetate = 4:1) on silica gel yielded the desired dialdehyde.
4-(4-Formylphenoxy)-3-methoxybenzaldehyde (7; proposed apteniol G)
According to the general coupling procedure, the coupling of vanillin (1.130 g, 7.40 mmol) and 4-bromobenzaldehyde (1.370 g, 6.60 mmol) produces compound 7 (proposed apteniol G) as a pale yellow powder with a 31.4% yield (530 mg, 2.07 mmol). Mp 127.0–127.3 °C; IR νmax (diamond ATR) cm−1: 1683, 1577, 1500, 1274, 1231, and 1149. The 1H and 13C NMR spectral data are reported in Table . HREIMS m/z (M+): Calcd. for C15H12O4: 256.0736, Found: 256.0739.
Table 1. 1H and 13C NMR dataTable Footnotea for synthesized and reported apteniol G (CD3OD, 500/125 MHz).
Ethyl (E)-3-(4-(4-((E)-3-ethoxy-3-oxoprop-1-en-1-yl)-2-methoxyphenoxy)phenyl) acrylate (8)
Sodium hydride (60% dispersion in mineral oil, 253 mg, 6.33 mmol) was washed with dry n-hexane and suspended in dry benzene (10 mL). This suspension was added dropwise to a solution of triethyl phosphonoacetate (1.16 g, 5.17 mmol) at ice-cooled temperature. The solution was then stirred at room temperature until gas evolution ceased. The resultant yellow solution was added dropwise to a solution of 7 (521 mg, 2.00 mmol) in dry benzene at ice-cooled temperature. The solution was then stirred at room temperature for 4 h and subsequently poured into water and extracted three times with ethyl acetate. The combined organic layers were washed with brine and dried over Na2SO4. Evaporation of the extract and purification of the residue by column chromatography (CHCl3/benzene = 1:1) on silica gel yielded diester 8 (790 mg, 2.00 mmol, 99.6%). IR νmax (diamond ATR) cm−1: 1704, 1634, 1591, 1499, 1259, 1155. 1H NMR δ (400 MHz in CD3OD): 1.30 (3H, t, J = 7.1 Hz), 1.32 (3H, t, J = 7.1 Hz), 3.79 (3H, s), 4.21 (2H, q, J = 7.1 Hz), 4.24 (2H, q, J = 7.1 Hz), 6.38 (1H, d, J = 16.1 Hz), 6.51 (1H, d, J = 15.9 Hz), 6.86 (2H, d, J = 8.5 Hz), 7.04 (1H, dd, J = 8.3, 2.0 Hz), 7.20 (1H, d, J = 8.3 Hz), 7.35 (1H, d, J = 2.0 Hz), 7.52 (2H, d, J = 8.5 Hz), 7.62 (1H, d, J = 16.1 Hz), 7.66 (1H, d, J = 15.9 Hz). 13C NMR δ (100 MHz in CD3OD): 14.7 (overlapped), 56.5, 61.6, 61.7, 113.4, 117.4, 117.7, 119.0, 123.0, 123.2, 130.1, 130.9, 133.7, 145.41, 145.43, 147.0, 153.3, 161.2, 168.6, 168.8. HREIMS m/z (M+): Calcd. for C23H24O6: 396.1573, Found: 396.1577.
