
Abstract
Extracellular glutamate concentration is a critical determinant of neuronal cell fate. We recently demonstrated that HT22 murine hippocampal cell viability was reduced by exposure to high concentrations of glutamate, whereas low concentrations promoted cell survival. Extracellular signal-regulated kinase (Erk)1/2 activation by glutamate is important for both glutamate-induced cell death and survival. In this study, we investigated the role of glutamate-induced or hydrogen peroxide (H2O2)-induced Erk1/2 activation in HT22 cell fate determination. Glutamate and H2O2 treatment similarly induced early (<1 h) Erk1/2 phosphorylation regardless of concentration. On the other hand, persistent Erk1/2 phosphorylation (16–24 h) was observed only in the presence of excess glutamate. Only the latter contributed to glutamate-induced cell death, which involved metabolic glutamate receptor 5. Our findings suggest that glutamate concentration modulates two distinct phases of Erk1/2 activation, which can explain the glutamate concentration-dependent determination of HT22 cell fate.
Graphical abstract
Glutamate concentration modulates two distinct phases of Erk1/2 activation, which can explain the glutamate concentration-dependent determination of HT22 cell fate.
Key words:
Glutamate is an excitatory neurotransmitter that is involved in many aspects of brain function, including cognition, memory, and learning.Citation1) However, the release of excessive amounts of glutamate by neurons after brain or spinal cord injury can induce neuronal apoptosis via activation of the caspase pathway.Citation2) This process is known as glutamate toxicity and is mediated by two distinct mechanisms. The first of these is excitotoxicity, in which excess glutamate released into the synaptic space over-stimulates postsynaptic N-methyl-d-aspartate (NMDA) glutamate receptors, causing a massive influx of Ca2+ ionsCitation3) that increases the depolarization of the postsynaptic membrane. This can lead to apoptosis by means of caspase cascade activation.Citation4,5) A second mechanism of glutamate-induced cell death is mediated via an increase in oxidative stress. This occurs when high extracellular glutamate concentrations disrupt the cysteine/glutamate exchange transporter, thereby reducing the level of intracellular glutathione, which normally functions to remove toxic reactive oxygen species (ROS).Citation6,7) The resultant overproduction of ROS by mitochondria, autophagosomes, and lysosome induces oxidative stress and consequently cell death.Citation8)
HT22 murine hippocampal cells do not express ionotropic glutamate receptors; as such, glutamate-dependent excitotoxicity is negligible.Citation9,10) Because of this characteristic, HT22 cells are often used to analyze the effects of glutamate-induced oxidative stress. For example, it has been reported that treatment of HT22 cells with glutamate-induced cell death resulting from ROS production.Citation9) Glutamate-dependent extracellular signal-regulated kinase (Erk)1/2 activation has been implicated in this process; however, the kinetics of Erk1/2 activation are complex. Glutamate was shown to induce a biphasic pattern of Erk1/2 activation that consists of an early phase occurring within 30–60 min that was mediated by metabolic glutamate receptors (mGluRs), and persistent activation that was mediated by oxidative inhibition of Erk1/2 phosphatase 6–8 h after glutamate treatment.Citation11) The latter was found to be directly involved in the accumulation of extracellular H2O2, which induced oxidative cell death.Citation12)
We recently found that administering low (non-toxic) concentrations of glutamate or H2O2 enhanced HT22 cell survival, indicating that the toxic effects of oxidative stress in these cells are dose-dependent.Citation13) Intriguingly, these protective effects were abolished by specific inhibition of mitogen-associated protein kinase kinase (MEK)1/2, an upstream regulator of Erk1/2.Citation13) Thus, Erk1/2 activation by glutamate activated signaling pathways involved in cell death as well as survival; however, the mechanistic basis for biphasic Erk1/2 activation is not fully understood. To address this issue, in this study, we investigated the detailed kinetics of Erk1/2 in the presence of different concentrations of glutamate in HT22 cells. Moreover, to investigate the contribution of oxidative stress on biphasic Erk1/2 activation, we also tested whether difference in kinetics of Erk1/2 activation by different concentrations of H2O2 is observed. Our findings indicate that early Erk1/2 activation is induced by lower concentration of glutamate or H2O2, whereas persistent Erk1/2 activation is observed only in the presence of excess glutamate, and that the latter is modulated by mGluR5 but not mGluR1 and contribute to glutamate-induced HT22 cell death.
