
Abstract
We obtained a novel glucose de-repressed mutant of Trichoderma reesei using disparity mutagenesis. A plasmid containing DNA polymerase δ lacking proofreading activity, and AMAI, an autonomously replicating sequence was introduced into T. reesei ATCC66589. The rate of mutation evaluated with 5-fluoroorotic acid resistance was approximately 30-fold higher than that obtained by UV irradiation. The transformants harboring incompetent DNA polymerase δ were then selected on 2-deoxyglucose agar plates with hygromycin B. The pNP-lactoside hydrolyzing activities of mutants were 2 to 5-fold higher than the parent in liquid medium containing glucose. Notably, the amino acid sequence of cre1, a key gene involved in glucose repression, was identical in the mutant and parent strains, and further, the cre1 expression levels was not abolished in the mutant. Taken together, these results demonstrate that the strains of T. reesei generated by disparity mutagenesis are glucose de-repressed variants that contain mutations in yet-unidentified factors other than cre1.
Graphical abstract
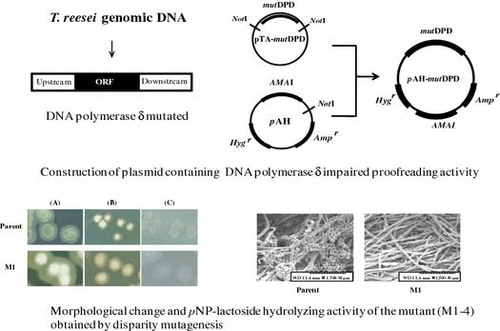
Generation of a glucose de-repressed mutant of Trichoderma reesei using disparity mutagenesis involving DNA polymerase δ impaired proofreading activity.
In recent years, the rapid depletion of fossil fuel resources is a matter of worldwide concern due to continuously increasing energy demands. Many alternatives to fossil fuels have been proposed, such as hydrogen, solar energy, wind energy, biodiesel, and bioethanol.Citation1) Among these ‘eco-friendly’ fuel types, bio-ethanol is a promising alternative source energy, as it can be produced by relatively simple and versatile conversion processes from renewable biomaterials.Citation2) However, as most lignocellulose has a rigid and complicated structure composed of cellulose, hemicellulose, and lignin, it must be hydrolyzed into component sugars for efficient ethanol fermentation by yeast. In this context, the enzymatic saccharification of lignocellulose is considered to be necessary for the development of cost-effective and environmentally friendly processes for large-scale bioethanol production.Citation3)
To date, a large number of saccharification enzymes have been identified in various organisms, including bacteria,Citation4) protists,Citation5) fungi,Citation6) and mushrooms.Citation7) Among them, the filamentous fungus Trichoderma reesei is well known to produce many potent carbohydrate-active enzymes that are useful for the breakdown of lignocellulose. For example, several mutants of T. reesei QM6aCitation8,9) obtained by UV irradiation and/or N-methyl-N′-nitro-N-nitrosoguanidine treatment were shown to produce large amounts of cellulase, ranging from 60 to 100 g/L depending on culture conditions.Citation10) Additionally, because the T. reesei genome sequence is publicly available, it is possible to perform comparative genomic analyses between mutant strains.Citation11)
In T. reesei QM6a wild type strain, crystalline cellulose induces the production of cellulase, whereas glucose acts as a repressor of cellulase production.Citation9) The mechanism of glucose repression was investigated using two glucose de-repressed mutants of T. reesei, Rut-C30Citation12) and KDR-27,Citation13) which were generated by UV-and N-methyl-N′-nitro-N-nitrosoguanidine-based mutagenesis in medium containing 2-deoxy-glucose that is a structural analogue of glucose. T. reesei ATCC66589, a mutant strain of KDR-27 was shown to possess a mutation in cre1 at position 78, changing threonine to proline,Citation14) whereas the mutant RutC-30 was found to have a truncated cre1 gene.Citation15) The cre1, the counterpart gene of that encoding the catabolite repressor CreA in Aspergillus nidulansCitation16) plays a key role in glucose repression in T. reesei.Citation17) In addition, the repressor ACEI, activators ACEII and XYRI, and CCAAT-binding complex HAP2/3/5 are also considered to act cooperatively via the transcription factor CRE1 to regulate carbon catabolite repression and cellulase production.Citation18–20) Glucose-derepressed mutants of T. reesei have also been generated by the UV-irradiated mutagenesis of strain derived from ATCC66589 in the medium containing high glucose concentrations.Citation21) However, despite characterization of these mutants, the mechanisms underlying glucose repression and cellulase induction in T. reesei have not been clearly elucidated due to the complexity of these processes.