Ethyl 3-(4-(2-methoxy-4-(3-methoxy-3-oxopropyl)phenoxy)phenyl)propanoate (9)
A suspension of diester 8 (790 mg, 2.00 mmol) and 5% Pd/C (50.0 mg) in dry methanol (8 mL) was stirred at room temperature under hydrogen for 2 h. The reaction mixture was then filtered and concentrated in vacuo. The dry residue was purified by preparative thin-layer chromatography (p-TLC) to produce diester 9 as a pale yellow oil (380 mg, 0.95 mmol, 47.5%). IR νmax (diamond ATR) cm−1: 1733, 1505, 1272, 1227, 1155. 1H NMR δ (400 MHz in CD3OD): 1.19 (3H, t, J = 7.1 Hz), 1.21 (3H, t, J = 7.1 Hz), 2.57 (2H, t, J = 7.6 Hz), 2.64 (2H, t, J = 7.6 Hz), 2.85 (2H, t, J = 7.6 Hz), 2.91 (2H, t, J = 7.6 Hz), 3.74 (3H, s), 4.08 (2H, q, J = 7.1 Hz), 4.10 (2H, q, J = 7.1 Hz), 6.72 (2H, d, J = 8.6 Hz), 6.77 (1H, dd, J = 8.1, 2.0 Hz), 6.85 (1H, d, J = 8.1 Hz), 6.95 (1H, d, J = 2.0 Hz), 7.08 (2H, d, J = 8.6 Hz). 13C NMR δ (100 MHz in CD3OD): 14.5, 14.6, 31.2, 31.8, 36.9, 37.1, 56.3, 61.5, 61.6, 114.5, 117.4, 121.9, 122.6, 130.4, 135.6, 139.4, 144.3, 152.9, 158.3, 174.66, 174.69. HREIMS m/z (M+): Calcd. for C23H28O6: 400.1886, Found: 400.1889.
3-(4-(4-(2-Carboxyethyl)-2-methoxyphenoxy)phenyl)propanoic acid (2; proposed apteniol B)
A solution of diester 9 (234 mg, 0.58 mmol) with a small amount of NaOH (100 mg, 2.5 mmol) in THF/H2O (1:2; 10 mL) was stirred at room temperature for 8 h. The solution was then poured into 4% aqueous HCl and extracted three times with ethyl acetate. The combined organic layers were washed with water and brine and then dried over Na2SO4. Evaporation of the extract and purification of the residue by preparative TLC (CHCl3/methanol = 10:1) yielded 2 as a white powder (172 mg, 0.50 mmol, 86.2%). Mp. 84.5–85.0 °C. IR νmax (diamond ATR) cm−1: 3001, 2933, 1700, 1505, 1445, 1414, 1274, and 1217. 1H and 13C NMR spectral data are reported in Table . HREIMS m/z (M+): Calcd. for C19H20O6: 344.1260, Found: 344.1262.
Table 2. 1H and 13C NMR dataTable Footnotea for synthesized and reported apteniol B (CD3OD, 500/125 MHz).
Methyl 3-(4-(2-methoxy-4-(3-methoxy-3-oxopropyl)phenoxy)phenyl)propanoate (3; proposed apteniol C)
A solution of the proposed apteniol B (2) (50 mg, 0.15 mmol) was refluxed for 24 h in the presence of a catalytic amount of H2SO4 in dry methanol (5 mL). The cooled solution was then evaporated in vacuo. The dry residue was diluted with ethyl acetate, and this solution was washed with saturated aqueous NaHCO3 and brine and then dried over Na2SO4. Evaporation of the solvent and purification by preparative TLC (n-hexane/ethyl acetate = 2:1) yielded diester 3 (proposed apteniol C) as a pale yellow oil (42 mg, 0.11 mmol, 73%). IR νmax (diamond ATR) cm−1: 1734, 1505, 1271, 1226, 1167, and 1155. 1H and 13C NMR spectral data are reported in Table . HREIMS m/z (M+): Calcd. for C21H24O6: 372.1573, Found: 372.1575.
Table 3. 1H and 13C NMR dataTable Footnotea for synthesized and reported apteniol C (CDCl3, 500/125 MHz).
4-(4-Formylphenoxy)-2-methoxybenzaldehyde (10)
According to the general coupling procedure, the coupling of 4-bromo-2-methoxybenzaldehyde (1.420 g, 6.60 mmol) and 4-hydroxybenzaldehyde (900 mg, 7.40 mmol) produced compound 10 as a colorless crystals with a 14.5% yield (246 mg, 0.96 mmol). Mp. 107.5–109.3 °C. IR νmax (diamond ATR) cm−1: 1691, 1576, 1222, 1159, and 838. 1H NMR δ (400 MHz in CD3OD): 3.95 (3H, s), 6.68 (1H, dd, J = 9.5, 2.0 Hz), 6.86 (1H, d, J = 2.0 Hz), 7.25 (2H, d, J = 8.6 Hz), 7.82 (1H, d, J = 9.5 Hz), 7.98 (2H, d, J = 8.6 Hz), 9.95 (1H, s), 10.31 (1H, s). 13C NMR δ (100 MHz in CD3OD): 55.2, 102.7, 110.6, 119.2, 119.7, 130.2, 131.7, 132.8, 161.0, 162.8, 164.0, 188.4, and 191.2. HRFABMS m/z (MH+): Calcd. for C15H13O4: 257.0814, Found: 257.0818.