Materials and methods
Materials
Dulbecco’s modified Eagle’s medium (DMEM), penicillin/streptomycin, and trypsin-EDTA were purchased from Nacalai Tesque (Kyoto, Japan). Fetal bovine serum (FBS) was from BioWest (Nuaille, France). 1-Aminoindan-1,5-dicarboxylic acid (AIDA) was from Santa Cruz Biotechnology (Santa Cruz, CA, USA). 2-Methyl-6-(phenylethynyl)pyridine (MPEP) hydrochloride was from Abcam (Cambridge, UK). Cell culture equipment was from Corning (Corning, NY, USA). Immobilon-P™ was from Millipore (Bedford, MA, USA). The Western blot detection kit (ECL Prime™) was purchased from GE Healthcare (Rockford, IL, USA). Unless otherwise noted, all chemicals were of the purest grade available from Nacalai Tesque, Sigma-Aldrich (St. Louis, MO, USA), or Wako Pure Chemical Industries (Osaka, Japan).
Cell culture
HT22 mouse hippocampal cells were maintained in DMEM supplemented with 10% FBS, 30 μg/mL penicillin, and 100 μg/mL streptomycin (growth medium) at 37 °C in a 5% CO2 atmosphere. For experiments, HT22 cells were seeded in 6-well plates (Corning) at a density of 1 × 105 cells/well in 2 mL growth medium or in 96-well plates (Corning) at a density of 2 × 103 cells/well in 0.2 mL growth medium.
Cell viability assay
Cell viability was assessed with the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay. HT22 cells were seeded in 96-well plates at a density of 2 × 103 cells/0.2 mL. After 24 h, cells were treated with different concentrations of H2O2 (0–1000 μM) or glutamate (0–10 mM) for 15 h, and cell viability was measured using the MTT Cell Count kit (Nacalai Tesque) according to the manufacturer’s protocol. At least three independent experiments were performed for each condition.
Western blotting
Protein expression and phosphorylation were analyzed by Western blotting. Briefly, cells were seeded in 6-well plates at a density of 1 × 105 cells/well for 24 h and then treated with different concentrations of H2O2 (100 μM or 300 μM) or glutamate (1 or 5 mM) for 0–24 h. Cell lysates were prepared with lysis buffer that consists of 2% sodium dodecyl sulfate (SDS), 1% 2-mercaptoethanol, 10% glycerol, 0.0033% bromophenol blue, and 50 mM Tris–HCl (pH 6.8). Protein concentration was determined using the Pierce bicinchoninic acid protein assay (Thermo Scientific). Equal amounts of protein were resolved by 10% SDS–polyacrylamide gel electrophoresis (bis:acrylamide, 1:30) and proteins were transferred to an Immobilon-PTM polyvinylidene difluoride membrane, which was blocked for 30 min in Tris-buffered saline containing 3% bovine serum albumin and 0.1% Tween-20. For protein detection, the membrane was incubated for 1 h in 1:1000 dilutions of primary antibodies against phosphorylated Erk1/2 and total Erk1/2 (Cell Signaling Technology, Danvers, MA, USA), followed by incubation in horseradish peroxidase-conjugated rabbit immunoglobulin G diluted 1:5000 and detection using ECL PrimeTM. At least three independent experiments were performed for each condition. The intensity of protein bands was quantified using ImageJ software (National Institutes of Health, Bethesda, MD, USA).
Statistical analysis
Group means were compared using one-way analysis of variance or Student’s t-test. Differences for which the P value was < 0.05 were considered statistically significant.
Results and discussion
Excess glutamate induces early and persistent activation of Erk1/2
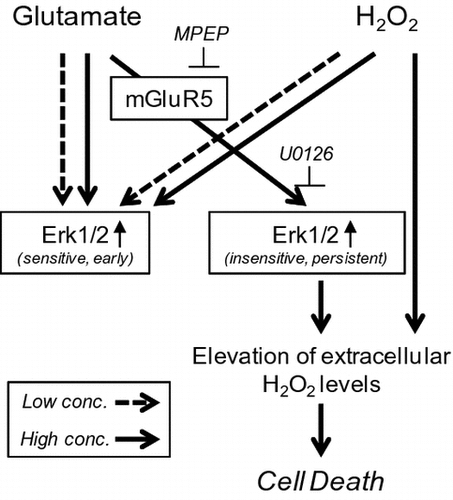
We first investigated the toxicity of glutamate and oxygen radicals by evaluating HT22 cell viability upon treatment with various concentrations of glutamate and H2O2 with the MTT assay. Consistent with our previous findings,Citation13) glutamate and H2O2 similarly induced cell death in a concentration-dependent manner (Fig. (A) and (B)). In particular, viability was markedly reduced by exposure to concentrations of 5 mM glutamate or 250 μM H2O2.