As T. reesei ATCC66589 was generated by UV-irradiation and mutagenesis treatment, it is likely that further alleviation of glucose-repression is not possible using similar mutational approaches. Hence, in the present study, we applied disparity mutagenesis technology to strain ATCC66589 in an attempt to generate glucose-derepressed mutants with novel mutations. This mutagenesis approach is based on the introduction of random mutations during lagging strand synthesis by a mutated form of DNA polymerase δ that still synthesizes Okazaki fragments, but lacks proofreading activity.Citation22–24) However, because DNA polymerase ε which is involved in leading strand synthesis, has normal proofreading function, mutations generated in DNA replicons are not infinitely expanded. Even if an unwanted mutation is introduced, it can be removed easily during targeted mutant selection. To date, disparity mutagenesis has been used to generate various types of mutant strains in a number of organisms, including antibiotic-resistant Escherichia coli,Citation25) thermostable Saccharomyces cerevisiae,Citation26) and riboflavin productivity-improved Ashbya gossypii.Citation27)
Here, we attempted to generate mutants of 2-deoxy-glucose-tolerant T. reesei ATCC66589 using a disparity mutagenesis technique. The characteristics of the obtained mutants were analyzed to better understand the glucose repression-releasing in T. reesei.
Materials and methods
Strain
Fungal strain T. reesei ATCC66589 was obtained from the American Type Culture Collection (ATCC; Manassas, VA, USA).
Chemicals
p-nitrophenyl β-D-lactopyranoside (pNP-L), Avicel-PH101, hygromycin B, and 5-Fluoroorotic acid hydrate (5′-FOA) were purchased from Sigma-Aldrich Chemical Co. (St. Louis, MO, USA). Carboxymethyl cellulose (CMC) was purchased from Nacalai Tesque (Kyoto, Japan). Restriction enzymes were obtained from Toyobo (Osaka, Japan). All other reagents were purchased from Wako Chemicals (Osaka, Japan).
Media and culture conditions
A minimal medium (MM) composed of: 5 mg/mL glucose or CMC, 5 mg/mL (NH4)2SO4,15 mg/mL KH2PO4, 0.6 mg/mL MgSO4, 0.06 mg/mL CaCl2, 0.005 mg/mL FeSO4・7H2O, 0.0016 mg/mL MnSO4・H2O, 0.0014 mg/mL ZnSO4・7H2O, and 0.002 mg/mL CoCl2 was used for mutant selection. For cellulase production, conidia of mutants were pre-cultured in 100 mL medium composed of 3% glucose, 0.6% (NH4)2SO4, and 0.6% corn steep liquor in a 500-mL shake flask at 30 °C for 3 days. Five milliliters of the preculture was transferred into 50 mL medium composed of 5% (w/v) Avicel or glucose, 1% (NH4)2SO4, and 1% corn steep liquor in a 500 mL shake flask, which was incubated at 30 °C for 5 days on a rotary shaker at 200 rpm.
Plasmid construction
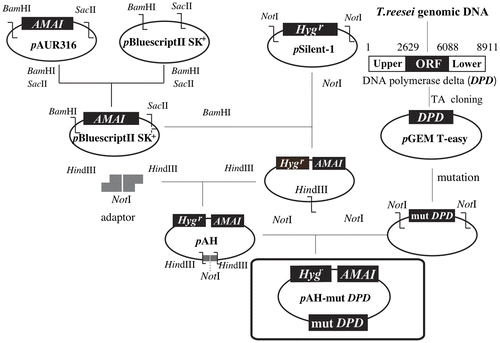
A plasmid carrying DNA polymerase δ lacking proofreading activity was constructed as follows (Fig. ). The pBluescript II SK+ was digested with BamHI and SacII, and then ligated with an AMAI fragment, autonomously replicating sequence of fungi that had been excised from pAUR316. A hygromycin B resistant gene from pSilent-1Citation28) was excised by digestion with NotI, blunting with Klenow fragment (Toyobo), and then, ligated with resulting plasmid digested with NotI, blunt-ended. The obtained plasmid was digested with HindIII and ligated with a phosphorylated adaptor fragment, which was generated by annealing the oligonucleotides 5′-AGCTTGCGGCCGCAATATTA-3′ and 5′-AGCTTAATATTGCGGCCGCA-3′, generating a plasmid designated pAH. The nucleotide sequence of DNA polymerase δ lacking proofreading activity (mutDPD) was cloned into pAH as follows. Mutation points introduced into DNA polymerase δ are as follows:D312A, E314, and L603G.Citation29) First, the nucleotide sequence of DNA polymerase δ was amplified in a 50 μL reaction mixture containing 300 pmol of upstream and downstream primers (5′-CTCAAAGATTCGCCCAAGAG-3′and 5′-GTCATCACAAAGGCCCTCTA-3′), 100 ng of T. reesei genomic DNA, 1 × PCR Buffer for KODplus (Toyobo), 0.2 mM dNTPs, 1.0 mM MgSO4, and 1 U KODplus. After a denaturation step at 95 °C for 10 min, the PCR consisted of 30 cycles of 95 °C for 30 s, followed by 68 °C for 10 min, with a final extension step at 68 °C for 15 min. The amplified PCR product was modified with deoxyadenine triphosphate (dATP) using Go Taq DNA polymerase (Promega KK, Tokyo, Japan) and was then ligated into pGEM-T Easy vector (Promega KK, Tokyo, Japan), generating pTA-DPD). A plasmid containing mutated DNA polymerase δ (pTA-mutDPD) was then constructed by site-directed mutagenesis using pTA-DPD as a template. pTA-mutDPD was digested with NotI and was then inserted into NotI-digested pAH, generating pAH-mutDPD.
