
Abstract
Marine glycoside hydrolases hold enormous potential due to their habitat-related characteristics such as salt tolerance, barophilicity, and cold tolerance. We purified an α-glucosidase (PYG) from the midgut gland of the Japanese scallop (Patinopecten yessoensis) and found that this enzyme has unique characteristics. The use of acarbose affinity chromatography during the purification was particularly effective, increasing the specific activity 570-fold. PYG is an interesting chloride ion-dependent enzyme. Chloride ion causes distinctive changes in its enzymatic properties, increasing its hydrolysis rate, changing the pH profile of its enzyme activity, shifting the range of its pH stability to the alkaline region, and raising its optimal temperature from 37 to 55 °C. Furthermore, chloride ion altered PYG’s substrate specificity. PYG exhibited the highest Vmax/Km value toward maltooctaose in the absence of chloride ion and toward maltotriose in the presence of chloride ion.
Graphical abstract
An α-glucosidase was purified from the midgut gland of the scallop. The acarbose affinity chromatography was effective for the purification. The enzyme showed chloride ion dependency.
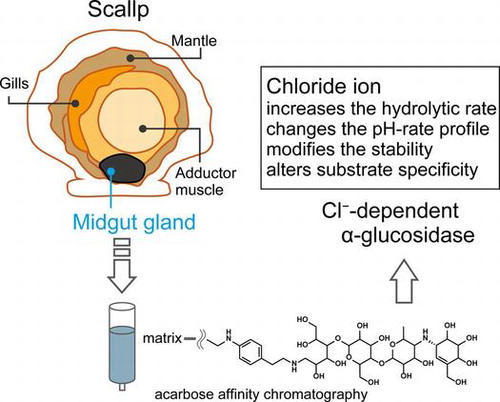
Marine organisms are attracting attention as a biological resource because they have adapted to the diversity of the oceanic environment, such as high salinity, high pressure, and low temperature, which allows enzymes from marine sources to hold enormous potential.Citation1,2) Knowledge of these enzymes is certainly beneficial from a biotechnological perspective. Most of the useful materials obtained from marine resources thus far have been derived from microbesCitation1); however, animals, plants, and algae may have hidden potential.
Marine glycoside hydrolases can have just as much potential as any other enzymes. However, studies of the glycoside hydrolases of marine organisms have been inadequate compared with the rigorous studies of the enzymes from land-dwelling creatures. Shelled invertebrates in the marine environment have been shown to possess various polysaccharide-degrading enzymes, such as alginate lyase, cellulase, and mannanase, in their digestive fluids.Citation3–8) This is because they feed on seaweed and need the ability to digest the abundant polysaccharides, such as alginic acid, cellulose, laminarin, and mannan present in seaweed. Thus, studies on alginate lyase, cellulase, laminarinase, and mannanase can often be found in the literature, whereas there is little information on enzymes that digest α-glucans.Citation9)
α-Glucosidase (α-glucoside glucohydrolase, EC 3.2.1.20) catalyzes the liberation of α-glucose from the non-reducing termini of substrates such as maltooligosaccharides and α-glucan. In mammals, α-glucosidase, existing in the small intestine as sucrase–isomaltase and the maltase–glucoamylase complex, is a terminal enzyme in the degradation of starch and its related compounds. These carbohydrates are digested into maltooligosaccharides by salivary and pancreatic α-amylases, and maltooligosaccharides are digested into glucose by α-glucosidases localized to the small intestine. Thus, α-glucosidase is one of the most important enzymes of α-glucan utilization in mammals.
The digestive glands of several bivalves, including Pecten maximus, Placopecten magellanicus, and Perna viridis, possess α-glucosidase as well as other carbohydrate metabolic enzymes such as amylase, laminarinase, and cellulase.Citation10–12) Accordingly, α-glucosidase likely plays an important role in the degradation of α-glucan in bivalves. However, to date, there has been no detailed examination of α-glucosidases from bivalves. In this study, we purified an α-glucosidase from the midgut gland of the Japanese scallop Patinopecten yessoensis (PYG) and characterized its enzymatic properties. Kinetic studies indicated that PYG is a chloride ion-dependent enzyme. Chloride ion significantly increases the catalytic activity and alters the substrate specificity of PYG.