3-(4-Formylphenoxy)-6-methoxybenzaldehyde (11)
According to the general coupling procedure, the coupling of 5-hydroxy-2-methoxybenzaldehyde (1.130 g, 7.40 mmol) and 4-bromobenzaldehyde (1.370 g, 6.60 mmol) produced compound 11 as a white powder with a 7.8% yield (131 mg, 0.51 mmol). Mp. 89.0–90.5 °C. IR νmax (diamond ATR) cm−1: 1677, 1485, 1261, 1229, 1158, 1023, and 826. 1H NMR δ (400 MHz in CD3OD): 3.99 (3H, s), 7.07 (2H, d, J = 8.9 Hz), 7.28 (1H, d, J = 9.1 Hz), 7.41 (1H, dd, J = 9.1, 3.1 Hz), 7.46 (1H, d, J = 3.1 Hz), 7.90 (2H, d, J = 8.9 Hz), 9.89 (1H, s), 10.40 (1H, s). 13C NMR δ (100 MHz in CD3OD): 56.9, 102.7, 115.4, 118.0, 118.3, 120.3, 129.7, 133.2, 154.6, 164.8, 168.9, 190.2, and 192.7. HRFABMS m/z (MH+): Calcd. for C15H13O4: 257.0814, Found: 257.0815.
2-(4-Formylphenoxy)-5-methoxybenzaldehyde (12)
According to the general coupling procedure, the coupling of 2-hydroxy-5-methoxybenzaldehyde (1.130 g, 7.40 mmol) and 4-bromobenzaldehyde (1.370 g, 6.60 mmol) produced compound 12 as a white powder with a 64.7% yield (1.090 g, 4.27 mmol). Mp. 71.5–72.5 °C. IR νmax (diamond ATR) cm−1: 1676, 1482, 1223, 1156, 1026, and 827. 1H NMR δ (400 MHz in CD3OD): 3.88 (3H, s), 7.12 (2H, d, J = 8.8 Hz), 7.14 (1H, d, J = 9.0 Hz), 7.30 (1H, dd, J = 9.0, 3.3 Hz), 7.44 (1H, d, J = 3.3 Hz), 7.93 (2H, d, J = 8.8 Hz), 9.90 (1H, s), 10.19 (1H, s). 13C NMR δ (100 MHz in CD3OD): 54.9, 111.0, 111.5, 115.0, 116.9, 122.8, 123.0, 131.8, 157.3, 163.7, 164.4, 188.2, and 191.3. HRFABMS m/z (MH+): Calcd. for C15H13O4: 257.0814, Found: 257.0817.
3-(4-Formylphenoxy)-4-methoxybenzaldehyde (13)
According to the general coupling procedure, the coupling of 3-hydroxy-4-methoxybenzaldehyde (1.130 g, 7.40 mmol) and 4-bromobenzaldehyde (1.370 g, 6.60 mmol) produced compound 13 as a white powder with a 68.4% yield (1.160 g, 4.51 mmol). Mp. 63.8–64.2 °C. IR νmax (diamond ATR) cm−1: 1677, 1482, 1224, 1157, 858, and 827. 1H NMR δ (400 MHz in CD3OD): 3.87 (3H, s), 6.99 (2H, d, J = 8.6 Hz), 7.35 (1H, d, J = 8.3 Hz), 7.65 (1H, d, J = 1.7 Hz), 7.87 (2H, d, J = 8.6 Hz), 7.88 (1H, dd, J = 8.3, 1.7 Hz), 9.86 (1H, s), 9.87 (1H, s). 1H NMR δ (400 MHz in CDCl3): 3.90 (3H, s), 7.01 (2H, d, J = 8.8 Hz), 7.16 (1H, d, J = 8.6 Hz), 7.63 (1H, d, J = 1.8 Hz), 7.79 (1H, dd, J = 8.6, 1.8 Hz), 7.85 (2H, d, J = 8.8 Hz), 9.89 (1H, s), 9.93 (1H, s). {lit. Citation8), 1H NMR δ (400 MHz in CDCl3): 3.90 (3H, s), 7.01 (2H, d, J = 8.7 Hz), 7.16 (1H, d, J = 8.4 Hz), 7.63 (1H, d, J = 2.0 Hz), 7.79 (1H, dd, J = 8.4, 2.0 Hz), 7.85 (2H, d, J = 8.7 Hz), 9.88 (1H, s), 9.92 (1H, s)} 13C NMR δ (100 MHz in CD3OD): 56.8, 114.3, 117.3, 123.6, 131.2, 132.0, 132.8, 133.1, 144.8, 158.6, 164.6, 192.2, and 192.7. 