Fig. 1. Scheme for the construction of pAH and pAH-mutDPD.
Protoplast preparation and transformation
A spore suspension (1 × 101 spore per mL) was inoculated onto a sterilized cellophane disc on potato dextrose agar (PDA) plate, which was then incubated for 20 h at 30 °C. The propagated mycelia were collected and suspended in 10 mL buffer A (100 mM potassium phosphate [pH 5.6], 1.2 M sorbitol and 2 mg/mL Yatalase [Takara, Shiga, Japan]) and incubated at 30 °C with gentle shaking for approx. 1.5 h. The obtained protoplasts were separated from undigested mycelial debris by filtering through sintered glass (porosity grade 3; Iwaki, Shizuoka, Japan), washed twice with buffer B [10 mM Tris-HCl (pH 7.5) and 1.2 M sorbitol], washed once with buffer C [10 mM Tris-HCl (pH 7.5), 10 mM CaCl2 and 1.0 M sorbitol], and were then resuspended in 0.4 mL buffer C.
Plasmid DNAs of pAH-mutDPD and pAH were purified using an alkaline lysis method and an EZ-10 Spin Column PCR Product Purification kit (Biouniverse, Tokyo, Japan). For the transformation of protoplasts, 20 μg of purified plasmid DNA was mixed with 200 μL of the prepared protoplast suspensions, which were then mixed with 50 μL PEG buffer (10 mM Tris-HCl [pH 7.5], 50 mM CaCl2 and 25% polyethylene glycol 6000) and incubated in an ice-cooled bath for 20 min. The mixture was further incubated at 25 °C for 5 min after the addition of 2 mL PEG buffer. Following the addition of 4 mL buffer C to the mixture, the protoplasts were collected and plated in 1.2% agar overlayed onto MM with 200 μg/mL of hygromycin B. Plates were incubated in the dark at 30 °C for 7 days. More than 40 transformants were selected and transfered onto MM agar plate with 200 μg/mL of hygromycin. After 7 days incubation at 30 °C, conidia formed were suspended in solution containing 10% glycerol and 0.9% NaCl.
Transformation was verified as follows. Genomic DNA was extracted from the tranformant cells grown in liquid MM medium containing 5% glucose with urea-phenol method. Using this DNA as a template, PCR was performed to amplify the hygromycin resistant gene (199 bp) with primers set of 5′-AACGTCGTGACTGGGAAAAC and 5′-CTCCTTTCGCTTTCTTCCCT, and also to amplify genomic DNA with primer set of DNA polymerase δ as described above in plasmid formation. Optimized PCR condition consisted of 95 °C for 2 min, 30 cycles at 95 °C at 30 s, 55 °C for 30 s, and 72 °C for 30 s.
Mutant selection
For the selection of 5′-FOA resistant and 2-deoxy-glucose-tolerant mutants of ATCC66589, the conidia harboring pAH-mutDPD or pAH were inoculated onto MM agar plates containing 5 g/L 5-fluoroorotate with 100 μg/mL uridine or 10 g/L 2-deoxy-glucose with 200 μg/mL of hygromycin. A number of 5′-FOA resistant auxotrophs was counted according to the method previously described.Citation30) 2-deoxy-glucose-tolerant mutants were subjected to further investigation as described below. After 5 days of incubation at 30 °C, colonies grown on the plate were transferred to PDA plates, a nutrient-rich medium that was used for curing pAH-mutDPD. Deletion of plasmid DNA was evaluated by growth on PDA plates containing 200 μg/mL hygromycin B. After cultivation on PDA agar plate, single spore of the mutant thus selected and cured successively was inoculated on MM agar plate containing 200 μg/mL hygromycin B. The spore unable to grow on that plate was picked up as a plasmid deleted transformant.