Materials and methods
Materials
Midgut glands of scallop (P. yessoensis) were collected in Mutsu Bay of Aomori prefecture, Japan, in 2005. Maltose, maltotetraose, maltopentaose, maltohexaose, maltoheptaose, and maltooctaose were kindly gifted by Nihon Shokuhin Kako (Tokyo, Japan). Maltotriose, kojibiose, and nigerose were purchased from Wako Pure Chemical Industries (Osaka, Japan). Other chemicals used were analytical grade and obtained from Nacalai Tesque (Kyoto, Japan) if not otherwise specified.
Enzyme activity assay
Reaction mixtures containing 0.2% (w/v) maltose, 60 mM sodium acetate buffer (pH 5.0), 0.03% (v/v) Triton X-100, and 300 mM NaCl were incubated at 37 °C. Reactions were terminated by the addition of two volumes of 2 M Tris-HCl buffer (pH 7.0). Glucose liberated from the substrate was quantified using the glucose oxidase-peroxidase methodCitation13) performed with the Wako Glucose CII Test (Wako Pure Chemical Industries). One unit of maltase activity was defined as the amount of enzyme hydrolyzing 1 μmol of maltose per min under the conditions described above.
Preparation of acarbose Sepharose 4 fast flow
Acarbose (0.1 mmol) (Sigma-Aldrich, St. Louis, MO, USA) was stirred overnight with 3.5 mmol 2-(4-aminophenyl)ethylamine (Tokyo Chemical Industry, Tokyo, Japan). The resulting aminoethylated acarbose was reduced with sodium borohydride (0.63 mmol) in ethanol (1.5 mL) for 5 h at room temperature. The mixture was titrated with 1 M acetic acid to pH 5.6 and then applied to a Sephadex G-25 column equilibrated with 1 M sodium acetate buffer (pH 5.0). Upon isocratic elution with 1 M sodium acetate buffer (pH 5.0), the fractions containing modified acarbose were identified by using thin-layer chromatography on silica gel 60 (nitromethane/1-propanol/water, 4/10/3). The plates were visualized by charring with a methanolic solution of sulfuric acid containing 0.02% α-naphthol. The modified acarbose was coupled with NHS-activated Sepharose 4 Fast Flow (GE Healthcare, Buckinghamshire, England) by following the supplier’s instruction.
Purification of PYG
Midgut glands (52 g) were stirred in 10 mM sodium acetate buffer (pH 5.0) containing 5 mM MgCl2 and 500 mM NaCl overnight at 4 °C to extract protein. The supernatant (crude enzyme) was recovered by centrifugation at 18,700 × g for 20 min at 4 °C. Ammonium sulfate was added to the crude enzyme until its concentration reached 70% saturation, and the suspension was left overnight at 4 °C. The precipitate was collected by centrifugation at 18,700 × g for 20 min. The precipitate was dissolved in 10 mM sodium acetate buffer (pH 5.0) containing 5 mM MgCl2 and 30% saturated ammonium sulfate and then loaded onto a TOYOPEAL Butyl-650 M (Tosoh, Tokyo, Japan) column equilibrated with the same buffer. Absorbed proteins were eluted with a linear gradient of 30%–0% saturated ammonium sulfate in 10 mM sodium acetate buffer (pH 5.0) containing 5 mM MgCl2. The active fractions were pooled and subjected to acarbose affinity chromatography at room temperature. An acarbose Sepharose 4 fast flow column (1.5 cm × 11 cm) was equilibrated with 10 mM sodium acetate buffer (pH 5.0) containing 5 mM MgCl2 and 200 mM NaCl. Bound enzyme was eluted with 100 mL of 4 M Tris-HCl buffer (pH 7.0). The eluted fractions were pooled and dialyzed against 10 mM sodium acetate buffer (pH 5.0) containing 5 mM MgCl2. The dialysate was concentrated using Amicon Ultracel-30 K Ultra Centrifuge Filters (Merck Millipore, Billerica, MA, USA). The concentrated solution was loaded onto a Sephacryl S-200HR (GE Healthcare) column (1.6 cm × 55 cm) equilibrated with 10 mM sodium acetate buffer (pH 5.0) containing 5.0 mM MgCl2 and 200 mM NaCl. The active fractions were pooled and dialyzed against a 10 mM sodium acetate buffer (pH 5.0). Purification procedures, except acarbose affinity chromatography, were performed at 4 °C. The protein concentration of the purified enzyme was calculated from its absorbance at 280 nm in a 1-cm cuvette using an extinction coefficient of (1.0 g/L)−1 cm−1.