13C NMR δ (100 MHz in CDCl3): 56.3, 112.5, 116.6, 122.3, 129.5, 130.4, 131.5, 131.9, 143.8, 156.8, 162.7, 190.0, and 190.7. {lit. Citation8), 13C NMR δ (100 MHz in CDCl3): 56.3, 112.5, 116.6, 122.3, 129.5, 130.4, 131.5, 132.0, 143.8, 156.8, 162.6, 190.0, and 190.7} HRFABMS m/z (MH+): Calcd. for C15H13O4: 257.0814, Found: 257.0811.
2-(4-Formylphenoxy)-4-methoxybenzaldehyde (14)
According to the general coupling procedure, the coupling of 2-hydroxy-4-methoxybenzaldehyde (1.130 g, 7.40 mmol) and 4-bromobenzaldehyde (1.370 g, 6.60 mmol) produced compound 14 as colorless crystals with a 27.7% yield (470 mg, 1.83 mmol). Mp. 90.0–91.5 °C. IR νmax (diamond ATR) cm−1:1678, 1601, 1214, 1155, 1023, and 826. 1H NMR δ (400 MHz in CD3OD): 3.83 (3H, s), 6.61 (1H, d, J = 2.2 Hz), 6.95 (1H, dd, J = 8.8, 2.2 Hz), 7.21 (2H, d, J = 8.6 Hz), 7.92 (1H, d, J = 8.8 Hz), 7.96 (2H, d, J = 8.6 Hz), 9.93 (1H, s), 10.12 (1H, s). 13C NMR δ (100 MHz in CD3OD): 55.1, 105.4, 111.4, 117.9, 121.3, 130.6, 131.8, 132.4, 159.7, 162.5, 166.5, 187.3, and 191.2. HRFABMS m/z (MH+): Calcd. for C15H13O4: 257.0814, Found: 257.0813.
Results and discussion
The synthetic plan for the preparation of the proposed apteniols B, C, and G is shown in Fig. . Formation of the diphenyl ether, which was the key step in this synthesis, was performed via Ullmann ether synthesis, which was previously used for the preparation of apteniol A (1).Citation4) Ullmann etherification of vanillin and 4-bromobenzadehyde produced the compound with the proposed structure for apteniol G (7) with a 31.4% yield. Both of the formyl groups in 7 were then converted to α,β-unsaturated diethyl ester substituents in 8 via the Horner–Wadsworth–Emmons reaction. Catalytic hydrogenation and subsequent hydrolysis of these ester groups afforded the desired dicarboxylic acid corresponding to the proposed structure for apteniol B (2), and methylation of 2 gave the proposed structure for apteniol C (3). The chemical structures of all of the synthesized compounds were determined by 1H and 13C NMR spectroscopy and high-resolution mass spectrometry (HRMS) analyses; the 1H and 13C NMR spectral data for the synthesized compounds and isolated apteniols B (2), C (3), and G (7) are shown in Tables , respectively.
Fig. 2. Synthesis of proposed apteniols B (2), C (3), and G (7).