Fig. 1. Glutamate and H2O2 treatment similarly induced cell death via temporally distinct Erk1/2 activation patterns.
It has been previously reported that glutamate administration induced biphasic Erk1/2 activation which consists of early and persistent phases.Citation11) To elucidate the detailed kinetics of Erk1/2 activation, we evaluated time-dependent changes in Erk1/2 phosphorylation following the administration of high or low glutamate or H2O2 concentrations. In order to analyze long-term effects of glutamate or H2O2 on Erk1/2 phosphorylation, we used 5 mM glutamate or 300 μM H2O2 as high concentrations that were minimum effective concentrations required to induce cell death. Treatment of HT22 cells with glutamate or H2O2 did not change the levels of total Erk1/2 protein (Fig. ). Toxic levels of glutamate (5 mM) induced early (1 h) and persistent (16–24 h) Erk1/2 activation (Fig. (C)). On the other hand, early activation was observed but the persistent phase almost completely disappeared in the presence of 1 mM glutamate (Fig. (D)). Erk1/2 phosphorylation was decreased relative to basal states after 1 mM glutamate treatment for 24 h. Time-dependent changes in Erk1/2 phosphorylation in response to non-toxic or toxic levels of H2O2 treatment were similar to those induced by low concentrations of glutamate (Fig. (E) and (F)). Early Erk1/2 phosphorylation was observed under all treatment conditions (toxic and non-toxic concentrations of either glutamate or H2O2); however, persistent Erk1/2 activation was only observed upon exposure to a toxic concentration of glutamate. In agreement with these findings, many studies have demonstrated that the kinetics of Erk1/2 activation are modulated by different types of extracellular stimulus; for instance, in PC12 cells, nerve growth factor (NGF) and epidermal growth factor induce rapid Erk1/2 activation that stimulates cell proliferation, whereas NGF and fibroblast growth factor induce persistent Erk1/2 activation that triggers differentiation.Citation14) Interestingly, both non-toxic and toxic levels of H2O2 effectively stimulated early Erk1/2 activation. It was also reported that phosphatidylinositol 3-kinase (PI3 K) plays a role in H2O2-dependent Erk1/2 phosphorylation in SH-SY5Y human neuroblastoma cellsCitation15); however, in HT22 cells, we did not observe PI3 K activation—as assessed by Akt phosphorylation—upon H2O2 treatment (data not shown).
Persistent Erk1/2 activation reduces cell viability
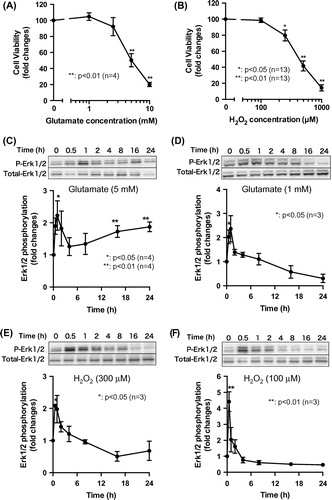
We investigated the role of biphasic Erk1/2 activation in HT22 cell viability. We first examined whether glutamate-induced cell death could be blocked by inhibiting Erk1/2 signaling. Treatment with the MEK1/2 inhibitor U0126 completely abolished glutamate-induced cytotoxicity (Fig. (A)), although it had no effect on H2O2-induced cell death (Fig. (B)). These results are consistent with an earlier finding that Erk1/2 activation in the presence of glutamate resulted in the accumulation of extracellular H2O2, which decreased cell viability.Citation12) To determine whether early or persistent Erk1/2 activation is responsible for glutamate-induced cell death, we treated cells with 10 μM U0126 at different time points after administering toxic levels of glutamate and then evaluated cell viability with the MTT assay. Viability was reduced by 5 mM glutamate; however, this effect was abrogated when U0126 was added to the cells within 6 h of glutamate treatment (Fig. (C)). Glutamate-induced cell death was blocked even when U0126 was added 3 or 6 h after glutamate administration, at which time early Erk1/2 activation would not be affected. Although U0126 has been widely used as a selective inhibitor for MEK1/2,Citation16) Son et al. suggested this inhibitor also affects p38 MAP kinase activity, either directly or indirectly.Citation17) However, our preliminary experiment suggested that the apparent effects of 10 μM SB203580, a specific inhibitor for p38 MAP kinase, on glutamate-induced cell death were not observed (data not shown). Consequently, we speculate that persistent but not early Erk1/2 activation in response to toxic levels of glutamate is important for inducing HT22 cell death.