Polyacrylamide gel electrophoresis
SDS-PAGE and Blue Native-polyacrylamide gel electrophoresis (BN-PAGE) were performed using a Mini-Protean III cell apparatus (Bio-Rad, Richmond, CA, USA). SDS-PAGE was performed as described by LaemmliCitation14) and BN-PAGE as described by Schagger and von Jagow.Citation15) Protein was stained with Coomassie Brilliant Blue (CBB) R-250 using a Rapid CBB staining kit (Kanto Chemical, Tokyo, Japan). Mark12 unstained protein standards and Native mark unstained protein standard (Invitrogen, Life Technologies, Carlsbad, CA, USA) were used as molecular size markers in SDS-PAGE and BN-PAGE, respectively. Activity staining after BN-PAGE was done by incubating the gel in 100 mM sodium acetate buffer (pH 5.0) containing 1 mg/ml 4-methylumbelliferyl α-d-glucopyranoside (Sigma-Aldrich) and 500 mM NaCl at 37 °C for 10 min. Fluorescence from the 4-methylumbelliferone produced by hydrolysis was detected using a UV transilluminator.
In-gel digestion with trypsin
Purified PYG (15 μg) was subjected to SDS-PAGE, and a CBB-stained protein band was excised. The gel piece was washed three times with 100 μL of 50 mM NH4HCO3 in 50% (v/v) acetonitrile for 20 min at 37 °C and then dried using a vacuum centrifugation. The dried gel was swollen in 5 μL trypsin solution (1 mg/mL) containing 1 mM HCl and 20 mM CaCl2 at 4 °C for 10 min. After reswelling the gel, 15 μL of 50 mM NH4HCO3 was added and digestion was continued for 24 h at 37 °C. Peptides were extracted three times in 100 μL of 60% (v/v) acetonitrile, lyophilized, and redissolved in 0.1% trifluoroacetic acid containing 50% (v/v) acetonitrile.
Matrix-assisted laser desorption ionization time-of-flight mass spectrometric analysis
The tryptic peptides were desalted using a Zip-Tip C-18 (Merck Millipore, Billerica, MA, USA) according to the manufacturer’s instructions. The eluate (2 μL) was mixed with 0.8 μⅬ of matrix solution [10 mg/mⅬ α-cyano-4-hydroxycinnamic acid (Sigma-Aldrich) in 0.1% trifluoroacetic acid/50% (v/v) acetonitrile], and dried at room temperature on the sample plate. Mass spectra were recorded with a matrix-assisted laser desorption ionization time-of-flight mass spectrometer (MALDI-TOF-MS)(Voyager-DE STR, Applied Biosystems, Foster City, CA, USA) with delayed ion extraction technology. All mass spectra were obtained in reflector mode and calibrated with GRAMS software (Applied Biosystems) using angiotensin II as an external mass standard. We used the MALDI-TOF-MS in the facilities of the Research Faculty of Agriculture, Hokkaido University.
Effect of various salts on enzyme activity
The enzyme reactions were performed in 60 mM sodium acetate buffer (pH 5.0) containing 0.03% (v/v) Triton X-100, 0.2% maltose, and diluted enzyme (102 ng/mL) at 37 °C while varying the concentrations of various salts (0 mM–400 mM MgSO4, NaCl, KCl, MgCl2 or CaCl2). The liberated glucose was measured as described above.