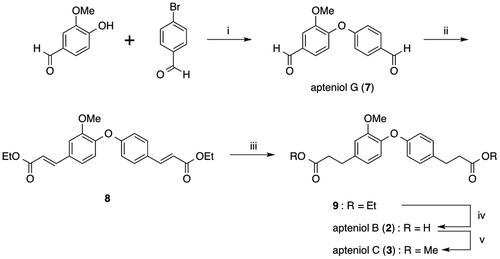
It can be seen from Table that the 1H and 13C NMR spectral data for the synthesized compound 7 with the proposed structure and isolated apteniol G do not agree but are similar. For example, the H-6 proton signal is observed at 7.61 ppm in the spectrum for synthesized 7 and at 6.91 ppm in the spectrum of the isolated compound proposed to have the structure in 7. Furthermore, the C-1, C-6, and C-1’ carbon signals differ by 3.5, 5.3, and 3.2 ppm, respectively, for synthesized 7 and the isolated compound, despite the fact that the same analysis conditions (solvent and measurement frequency) were used.
For synthesized compound 2 and isolated apteniol B, there are only slight differences of less than 0.2 ppm in the shifts of the 1H NMR signals. However, the C-1, C-3, and C-5 carbon signals for the two compounds each differ by more than 6 ppm, as shown in Table .
The 13C NMR spectral data for the three groups in apteniol C (3) are also listed in Table . In particular, the 13C NMR data for synthesized compound 3 are in complete agreement with those previously reported for the apteniol C (3) that was synthesized by Jung and Bräse using a different synthetic approach.Citation5) However, the spectral data for both synthetic compounds (3) differ from those reported for natural apteniol C (3). Furthermore, DellaGreca et al. reported that apteniol C was obtained via methylation of apteniol B.
The results described above suggest that the structures of isolated apteniols B, C, and G differ from the proposed structures. Therefore, additional compounds that could potentially have the actual structure of apteniol G were synthesized. As shown in Fig. , bisaldehyde ethers 10 to 14 were prepared from 1,2,4-trisubstituted benzaldehydes and 4-bromo(or 4-hydroxy)benzaldehyde. In the case of preparation of compounds 10, 11 and 14, the yields decreased due to the formation of uncharacterized polar by-products.
Fig. 3. Synthesis of compounds 10–14.
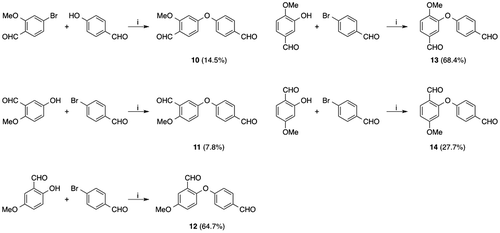
The chemical shifts of the typical functional groups of these compounds are listed in Table . Unfortunately, the spectral data for all of compounds 10–14 did not completely agree with those reported for isolated apteniol G. For example, DellaGreca et al. reported that the chemical shifts of both aldehyde carbons in the natural compound are 193.3 ppm; however, the chemical shifts for the two aldehyde carbons in each of compounds 10–14 differed by more than 0.5 ppm. Furthermore, for compound 13, while the aldehyde carbon chemical shifts were relatively close to one another, the chemical shifts for many hydrogen atoms were very different than those in the spectrum of natural apteniol G. For example, the H-2, H-5, and H-6 proton signals observed at 7.66, 7.35, and 7.87 ppm in the spectrum for compound 13 were instead observed at 7.43, 6.91, and 7.41 ppm, respectively, in the 1H NMR spectrum of the isolated compound proposed to have structure 7. Based on the foregoing results, it can be concluded that the actual structure of the “apteniol G” that was isolated by DellaGreca et al. is neither that of compound 7 nor those of compounds 10–14. Furthermore, the proposed structures for apteniols B and C, which are related to apteniol G by a biosynthetic pathway, are also incorrect.
Table 4. Comparison of chemical shifts of 7- and 7ʹ-aldehyde and methoxy group.Table Footnotea
Next, since the synthesized compounds matching the proposed structures for apteniols B, C, and G had already been produced, their degranulation-inhibiting activities were evaluated, because the methyl ester of the compound with the structure proposed for apteniol A had been reported to show potent degranulation-inhibiting activity.Citation4) As shown in Fig. , synthesized proposed apteniol B (2) did not show such activity in the concentration range from 50 to 150 μM. On the other hand, the proposed apteniols C (3) and G (7) showed activities that were considerably weaker than that of the methyl ester of proposed apteniol A at the same concentration (the release ratio of β-hexosaminidase at 150 μM for the methyl ester of proposed apteniol A is ca. 5%, according to ref.Citation4). It should be noted that the hydrophobic compounds showed activity, while the hydrophilic compound did not, suggesting a characteristic behavior. However, because of the limited number of compounds evaluated, this characteristic can only be postulated.