Morphological analysis of spore and mycelia
Morphological analysis of the 2-deoxy-glucose-tolerant mutants was performed according to the method previously described.Citation31) M1, one of the selected mutants, and the parent strain were inoculated on PDA, 2% Avicel agar plate containing 0.2% peptone (BD, Tokyo, Japan) as a nitrogen source, or 2% glucose agar plate containing 0.2% peptone (BD, Tokyo, Japan). Following inoculation, 0.1% Triton X-100 was added to the plates to enhance spore formation. Colonies propagated on the plates were photographed with a GT-X820 image scanner (Epson, Tokyo, Japan).
For scanning electron microscopic analysis, one loopful of mycelia from the mutant and parent strains was inoculated onto PDA plates and cultured at 30 °C for 7 days. Mycelia of the mutant and parent strains were soaked in 2% glutaraldehyde, washed three times with 0.1 M cacodylate buffer (pH 7.2), and then suspended in 1% osmium. Following sequential dehydration for 10 min in 50, 70, 80, 90, and 99.8% acetone, the mycelia were further dehydrated twice in 100% acetone for 30 min. The obtained samples were immersed for 30 min in a mixture (1:1) of 100% acetone and isoamylacetone, followed by 100% isoamylacetone for 90 min, and were then stored at 4 °C. For the final preparation step, the mycelia were dried using a critical point drier (HCP-2; Hitachi High-Tech. Co., Tokyo, Japan) and observed with a scanning electron microscope (S-3500 N; Hitachi High-Tech. Co.) after vacuum deposition.
Sequence and expression analyses of cre1
Total RNA (1 μg) was extracted using TRIZOL reagent (Invitrogen, Tokyo, Japan) from cells grown in liquid MM medium containing 5% glucose and 0.2% sorbitol as a carbon source and then, a single-stranded cDNA was synthesized using PrimeScript Reverse Transcriptase (Takara Bio, Osaka, Japan). cre1 gene was amplified using cDNA obtained as a template with Go Taq DNA polymerase (Promega, Tokyo, Japan). The primers 5′-ATGCAACGAGCACAGTCTG -3′ and 5′-CTACATCCGATCCATGAGG -3 were designed to amplify full-length cDNA of the cre1 with Go Taq DNA polymerase (Promega, Tokyo, Japan). After denaturation of the template DNA for 5 min at 95 °C, PCR was performed for 30 cycles at 95 °C for 30 s, 55 °C for 30 s, 72 °C for 90 s. The obtained PCR products were purified using an EZ-10 Spin Column PCR Product Purification kit (BIOBasic Inc., Toronto, Canada) and were analyzed for the nucleotide sequences of the cre1 using BigDye Terminator ver. 1.1/3.1 (Applied Biosystems, Tokyo, Japan) and an ABI Automatic Sequencer (PerkinElmer, Tokyo, Japan) according to the manufacturers’ instructions.
Total RNA (1 μg) was extracted using TRIZOL reagent (Invitrogen, Tokyo, Japan) from the same cells as used for sequence analysis, and was then used as template for PCR using PrimeScript Reverse Transcriptase (Takara Bio, Osaka, Japan). The primers, 5′-TTTTTGACTGCCGCTCGATC -3′and 5′-GGATCGGATGTAGAATGTCC-3′, were designed to amplify part of the cre1 gene. PCR was performed under the following optimized conditions: 95 °C 2 min, followed by 30 cycles at 95 °C for 30 s, 55 °C for 30 s, and 72 °C for 30 s.
Measurement of pNP-lactoside hydrolyzing activity
Cellulase activity of culture filtrates was assayed using pNP-lactoside as a substrate. Reaction mixtures consisting of 50 μL of 4 mM pNP-lactoside, 200 mM acetate buffer (pH 5.0), and 50 μL of culture filtrate was incubated for 10 min at 50 °C, and 100 μL of 1 M sodium carbonate was then added to stop the reaction. Absorbance at 415 nm was measured using a DU 730 spectrophotometer (Beckman Coulter, Inc., Brea, CA, USA). One unit of enzyme was defined as 1 μmol of pNP released per 1 min.