Effects of pH and temperature
The effect of pH on maltose hydrolysis activity was determined using the standard assay conditions with the exception that 60 mM sodium acetate (pH 3.6–pH 5.6), 60 mM sodium phosphate (pH 5.4–pH 8.2), or Britton–Robinson (pH 2.2–pH 6.9) buffers were used. Enzyme concentrations used were 406 ng/mL (in the absence of NaCl) and 81.2 ng/mL (in the presence of 300 mM NaCl). For pH–stability experiments, the purified enzyme (203 μg/mL) diluted with 40 mM Briton–Robinson buffer (pH 2.0–pH 11.0) containing 0.05% (v/v) Triton X-100 was kept at 4 °C for 24 h in either the absence or presence of 300 mM NaCl. The remaining activity was measured under the standard assay conditions except that the remaining activity of samples treated in the absence of NaCl was measured using the NaCl-free buffer. The optimal temperatures for the hydrolytic activity of the enzyme in the absence of NaCl or in the presence of 300 mM NaCl were measured under the standard assay condition except that the temperature was varied from 25 to 65 °C. Enzyme concentrations used were 406 ng/mL (in the absence of NaCl) and 102 ng/mL (in the presence of 300 mM NaCl). For thermal stability experiments, the purified enzyme (508 ng/mL in the absence of NaCl and 102 ng/mL in the presence of 300 mM NaCl) in 10 mM sodium acetate buffer (pH 5.0) containing 0.05% (v/v) Triton X-100 was incubated at 30 to 65 °C for 15 min. The residual activity was assayed under the standard assay conditions except that the remaining activity treated without NaCl was measured using NaCl-free buffer.
Hydrolysis of maltooligosaccharides and various disaccharides
The initial velocities of the hydrolysis reactions performed using 2 mM concentrations of various substrates (maltose, maltotriose, maltotetraose, maltopentaose, maltohexaose, maltoheptaose, maltooctaose, trehalose, kojibiose, and nigerose) were measured in the absence or presence of 300 mM NaCl under standard assay conditions. Enzyme concentrations used were 0.102 to 40.6 μg/mL. The initial velocity was expressed as the number of μmol of glucose released from the non-reducing end of the substrate per min per mg protein. When determining the hydrolysis rate for disaccharides, the concentration of liberated glucose was divided by two.
Kinetic parameters
The kinetic parameters (Vmax and Km) for kojibiose, nigerose, and maltooligosaccharides in the absence or presence of 300 mM NaCl were calculated using the initial velocities of reactions containing six to eight different substrate concentrations (0.2–32 mM). These initial rates were fitted to the Michaelis–Menten equation using the computer program Kaleidagraph 3.6. The initial velocities were measured in the standard assay conditions except for the NaCl concentration. The enzyme concentration used was 127 ng/mL.
Results and discussion
Purification of PYG
PYG was purified using a combination of ammonium sulfate precipitation and several column chromatographic steps, including acarbose affinity chromatography, which was performed using acarbose covalently cross-linked to a Sepharose 4 fast flow column. The enzyme preparation was subjected to electrophoresis to confirm the purity of the enzyme. The enzyme migrated as a single protein band with a molecular mass of about 66 kDa on BN-PAGE, stained by CBB R-250 or visualized by activity staining (Fig. (A)). However, on SDS-PAGE, the enzyme separated into two components (Fig. (B)). The molecular masses of the major and minor components were 51 and 40 kDa, respectively. The results of PAGEs imply that these components do not dimerize, and exist as a monomer. The components could not be separated using further purification procedures including ion-exchange, gel filtration, and hydrophobic interaction chromatographies. MALDI-TOF-MS of the mixture of tryptic fragments generated by in-gel digestion of the 51- and 40-kDa components indicated that nine peptides derived from each component were identical (Fig. ). It was likely that the 51- and 40-kDa components were derived from the same peptide, which underwent different proteolysis. The midgut gland contains proteases since it is a digestive tract with functions similar to those of the liver and pancreas in mammals.Citation16) It is possible that PYG undergoes a proteolytic process that splits it into two components. The enzyme preparation was regarded as the purified enzyme (PYG) and was thus used for further analysis. Peptide masses determined by MALDI-TOF-MS were used in database searches by Mascot; however, no glycoside hydrolase was identified as a significant match. The purification process yielded 0.427 mg of purified PYG, whose specific activity was 122 U/mg, from 52 g of midgut glands with a yield of 39.8%. The PYG purification procedures are summarized in Table . Acarbose affinity chromatography was the most effective step, increasing the specific activity 570-fold.