Fig. 2. Effect of biphasic Erk1/2 activation on glutamate-induced cell death.
Our previous work showed that non-toxic levels of glutamate or H2O2 protected HT22 cells via an Erk1/2 activation-dependent mechanism.Citation13) We therefore speculated that early Erk1/2 activation by glutamate or H2O2 may have a protective function with respect to HT22 cells. Consistent with this notion, a previous study showed that early glutamate-induced Erk1/2 activation restores intracellular glutathione levels reduced by glutamate treatment.Citation11) H2O2 also induced acute Erk1/2 activation in SH-SY5Y cells; however, in this case, treatment with the MEK1 inhibitor PD 98059 suppressed H2O2-induced cell death.Citation15) Zinc is known to have a similar biphasic effect on Erk1/2 phosphorylation, but cell viability was only dependent on early Erk1/2 activation.Citation18) These findings suggest that the duration of Erk1/2 activation as well as other factors such as the subcellular distribution of related proteins is important for cell fate determination.
mGluR5 mediates persistent Erk1/2 activation
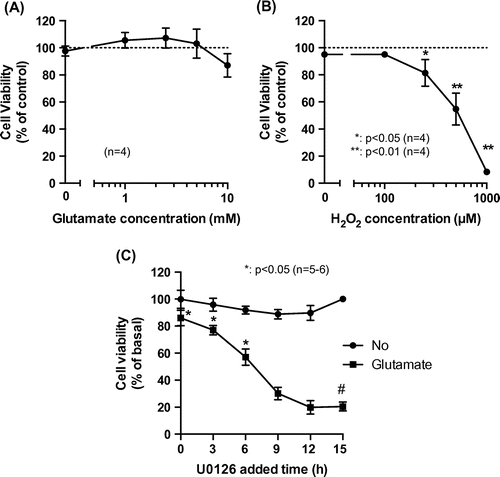
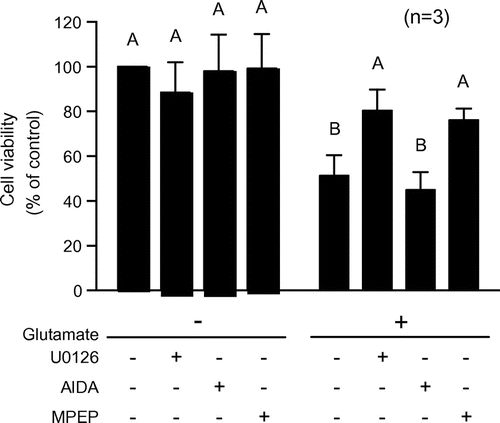
Eight mGluR subtypes (mGluR1–8) have been identified to date, which are classified into three groups.Citation19) Group 1 mGluRs (mGluR1 and 5) are expressed in HT22 cells.Citation20) The activation of either of these receptors increases inositol triphosphate (IP3) levels via phospholipase β activation.Citation21) However, the effects of mGluR activation on the biphasic glutamate-induced activation of Erk1/2 and consequent cell death are not well understood. We verified whether treatment with AIDA and MPEP—antagonists of mGluR1 and mGluR5, respectively—affected Erk1/2 phosphorylation state in HT22 cells and found that 50 μM AIDA and 200 μM MPEP induced early Erk1/2 phosphorylation (Fig. (A) and (B)), indicating that mGluR may negatively regulate Erk1/2 phosphorylation.
Fig. 3. Role of mGluR on biphasic Erk1/2 activation and glutamate-induced cell death.