Results
Construction of T. reesei strain harboring mutated DNA polymerase δ gene
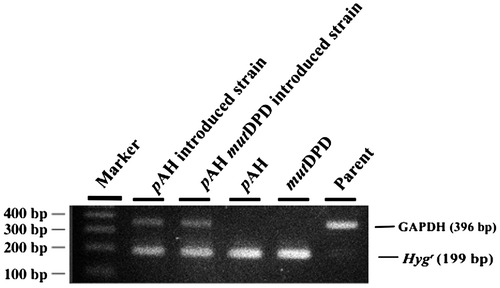
The plasmids pAH and pAH-mutDPD were individually introduced into ATCC66589 by the protoplast-PEG method. After a 7-day incubation, colonies appeared on MM agar plates supplemented with 200 μg/mL hygromycin B, indicating these were mutated DNA polymerase δ harboring transformants. To verify transformation efficiency of T. reesei, PCR with primers targeting the hygromycin resistant gene (199 bp) was performed with transformant DNA using primer set indicated in materials and method. As shown in Fig. ., the hygromycin resistant gene of pAH-mutDPD was clearly detected in all selected mutants.
Fig. 2. Plasmid insertion confirmed by PCR.
Evaluation of mutation frequency based on 5′-FOA-resistant mutant generation
To evaluate the mutation frequency of strain ATCC66589 using the disparity mutagenesis approach, the number of 5′-FOA-resistant auxotrophs was counted according to the method previously described.Citation30) 89 colonies of 5′-FOA resistant, and 8 colonies of 5′-FOA resistant mutant were generated respectively from 1 × 108 conidia, and 1 × 107 conidia harboring pAH-mutDPD on MM agar plates supplemented with uridine. Whereas, no such mutants were obtained from conidia transformed with pAH as a control (data not shown). The frequency of 5′-FOA-resistant mutant generation by disparity mutagenesis was approx. 30-fold higher than that by UV-induced mutagenesis (Table ).
Table 1. Mutation ratio of disparity mutagenesis evaluated by colony formation in comparison to UV-irradiation.
Selection of 2-deoxyglucose-tolerant mutants
Mutants were selected for 2-deoxyglucose tolerance by plating of conidia on MM consisting of 1% CMC and 1% 2-deoxyglucose. The concentration of 2-deoxyglucose used here was two times higher than in media described in a previous report examining resistance to catabolite repression in T. reesei.Citation13) The selected mutants were further cultured in PDA, and six variants that exhibited good growth were randomly selected and named M1-M6. The colony size of these variants were 2–3 cm in a week on agar plates containing 1% 2-deoxyglucose, which severely inhibited the growth of the parent strain.
Morphological analysis
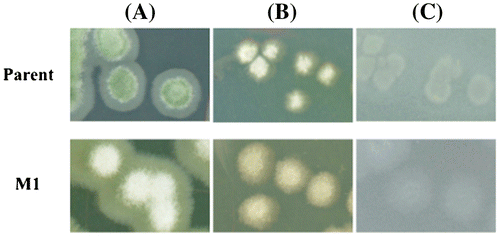
The parent strain formed green conidia on PDA, whereas the conidia of mutant strain M1 appeared weakly yellowish-white, as shown in Fig. (A). In contrast, both the parent strain and M1 formed light creamy or white conidia on Avicel and glucose agar plates (Fig. (B) and (C)). The colony size of the mutant strain was similar to that of the parent on each type of plate. All other mutant strains (M2-M6) appeared morphologically similar to M1 (data not shown).
Fig. 3. Colony formation of glucose de-repressed mutant and parent.
Spore formation and mycelial morphology were also observed by scanning electron microscopic analysis (Fig. ). The formation of both conidia and conidiospores by the mutant strains was markedly reduced compared to the parent strain, despite the fact the mycelia of each strain had highly similar morphologies (Fig. ).
Fig. 4. Scanning electron microscopy of conidia and mycelia.

Sequence and expression analyses of cre1
The nucleotide sequences of cre1 from the six mutants were identical with that of the parent strain (data not shown). In addition, the cre1 gene expression level of the mutants was not abolished in liquid medium containing 5% glucose (Fig. ).
Fig. 5. Expression of cre1 analyzed by RT-PCR.
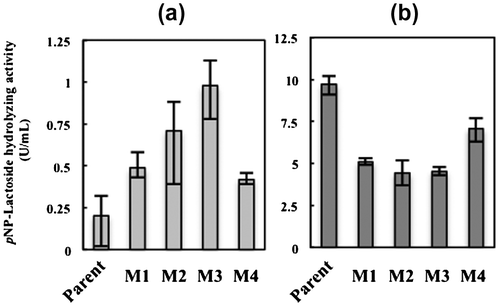
pNP-lactoside hydrolyzing activity of 2-deoxyglucose-tolerant mutants
pNP-lactoside hydrolyzing activity was compared between the parent and four 2-deoxyglucose-tolerant mutants (M1-M4) arbitrarily selected from the six mutant strains. The pNP-lactoside hydrolyzing activity of the four mutants was 2–5-fold higher than that of the parent strain when cultivated in medium containing 5% glucose as the sole carbon source (Fig. ). Among the examined strains, M2 and M3 showed 4- and 5-fold higher hydrolyzing activities, respectively, than that of the parental strain. In contrast, the pNP-lactoside hydrolyzing activity of all mutants was slightly lower, 0.5–0.7-fold, than that of the parent strain when cultivated in medium containing 5% avicel as the carbon source (Fig. ).