Fig. 1. Electrophoretic analysis of PYG.
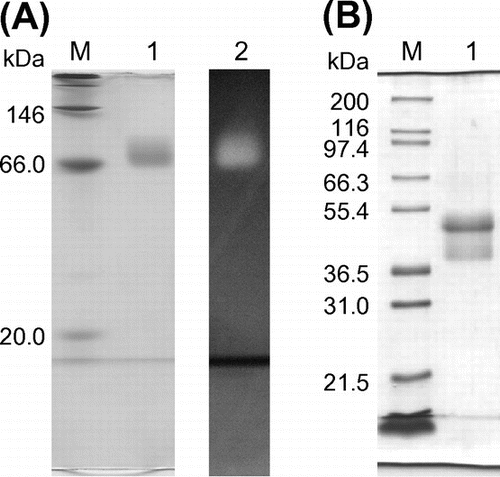
Fig. 2. MALDI-TOF-MS analysis of PYG tryptic peptides.
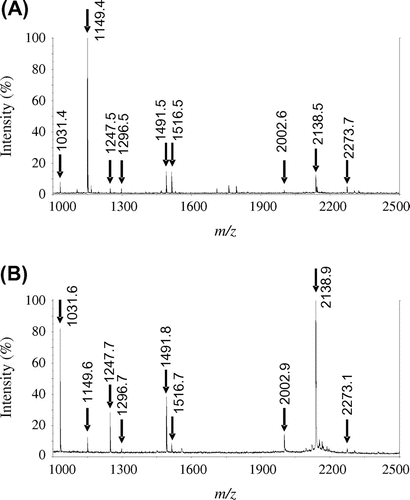
Fig. 3. Effect of various salts on enzyme activity.
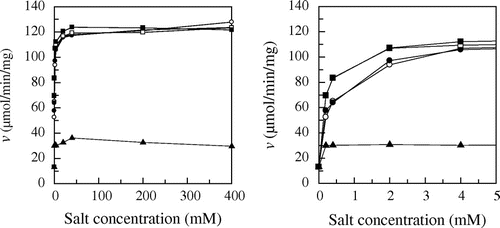
Table 1. PYG purification summary.
Effect of various salts on the hydrolysis rate
The effect of MgCl2 and MgSO4 on PYG activity was investigated by measuring the rate of maltose hydrolysis (Fig. ). The hydrolysis rate in 60 mM sodium acetate buffer without any salts was 13.1 μmol/min/mg. MgSO4 doubled the hydrolysis rate: the hydrolysis rate in the presence of 400 mM MgSO4 was 26.4 μmol/min/mg. MgCl2 increased more efficiently than the sulfate ion: the hydrolysis rate in the presence of 400 mM MgCl2 was 124 μmol/min/mg. The effect of MgCl2, NaCl, KCl, and CaCl2 on PYG activity was investigated (Fig. ). Every salt increased the enzyme activity in the same manner and the activity was saturated at the concentration of 40 mM. The hydrolysis rates in the presence of sufficient concentration (400 mM) of salts were 124 (MgCl2), 122 (NaCl), 123 (KCl), and 128 (CaCl2) μmol/min/mg, respectively. These results indicate that the chloride ion increased the hydrolytic activity efficiently, but the identity of the cation had little effect on the activity.
pH and thermal properties of PYG
The optimal pH of maltose hydrolytic activity and pH stability of PYG in the presence or absence of NaCl were examined. We used 300 mM NaCl, in which the activity was adequately enhanced, as the salt-existence condition. The optimal pH values in the absence and presence of 300 mM NaCl were 3.9 and 4.0, respectively (Fig. (A)). The pH–activity profiles showed a bell-shaped curve with or without NaCl. The enzyme was incubated at various pHs in the presence or absence of NaCl at 4 °C for 24 h, and its residual activity was measured. The stable range was defined as the pH range exhibiting more than 90% residual activity. NaCl shifted the pH–stability range to the alkaline region. PYG without NaCl was stable in a pH range from 3.2 to 6.3, whereas PYG with NaCl was stable in a pH range from 4.1 to 7.3 (Fig. (B)). In addition, when the enzyme was treated at pH 10.0, the residual activity was 50% in the presence of NaCl, whereas the enzyme was almost inactive in the absence of NaCl.