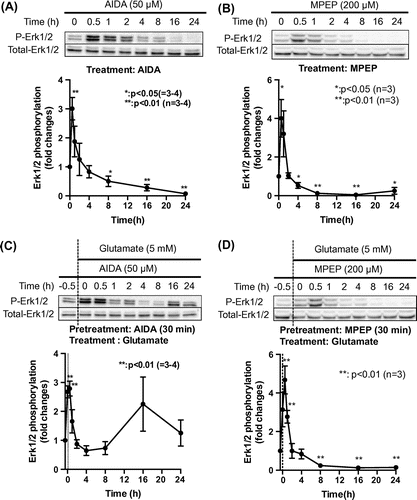
To determine whether mGluRs are involved in both phases of Erk1/2 phosphorylation induced by subtoxic levels of glutamate, we preincubated HT22 cells with 50 μM AIDA or 200 μM MPEP, followed by treatment with 5 mM glutamate for 0.5–24 h. The persistent glutamate-induced activation of Erk1/2 was unaffected by AIDA, but was markedly reduced by MPEP pretreatment (Fig. (C) and (D)). We also examined whether mGluRs are involved in glutamate-induced cell death. Although 50 μM AIDA had no effect, the decrease in cell viability resulting from glutamate treatment was mitigated by 200 μM MPEP as well as by 10 μM U0126 (Fig. ). These results suggest that mGluR5 but not mGluR1 is involved in the persistent activation of Erk1/2 and cell death induced by subtoxic levels of glutamate.
Fig. 4. Role of mGluR in glutamate-induced cell death.
Several reports indicate that pharmacological blockade of mGluR5 has neuroprotective effects. For example, MPEP protects against glutamate-induced injury in mouse and rat cortical neurons,Citation2,22,23) and mGluR5 gene transfer into the hippocampal CA1 region of 2XTgα-syn/amyloid precursor protein mice resulted in neurodegeneration.Citation24) Given that mGluR5 is highly expressed in the cortex and hippocampus,Citation25) we speculate that mGluR5-mediated glutamate toxicity plays a significant role in cognitive dysfunction, for instance in Alzheimer’s disease.Citation26) On the other hand, since mGluR5 expression is required for NMDA receptor-mediated neuronal plasticity, appropriate mGluR5 expression/activation is critical. It was recently shown that treatment with the selective mGluR5 agonist (±)-2-chloro-5-hydroxyphenylglycine protects cortical neurons in traumatic brain injury via Erk and Akt signaling pathways.Citation27) It was also demonstrated that Group 1 mGluR antagonists potentiate glutamate toxicity.Citation20) Thus, whether mGluR5 activation promotes the maintenance of normal brain function may depend on glutamate levels. To this end, this study provides evidence that mGluR5-dependent cell death signaling pathways involving the persistent activation of Erk1/2 are induced by subtoxic levels of glutamate in HT22 cells.
Another intriguing finding of this study is that the mGluR5 antagonist MPEP, but not the mGluR1 antagonist AIDA, abolished glutamate-induced cytotoxicity. The activation of Group 1 mGluRs triggers similar intracellular signaling mechanisms, including the IP3/Ca2+ pathway.Citation21) However, distinct Ca2+ kinetics associated with mGluR1 and 5 have also been reportedCitation28–30); for instance, mGluR1 activation was shown to stimulate Ca2+ entry via transient receptor potential channels, whereas mGluR5 activation mobilized intracellular Ca2+ stores. Further studies are required to clarify the different effects of mGluR1 and 5 on glutamate-induced cell death.
Conclusion
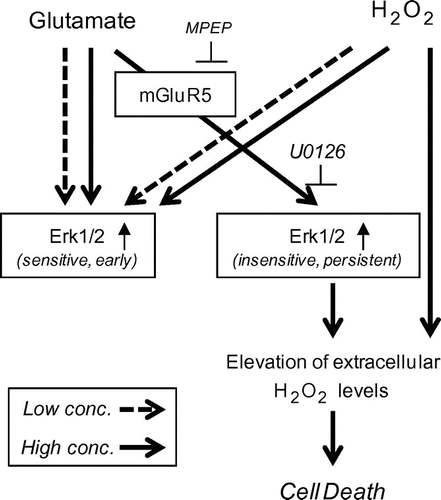
Our findings indicate that glutamate concentration modulates two distinct phases of glutamate-induced Erk1/2 activation. We also found that persistent Erk1/2 activation occurs via mGluR5 and that inhibiting this receptor was sufficient to block glutamate-induced cytotoxicity (Fig. ). This biphasic activation of Erk1/2 may explain the dependence of cell fate on glutamate levels.