Fig. 4. Inhibitory effects of compounds on antigen-induced degranulation in RBL-2H3 cells.
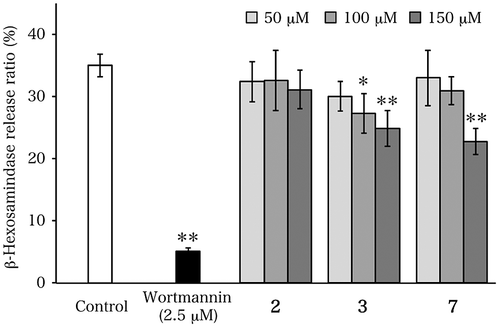
In conclusion, the proposed apteniols B (2), C (3), and G (7) were synthesized, and their 1H and 13C NMR data were found to be different from the corresponding spectral data for the natural products. Therefore, the results of this study indicate that the actual structures of the isolated compounds referred to as apteniols B, C, and G differ from the proposed structures.
The synthesized proposed apteniol B did not show degranulation-inhibiting activity, while the prepared, proposed apteniols C and G exhibited activities considerably weaker than that of the methyl ester of proposed apteniol A. These results suggest that the hydrophobicity of the compounds in this series may influence their degranulation-inhibiting activities.
Author contribution
Study conception and design: TN. Acquisition of data: HN, KI, YH, and TY. Analysis and interpretation of data: AT, HO, and AS. Drafting of manuscript: TN. Critical revision: AT. And all authors read and approved the final manuscript.
Disclosure statement
No potential conflict of interest was reported by the authors.
Funding
This work was supported by a Sasakawa Scientific Research Grant from the Japan Science Society and BRAIN; Technology Research Promotion Program for Agriculture, Forestry, Fisheries, and Food Industry; Program for the Promotion of Basic and Applied Research for Innovations in Bio-oriented Industry.
Acknowledgments
The authors would like to thank Enago (www.enago.jp) for the English language review.
Related Research Data
References
- DellaGreca M, Di Marino C, Previtera L, Purcaro R, Zarrelli A. Apteniols A-F, oxyneolignans from the leaves of Aptenia cordifolia. Tetrahedron. 2005;61:11924–11929.10.1016/j.tet.2005.09.054
- DellaGreca M, Fiorentino A, Izzo A, Napoli F, Purcaro R, Zerrelli A. Phytotoxicity of secondary metabolites from Aptenia cordifolia. Chem. & Biodivers. 2007;4:118–128.
- Devi P, Wahidullah S, Rodrigues C, Souza LD. The sponge-associated bacterium Bacillus licheniformis SAB1: a source of antimicrobial compounds. Mar. Drugs. 2010;8:1203–1212.10.3390/md8041203
- Nishikawa H, Noshita T, Tai A, et al. Syntheses and biological activities of the proposed structure of apteniol A and its derivatives. Biosci. Biotechnol. Biochem. 2014;78:1485–1489.10.1080/09168451.2014.930322
- Jung N, Bräse S. Synthesis of natural products on solid phases via copper-mediated coupling: synthesis of the aristogin family, spiraformin A, and hernandial. Eur. J. Org. Chem. 2009;26:4494–4502.10.1002/ejoc.v2009:26
- Watanabe J, Shinmoto H, Tsushida T. Coumarin and flavone derivatives from estragon and thyme as inhibitors of chemical mediator release from RBL-2H3 cells. Biosci. Biotechnol. Biochem. 2005;69:1–6.10.1271/bbb.69.1
- Demo SD, Masuda E, Rossi AB, et al. Quantitative measurement of mast cell degranulation using a novel flow cytometric annexin-V binding assay. Cytometry. 1999;36:340–348.10.1002/(ISSN)1097-0320
- Jung N, Bräse S. Diaryl ether and diaryl thioether syntheses on solid supports via copper (I)-mediated coupling. J. Comb. Chem. 2009;11:47–71.10.1021/cc800032q