Fig. 4. Effect of NaCl on the pH–activity profile (A), the pH–stability (B), the temperature–activity profile (C), and the thermal stability (D) of PYG in the absence (○) and presence (●) of 300 mM NaCl.
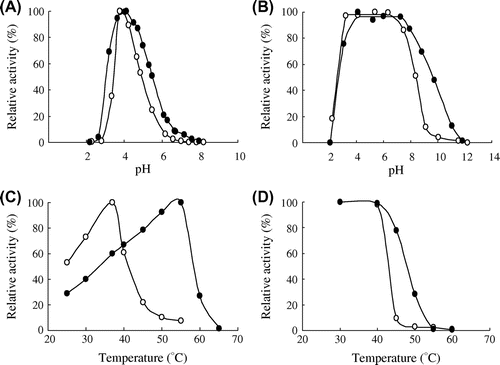
The optimal temperature and thermal stability in the presence or absence of NaCl were examined. The optimal temperature increased from 37 to 55 °C with the addition of NaCl (Fig. (C)). The thermal stability of the enzyme in the presence or absence was investigated based on the measurement of the residual activity after heat treatment for 15 min. The stable region was defined as the temperature range exhibiting more than 90% residual activity. PYG was stable up to 40 °C under both NaCl conditions. However, the residual activity at 45 °C was different in the presence or absence of NaCl. The enzyme exhibited 80% residual activity in the presence of NaCl, whereas the residual activity was 10% in the absence of NaCl. (Fig. (D)).
The increase in pH and thermal stabilities with the addition of NaCl might be explained by facilitating energetic interactions within the protein structure and by the stabilization of surface-exposed residues. Alteromonas haloplanctis α-amylase is stabilized by facilitating energetic interactions between the protein structure and the chloride ion.Citation17) Furthermore, monovalent cation, including potassium and sodium ions, enhances the overall thermal stability of several enzymes by stabilization of surface-exposed residues.Citation18) The thermal stabilization of PYG might be caused by the similar manner as these enzymes.
Substrate specificity of PYG
The kinetic parameters for the hydrolysis of maltooligosaccharides, kojibiose, and nigerose in the presence or absence of 300 mM NaCl were determined to evaluate substrate specificity (Table ). PYG readily hydrolyzed maltooligosaccharides, nigerose, and kojibiose, but hardly hydrolyzed trehalose and isomaltose. The hydrolytic rates of 2 mM isomaltose and 2 mM trehalose in the presence of 300 mM NaCl were 1.38 × 10−2 and 4.15 × 10−2 μmol/min/mg, respectively. NaCl affected Km values more effectively than Vmax values. The Vmax/Km values for disaccharides followed the order, maltose > nigerose > kojibiose in the presence or absence of NaCl. However, NaCl had an effect on the preference for maltooligosaccharide chain length. In the absence of NaCl, Vmax/Km for maltooligosaccharides increased with increasing chain length; the highest Vmax/Km (57.1 μmol/min/mg/mM) was observed for the hydrolysis of maltooctaose. On the other hand, in the presence of NaCl, PYG exhibited the highest Vmax/Km with maltotriose (370 μmol/min/mg/mM). Vmax/Km values for maltooligosaccharides longer than maltotriose were similar. On the other hand, the Vmax and Km values with maltohexaose were lower than those with other maltooligosaccharides. Although it is hard to give a feasible reason for the lower Vmax and Km, the lower Km seems to decrease the Vmax value. As mentioned above, the Vmax/Km value for maltohexaose is similar to that for other maltooligosaccharides. It suggests that the activation free energies for the reactions of the free enzyme and these maltooligosaccharide substrates may be similar, and the transition state of the enzyme–maltohexaose may be stabilized equally to other maltooligosaccharides. Meanwhile, the lower Km makes the reaction fall into a thermodynamic pit, and causes a rise in a free-energy barrier from the Michaelis complex to the transition state.