Fig. 5. Model for the mechanism of cell fate determination by glutamate in HT22 hippocampal cells.
Author contributions
K.S. and T.N. designed the study. K.S., Y.Y., and Y.A. carried out the experiments. K.S. and T.N. wrote the paper. All authors read and approved the manuscript.
Funding
This work was supported by a Grant-in-Aid for Scientific Research from the Japan Society for the Promotion of Science [grant number S-23228004].
Acknowledgments
The authors thank Dr. Gabriel Livera (Université Paris Diderot Paris 7, France), Dr. Céline Méhats (Institut Cochin, France), and Dr. Masugi Nishihara (The University of Tokyo, Japan) for many helpful comments.
Disclosure statement
No potential conflict of interest was reported by the authors.
Notes
Abbreviations: AIDA, 1-aminoindan-1,5-dicarboxylic acid; DMEM, Dulbecco’s modified Eagle’s medium; Erk, extracellular signal-regulated kinase; mGluR, metabolic glutamate receptors; MPEP, 2-methyl-6-(phenylethynyl)pyridine; MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5 diphenyl tetrazolium bromide; NGF, nerve growth factor; NMDA, N-methyl-d-aspartate; PI3 K, phosphatidylinositol 3-kinase; ROS, reactive oxygen species; SDS, sodium dodecyl sulfate.
Related Research Data
References
- Meldrum BS. Glutamate as a neurotransmitter in the brain: review of physiology and pathology. J. Nutr. 2000;130:1007S–1015S.
- Kritis AA, Stamoula EG, Paniskaki KA, et al. Researching glutamate-induced cytotoxicity in different cell lines: a comparative/collective analysis/study. Front Cell Neurosci. 2015;9:91.
- Bloom FE, editor. Neuroscience: from the molecular to the cognitive. Amsterdam: Elsevier; 1994. p. 39–51.
- Wang Y, Qin ZH. Molecular and cellular mechanisms of excitotoxic neuronal death. Apoptosis. 2010;15:1382–1402.10.1007/s10495-010-0481-0
- Lai TW, Zhang S, Wang YT. Excitotoxicity and stroke: identifying novel targets for neuroprotection. Prog. Neurobiol. 2014;115:157–188.10.1016/j.pneurobio.2013.11.006
- Tyurin VA, Tyurina YY, Quinn PJ, et al. Glutamate-induced cytotoxicity in PC12 pheochromocytoma cells: role of oxidation of phospholipids, glutathione and protein sulfhydryls revealed by bcl-2 transfection. Brain Res. Mol. Brain Res. 1998;60:270–281.10.1016/S0169-328X(98)00181-8
- Penugonda S, Mare S, Lutz P, et al. Potentiation of lead-induced cell death in PC12 cells by glutamate: Protection by N-acetylcysteine amide (NACA), a novel thiol antioxidant. Toxicol. Appl. Pharmacol. 2006;216:197–205.10.1016/j.taap.2006.05.002
- Kubota C, Torii S, Hou N, et al. Constitutive reactive oxygen species generation from autophagosome/lysosome in neuronal oxidative toxicity. J. Biol. Chem. 2010;285:667–674.10.1074/jbc.M109.053058
- Murphy TH, Miyamoto M, Sastre A, et al. Glutamate toxicity in a neuronal cell line involves inhibition of cystine transport leading to oxidative stress. Neuron. 1989;2:1547–1558.10.1016/0896-6273(89)90043-3
- Murphy TH, Schnaar RL, Coyle JT. Immature cortical neurons are uniquely sensitive to glutamate toxicity by inhibition of cystine uptake. FASEB J. 1990;4:1624–1633.