Fig. 6. Cellulase activity of the glucose de-repressed mutants (M1–4) and parent strain.
Discussion
In the present study, we attempted to generate glucose-derepressed mutants of T. reesei using disparity mutagenesis. Among the wide variety of possible T. reesei strains, ATCC66589 was selected for mutagenesis due to its high cellulase productivity and industrial usefulness.Citation9) For this mutagenesis approach, we constructed a plasmid encoding DNA polymerase δ lacking proofreading ability that can be temporarily introduced into T. reesei using a hygromycin B selection method. In contrast to auxotrophic selection, hygromycin B selection is applicable to any medium and is highly sensitive for T. reesei.Citation32) For the plasmid construction, we used a replication origin from A. nidulans, AMA1, which has been shown to function in T. reesei.Citation33) The nucleotide sequence of DNA polymerase δ in T. reesei ATCC66589 is composed of 3315 bp and is identical with that of QM6a, the grand parental strain of ATCC66589. In general, transformation efficiency declines with the size of the vector used. However, an autonomous plasmid containing AMA1 was shown to have an approx. 10-fold higher transformation efficiency than a typical integrative-type vector for fungi, despite its relatively large size of approx. 20 kb.Citation34)
5′-FOA inhibits the functions of both Ura3 (orotidine-5′-phosphate decarboxylase) and Ura5 (orotate phosphoribosyl transferase) in bacteria, yeast,Citation35) and fungi.Citation30) 5′-FOA-resistant mutants of T. reesei were reportedly obtained by UV irradiation in culture medium containing 5 g/L 5′-FOA.Citation30) Here, we therefore selected mutants using the same 5′-FOA concentration and found that 5′-FOA-resistant mutants were generated at a 30-fold higher rate compared to UV-induced mutagenesis (Table ). Thus, disparity mutagenesis technology is considered to be much more efficient and convenient compared with UV irradiation or mutagen treatment, which require many laborious steps for the conditioning of cells. Error-prone rate of DNA polymerase δ is suggested to be increased by introducing mutations into critical points for proofreading. As the mutated DNA polymerase δ that is introduced into the host strain can be removed by simple inoculation on any nutrient-rich medium, such as PDA, disparity mutagenesis is thus considered to be a sophisticated method for generating mutants of T. reesei.
2-Deoxyglucose is a glucose isomer in which the 2-hydroxy group is replaced by hydrogen, and is a competitive inhibitor of hexokinase and phosphoglucose isomerase in the glycolytic pathway.Citation36) KDR-27, a parental strain of ATCC66589 treated with 5 g/L 2-deoxyglucose, is reported to produce only low levels of cellulase in medium containing glucose as the sole carbon source. For this reason, here, the more stressful condition of 10 g/L 2-deoxyglucose was used for the selection of glucose de-repressed mutants of T. reesei. Despite this severe culture condition, six 2-deoxyglucose tolerant mutants of ATCC66589 were obtained. Of these mutants, M2 and M3 showed considerably higher pNP-lactoside hydrolyzing activity than the parental strain when cultivated in glucose-supplemented medium, indicating that these mutants are glucose de-repressed variants of T. reesei. In contrast, pNP-lactoside hydrolyzing activity of the mutant was lowered in the avicel medium. Thus, pNP-lactoside hydrolyzing enzyme appears to be involved in glucose de-repression, which is strongly related to the cellulase induction regulating system of T. reesei.Citation21)
cre1 and its related transcriptional genes are known to up or down-regulating cellulase induction depending on the carbohydrate species present in the medium.Citation17) The cellulase productivity of a cre1-deleted mutant of T. reesei is higher than the QM6a parental strain in glucose medium.Citation31) We speculated that glucose de-repression would result from the mutation and reduced expression level of cre1 itself. However, the amino acid sequence and gene expression analyses of the generated mutants showed that cre1 was not involved in glucose de-repression. Hence, the glucose de-repression observed here is not considered to be directly associated with cre1 mutation, but rather with an unknown transcription factor and/or its regulatory system. The lower pNP-hydrolyzing activity of the mutant observed in avicel medium might be also due to this unknown system.