Table 2. PYG kinetic parameters in the absence and presence of 300 mM NaCl.
Possible mechanism for chloride ion dependence of PYG
The hydrolytic rate of PYG was significantly increased by chloride ion, suggesting PYG is chloride ion-dependent enzyme. A few chloride ion-dependent glycoside hydrolases have been reported thus far.Citation19–21) Human pancreatic α-amylase (HPA), a well-studied chloride ion-dependent enzyme,Citation22–24) catalyzes the same α-glucoside hydrolysis as PYG and could furnish information on the mechanism of the chloride ion dependency of PYG. Biochemical and structural studies of HPA have indicated that chloride ion makes the orientation of an acid/base catalyst accurate, thereby increasing its pKa and enhancing activity. The enhancement of PYG catalytic activity by chloride ion was associated with a small change in the bell-shaped pH-rate profile of PYG (Fig. (A)); thus, chloride ion might be connected to a catalytic residue as in HPA.
PYG underwent not only an increase in catalytic activity but also a change of substrate specificity toward maltooligosaccharides in the presence of chloride ion. In the case of HPA, chloride ion causes the conformational change of substrate-binding loop. It is possible that chloride ion also causes a conformational change in PYG and that the change in specificity is caused by a conformational change in the substrate-binding site.
In conclusion, we succeeded in finding a unique α-glucosidase from a marine source, purifying PYG from the midgut gland of the scallop, and determining the unique properties of this enzyme. The catalytic activity of PYG was evidently enhanced by chloride ion. Furthermore, substrate specificity was altered by the chloride ion. To the best of our knowledge, this is the first report of a chloride ion-dependent α-glucosidase. Unfortunately, we could not clarify the molecular mechanism of the chloride ion dependency at this stage because no structural information exists. It will be necessary to determine the structure of PYG to elucidate the molecular mechanism responsible for the chloride ion dependency.
Author contributions
Conceived and designed the experiments: Y. Masuda, M. Okuyama, H. Mori, and A. Kimura. Performed the experiments: Y. Masuda, T. Iizuka, T. Fukukawa, H. Nakai, and W. Saburi. Analyzed the data: Y. Masuda and M. Okuyama. Contributed reagents/materials/analysis tools: T. Naraoka, T. Tagami, and J. Maneesan. Wrote the paper: M. Okuyama, Y. Masuda, and A. Kimura.
Acknowledgment
We thank Mr K. Nishioka of Faculty of Agriculture, Hokkaido University for preliminary experiments for the purification of PYG.
Disclosure statement
No potential conflict of interest was reported by the authors.
Funding
This work was supported by the JSPS KAKENHI [grant number 26292049].
References
- Trincone A. Marine biocatalysts: enzymatic features and applications. Mar. Drugs. 2011;9:478–499.10.3390/md9040478
- Kobayashi H, Hatada Y, Tsubouchi T, et al. The hadal amphipod Hirondellea gigas possessing a unique cellulase for digesting wooden debris buried in the deepest seafloor. PLoS ONE. 2012;7:e42727. doi:10.1371/journal.pone.0042727.
- Kumagai Y, Ojima T. Enzymatic properties and the primary structure of a β-1,3-glucanase from the digestive fluid of the Pacific abalone Haliotis discus hannai. Comp. Biochem. Physiol. B. 2009;154:113–120.10.1016/j.cbpb.2009.05.005
- Mai K, Mercer JP, Donlon J. Comparative studies on the nutrition of two species of abalone, Haliotis tuberculata L. and Haliotis discus hannai Ino. III. response of abalone to various levels of dietary lipid. Aquaculture. 1995;134:65–80.10.1016/0044-8486(95)00043-2
- Ootsuka S, Saga N, Suzuki K, et al. Isolation and cloning of an endo-β-1,4-mannanase from Pacific abalone Haliotis discus hannai. J. Biotechnol. 2006;125:269–280.10.1016/j.jbiotec.2006.03.008
- Shimizu E, Ojima T, Nishita K. cDNA cloning of an alginate lyase from abalone, Haliotis discus hannai. Carbohydr. Res. 2003;338:2841–2852.10.1016/j.carres.2003.08.009
- Suzuki K, Ojima T, Nishita K. Purification and cDNA cloning of a cellulase from abalone Haliotis discus hannai. Eur. J. Biochem. 2003;270:771–778.10.1046/j.1432-1033.2003.03443.x
- Takami H, Kawamura T, Yamashita Y. Development of polysaccharide degradation activity in postlarval abalone Haliotis discus hannai. J. Shellfish Res. 1998;17:723–727.