- Luo Y, DeFranco DB. Opposing roles for ERK1/2 in neuronal oxidative toxicity: distinct mechanisms of ERK1/2 action at early versus late phases of oxidative stress. J. Biol. Chem. 2006;281:16436–16442.10.1074/jbc.M512430200
- Ha JS, Lim HM, Park SS. Extracellular hydrogen peroxide contributes to oxidative glutamate toxicity. Brain Res. 2010;1359:291–297.10.1016/j.brainres.2010.08.086
- Sato K, Yamanaka Y, Ishii M, et al. Dual cell protective mechanisms activated by differing levels of oxidative stress in HT22 murine hippocampal cells. Biosci. Biotechnol. Biochem. 2014;78:1495–1503.10.1080/09168451.2014.936343
- Marshall CJ. Specificity of receptor tyrosine kinase signaling: transient versus sustained extracellular signal-regulated kinase activation. Cell. 1995;80:179–185.10.1016/0092-8674(95)90401-8
- Ruffels J, Griffin M, Dickenson JM. Activation of ERK1/2, JNK and PKB by hydrogen peroxide in human SH-SY5Y neuroblastoma cells: role of ERK1/2 in H2O2-induced cell death. Eur. J. Pharmacol. 2004;483:163–173.10.1016/j.ejphar.2003.10.032
- Favata MF, Horiuchi KY, Manos EJ, et al. Identification of a novel inhibitor of mitogen-activated protein kinase kinase. J. Biol. Chem. 1998;273:18623–18632.10.1074/jbc.273.29.18623
- Son MH, Kang KW, Lee CH, et al. Potentiation of cadmium-induced cytotoxicity by sulfur amino acid deprivation through activation of extracellular signal-regulated kinase1/2 (ERK1/2) in conjunction with p38 kinase or c-jun N-terminal kinase (JNK). Biochem. Pharmacol. 2001;62:1379–1390.10.1016/S0006-2952(01)00780-8
- He K, Aizenman E. ERK signaling leads to mitochondrial dysfunction in extracellular zinc-induced neurotoxicity. J. Neurochem. 2010;114:452–461.10.1111/jnc.2010.114.issue-2
- Hollmann M, Heinemann S. Cloned glutamate receptors. Annu. Rev. Neurosci. 1994;17:31–108.10.1146/annurev.ne.17.030194.000335
- Sagara Y, Schubert D. The activation of metabotropic glutamate receptors protects nerve cells from oxidative stress. J. Neurosci. 1998;18:6662–6671.
- Lum EN, Campbell RR, Rostock C, et al. mGluR1 within the nucleus accumbens regulates alcohol intake in mice under limited-access conditions. Neuropharmacology. 2014;79:679–687.10.1016/j.neuropharm.2014.01.024
- Chen T, Cao L, Dong W, et al. Protective effects of mGluR5 positive modulators against traumatic neuronal injury through PKC-dependent activation of MEK/ERK pathway. Neurochem. Res. 2012;37:983–990.10.1007/s11064-011-0691-z
- O’Leary DM, Movsesyan V, Vicini S, et al. Selective mGluR5 antagonists MPEP and SIB-1893 decrease NMDA or glutamate-mediated neuronal toxicity through actions that reflect NMDA receptor antagonism. Br. J. Pharmacol. 2000;131:1429–1437.10.1038/sj.bjp.0703715
- Overk CR, Cartier A, Shaked G, et al. Hippocampal neuronal cells that accumulate α-synuclein fragments are more vulnerable to Aβ oligomer toxicity via mGluR5–implications for dementia with Lewy bodies. Mol. Neurodegener. 2014;9:18.10.1186/1750-1326-9-18
- D’Antoni S, Berretta A, Bonaccorso CM, et al. Metabotropic glutamate receptors in glial cells. Neurochem. Res. 2008;33:2436–2443.10.1007/s11064-008-9694-9
- Kumar A, Dhull DK, Mishra PS. Therapeutic potential of mGluR5 targeting in Alzheimer’s disease. Front Neurosci. 2015;9:215.
- Lea PM 4th, Movsesyan VA, Faden AI. Neuroprotective activity of the mGluR5 antagonists MPEP and MTEP against acute excitotoxicity differs and does not reflect actions at mGluR5 receptors. Br. J. Pharmacol. 2005;145:527–534.
- Anwyl R. Metabotropic glutamate receptor-dependent long-term potentiation. Neuropharmacology. 2009;56:735–740.10.1016/j.neuropharm.2009.01.002
- Kawabata S, Tsutsumi R, Kohara A, et al. Control of calcium oscillations by phosphorylation of metabotropic glutamate receptors. Nature. 1996;383:89–92.10.1038/383089a0
- Topolnik L, Azzi M, Morin F, et al. mGluR1/5 subtype-specific calcium signalling and induction of long-term potentiation in rat hippocampal oriens/alveus interneurones. J. Physiol. 2006;575:115–131.10.1113/jphysiol.2006.112896