The correlation between cre1 and conidia formation was reported previously by Nakali - SetäläCitation31) In comparison to the wild-type strain, the cre1-deleted mutant and Rut-C30 that contains a truncated cre1 sequence, were shown to form a small number of green conidia and broad mycelia on PDA.Citation31) In the present study, however, the mutants generated by disparity mutagenesis did not form green conidia, but did produce low numbers of yellowish-white conidia. Hence, the observed morphological changes in conidia might not be associated with cre1 mutation, because no changes in the expression or amino acid sequence of CRE1 were observed in our mutants.
In conclusion, we demonstrated that disparity mutagenesis is an advanced method for generating T. reesei mutants that involved the introduction of DNA polymerase δ lacking proofreading activity into cells. To clarify the mechanism of cellulase induction and glucose-repression releasing in T. reesei, it is necessary to determine what regulatory system(s) are targeted in the set of fungal mutants generated in the present study. Using disparity mutagenesis, we therefore expect to develop specific variants of T. reesei with desirable characteristics for basic and practical applications.
Authors contributions
T.K. conceived the study, designed the research and critical reading of the draft, H. I., Y.K., and A.Y. performed research and prepared the manuscript, M.N., H. M. and S.Y. participated in critical genetic experiments, and J.O. participated in interpretation of the data.
Disclosure statement
No potential conflict of interest was reported by the authors.
Funding
This work was supported by Biomass Utilization Project of Ministry of Agriculture, Forestry, and Fisheries of Japan.
Acknowledgments
We thank Dr. Hitoshi Nakayashiki, Faculty of Agriculture of Kobe University, for kindly providing the pSilent-1 vector.
Related Research Data
References
- Farrell AE, Plevin RJ, Turner BT, et al. Ethanol can contribute to energy and environmental goals. Science. 2006;311:506–508.
- Caniliha L, Anuj KC, Thais SSM, et al. Bioconversion of sugarcane biomass into ethnol: an overview about composition, pretreatment methods, detoxification of hydrolysates, enzyme saccharification and ethanol fermentation. J. Biomed. Biotechnol. 2012;doi:10.1155/2012/989572.
- Detroy RW, Lindenfelser LA, Sommer S, et al. Bioconversion of wheat straw to ethanol: chemical modification, enzymatic hydrolysis, and fermentation. Biotechnol. Bioeng. 1981;23:1527–1535.
- Gilbert HJ, Hazlewood GP. Bacterial cellulases and xylanases. J. Gen. Microbiol. 1993;139:187–194.
- Watanabe H, Tokuda G. Cellulolytic systems in insects. Annu. Rev. Entomol. 2010;55:609–632.
- Wood TM, McCrae SI, Bhat KM. The mechanism of fungal cellulase action. Synergism between enzyme components of Penicillium pinophilum cellulase in solubilizing hydrogen bond-ordered cellulose. Biochem. J. 1989;260:37–43.
- Baldrian P, Valásková V. Degradation of cellulose by basidiomycetous fungi. FEMS Microbiol. Rev. 2008;32:501–521.
- Mandels M, Reese ET. Induction of cellulase in Trichoderma viride as influenced by carbon sources and metals. J. Bacteriol. 1957;73:269–278.
- Kawamori M, Morikawa Y, Takasawa S. Induction and production of cellulases by L-sorbose in Trichoderma reesei. Appl. Microbiol. Biotechnol. 1986;24:449–453.
- Cherry JR, Fidantsef AL. Directed evolution of industrial enzymes: an update. Curr. Opin. Biotechnol. 2003;14:438–443.
- Martinez D, Berka RM, Henrissat B, et al. Genome sequencing and analysis of the biomass-degrading fungus Trichoderma reesei (syn. Hypocrea jecorina). Nat. Biotechnol. 2008;26:553–560.
- Peterson R, Nevalainen H. Trichoderma reesei RUT-C30 - thirty years of strain improvement. Microbiology. 2012;158:58–68.
- Kawamori M, Morikawa Y, Shinsha Y, et al. Preparation of mutants resistant to catabolite repression of Trichoderma reesei. Agric. Biol. Chem. 1985;49:2875–2879.
- Porciuncula Jde O, Furukawa T, Mori K, et al. Single nucleotide polymorphism analysis of a Trichoderma reesei hyper-cellulolytic mutant developed in Japan. Biosci. Biotechnol. Biochem. 2013; 77:534–543.
- Seidl V, Gamauf C, Druzhinina IS, et al. The Hypocrea jecorina (Trichoderma reesei) hypercellulolytic mutant RUT C30 lacks a 85 kb (29 gene-encoding) region of the wild-type genome. BMC Genomics. 2008.doi:10.1186/1471-2164-9-327.