- Nikapitiya C, Oh C, Whang I, et al. Molecular characterization, gene expression analysis and biochemical properties of α-amylase from the disk abalone, Haliotis discus discus. Comp. Biochem. Physiol. B. 2009;152:271–281.10.1016/j.cbpb.2008.12.007
- Wojtowicz MB. Carbohydrases of the digestive gland and the crystalline style of the atlantic deep-sea scallop (Placopecten magellanicus, gmelin). Comp. Biochem. Physiol. A. 1972;43:131–141.10.1016/0300-9629(72)90475-6
- Stark J, Walker R. Carbohydrate digestion in Pecten maximus. Comp. Biochem. Physiol. B. 1983;76:173–177.
- Teo L, Sabapathy U. Preliminary report on the digestive enzymes present in the digestive gland of Perna viridis. Mar. Biol. 1990;106:403–407.
- Miwa I, Okuda J, Maeua K, et al. Mutarotase effect on colorimetric determination of blood glucose with β-D-glucose oxidase. Clin. Chim. Acta. 1972;37:538–540.10.1016/0009-8981(72)90483-4
- Laemmli U. Cleavage of structural proteins during assembly of head of bacteriophage-T4. Nature. 1970;227:680–685.10.1038/227680a0
- Schägger H, von Jagow G. Blue native electrophoresis for isolation of membrane protein complexes in enzymatically active form. Anal. Biochem. 1991;199:223–231.10.1016/0003-2697(91)90094-A
- Le Chevalier P, Sellos D, Van Wormhoudt A. Purification and partial characterization of chymotrypsin-like proteases from the digestive gland of the scallop Pecten maximus. Comp. Biochem. Physiol. B. 1995;110:777–784.10.1016/0305-0491(94)00211-C
- Feller G, d'Amic D, Gerday C. Thermodynamic stability of a cold-active α-amylase from the Antarctic bacterium Alteromonas haloplanctis. Biochemistry. 1999;38:4613–4619.10.1021/bi982650+
- Page MJ, Di Cera E. Role of Na+ and K+ in enzyme function. Physiol. Rev. 2006;86:1049–1092.10.1152/physrev.00008.2006
- Rebuffet E, Barbeyron T, Jeudy A, et al. Identification of catalytic residues and mechanistic analysis of family GH82 ι-carrageenases. Biochemistry. 2010;49:7590–7599.10.1021/bi1003475
- Mizutani K, Toyoda M, Otake Y, et al. Structural and functional characterization of recombinant medaka fish alpha-amylase expressed in yeast Pichia pastoris. Biochim. Biophys. Acta. 2012;1824:954–962.10.1016/j.bbapap.2012.05.005
- Levitzki A, Steer ML. The allosteric activation of mammalian alpha-amylase by chloride. Eur. J. Biochem. 1974;41:171–180.10.1111/ejb.1974.41.issue-1
- Maurus R, Begum A, Kuo HH, et al. Structural and mechanistic studies of chloride induced activation of human pancreatic α-amylase. Protein Sci. 2005;14:743–755.10.1110/ps.041079305
- Maurus R, Begum A, Williams LK, et al. Alternative catalytic anions differentially modulate human α-amylase activity and specificity. Biochemistry. 2008;47:3332–3344.10.1021/bi701652t
- Numao S, Maurus R, Sidhu G, et al. Probing the role of the chloride ion in the mechanism of human pancreatic α-amylase. Biochemistry. 2002;41:215–225.10.1021/bi0115636