- David H, Krogh AM, Roca C, et al. CreA influences the metabolic fluxes of Aspergillus nidulans during growth on glucose and xylose. Microbiology. 2005;151:2209–2221.
- Portnoy T, Margeot A, Linke R, et al. The CRE1 carbon catabolite repressor of the fungus Trichoderma reesei: a master regulator of carbon assimilation. BMC Genomics. 2011;12/269:1–12.
- Kubicek CP, Mikus M, Schuster A, et al. Metabolic engineering strategies for the improvement of cellulase production by Hypocrea jecorina. Biotechnol. Biofuels. 2009.doi:10.1186/1754-6834-2-19.
- Portnoy T, Margeot A, Seidl-Seiboth V, et al. Differential Regulation of the cellulase transcription factors XYR1, AACE2, and ACE1 in Trichoderma reesei strains producing high and low levels of cellulose. Euk. Cell. 2011;10:262–271.
- Mach-Aigner AR, Pucher ME, Steiger MG, et al. Transcriptional regulation of xyr1, encoding the main regulator of the xylanolytic and cellulolytic enzyme system in Hypocrea jecorina. Appl. Environ. Microbiol. 2008;74:6554–6562.
- Ike M, Park JY, Tabuse M, et al. Cellulase production on glucose-based media by the UV-irradiated mutants of Trichoderma reesei. Appl. Microbiol. Biotechnol. 2010;87:2059–2066.
- Wada KN, Doi H, Tanaka S, et al. A neo-Darwinian algorithm: asymmetrical mutations due to semiconservative DNA-type replication promote evolution. Proc. Nat. Acad. Sci. USA. 1993;5:11934–11938.
- Furusawa M, Doi H. Promotion of evolution: disparity in the frequency of strand-specific misreading between the lagging and leading DNA strands enhances disproportionate accumulation of mutations. J. Theor. Biol. 1992;157:127–133.
- Iwaki T, Kawamura A, Ishino Y, et al. Preferential replication-dependent mutagenesis in the lagging DNA strand in Escherichia coli. Mol. Genet. Genomics. 1996;251:657–664.
- Tanabe K, Kondo T, Onodera Y, et al. A conspicuous adaptability to antibiotics in the Escherichia coli mutator strain, dnaQ49. FEMS Microbiol. Lett. 1999;176:191–196.
- Shimoda C, Itadani A, Sugino A, et al. Isolation of thermotolerant mutants by using proofreading-deficient DNA polymerase δ as an effective mutator in Saccharomyces cerevisiae. Genes Genet. Syst. 2006;81:391–397.
- Park EY, Ito Y, Nariyama M, et al. The improvement of riboflavin production in Ashbya gossypii via disparity mutagenesis and DNA microarray analysis. Appl. Microbiol. Biotechnol. 2011;91:1315–1326.
- Nakayashiki H, Hanada S, Nguyen BQ, et al. RNA silencing as a tool for exploring gene function in ascomycete fungi. Fungal Genet. Biol. 2005;42:275–283.
- Mutation points of DNA polymerase used here are as follows:D312A, E314, and L603G (Japan Patent 2011-524752 A 2011.9.8).
- Bergès T, Barreau C. Isolation of uridine auxotrophs from Trichoderma reesei and efficient transformation with the cloned ura3 and ura5 genes. Curr. Genet. 1991;19:359–365.
- Nakari-Setälä T, Paloheimo M, Kallio J, et al. Genetic modification of carbon catabolite repression in Trichoderma reesei for improved protein production. Environ. Microbiol. 2009;75:4853–4860.
- Mach RL, Schindler M, Kubicek CP. Transformation of Trichoderma reesei based on hygromycin B resistance using homologous expression signals. Curr. Genet. 1994;25:567–570.
- Gems D, Johnstone IL, Clutterbuck AJ. An autonomously replicating plasmid transforms Aspergillus nidulans at high frequency. Gene. 1991;98:61–67.
- Kubodera T, Yamashita N, Nishimura A. Transformation of Aspergillus sp. and Trichoderma reesei using the pyrithiamine resistance gene (ptrA) of Aspergillus oryzae. Biosci. Biotechnol. Biochem. 2002;66:404–406.
- Mudge DK, Hoffman CA, Lubinski TJ, et al. Use of a ura5+-lys7+ cassette to construct unmarked gene knock-ins in Schizosaccharomyces pombe. Curr. Genet. 2012;58:59–64.
- Chen W, Guéron M. The inhibition of bovine heart hexokinase by 2-deoxy-d-glucose-6-phosphate: characterization by 31P NMR and metabolic implications. Biochimie. 1992;74:867–873.