
Abstract
Tenascin-C (TN-C) is an extracellular matrix glycoprotein markedly upregulated during liver fibrosis. The study is performed to explore the role of TN-C during the growth and activation of hepatic stellate cells (HSCs). We found that TN-C was accumulated accompanying with the HSC activation. Our data on cell migration assay revealed that the rTN-C treatment enhanced HSC migration in a dose- and time-dependent manner, but did not influence their proliferation. HSCs transfected with pTARGET-TN-C overexpression vector displayed increased the type I collagen (Col I) production. TN-C overexpression enhanced the process of HSC activation through TGF-β1 signaling. Moreover, the anti-α9β1 integrin antibody treatment blocked the TN-C-driven Col I increase in rat HSCs. Collectively, TN-C had a positive role in activation of HSCs mediated by TGF-β1 and α9β1 integrin, manifesting elevation of Col I production and promotion of cell migration. Our results provide a potential insight for the therapy of hepatic fibrosis.
Graphical abstract
TN-C induced TGF-β1 and α9β1 expression to promote the cell migration and type I Collagen production in the activated HSCs.
Hepatic stellate cells (HSCs) are liver-specific mesenchymal cells, which are located between parenchymal cell plates and sinusoidal endothelial cells in the space of Disse.Citation1) They maintain close interactions with sinusoidal endothelial cells or hepatic epithelial cells and are necessary in liver physiology and fibrosis.Citation2) A growing body of evidence revealed that HSCs have a profound impact on the proliferation, differentiation, and morphogenesis of other hepatic cell types during liver development and regeneration.Citation2–4)
Hepatic fibrosis is a dynamic and sophisticatedly regulated wound-healing process response to chronic hepatocellular injury. It represents the final common pathway of virtually all types of chronic liver diseases, and has become a major public health concern.Citation5,6) Previous studies have demonstrated that HSCs play an important role in the response to multiple etiologies of liver injury.Citation7,8) More specifically, this fibrotic process results from the accumulation of the extracellular matrix (ECM) principally produced by HSCs, including proteoglycan, collagen, and adhesive glycoproteins.Citation9)
In physiological conditions, HSCs are quiescent cells. They play important roles in the regulation of retinoid homeostasis, hepatocyte interactions, and ECM remodeling by producing soluble factors and ECM components as well as matrix metalloproteases (MMP) and their inhibitors.Citation10,11) However, HSCs can be activated in response to different types of liver injury and differentiate into myofibroblasts.Citation8,11) Upon activation, HSCs can lead to the deposition of excessive scar matrix into the interstitium as a wound-healing response.Citation12–14) Recently, HSCs have been proposed as a component of the pro-metastatic liver microenvironment as they can trans-differentiate into highly proliferative and motile myofibroblasts.Citation15,16) These clues suggested them a potential role in the desmoplastic reaction and metastatic growth. Moreover, activated HSCs are the major subtype of stromal cells in the liver tumor microenvironment, which can promote the growth, migration, and invasion of hepatocellular carcinoma (HCC) cells.Citation17,18)
Tenascins are a family of four multimeric ECM glycoproteins, including tenascin-C (TN-C), tenascin-X, tenascin-R, and tenascin-W.Citation19,20) The expression of TN-C is developmentally regulated during many diseases and intensively induced by inflammation and infections.Citation21) All tenascins have the potential to modify cell adhesion either directly or through an interaction with fibronectin, and cell-tenascin interactions typically lead to increased cell motility.Citation19) For TN-C, there is a correlation between its elevated expression and increased cell metastasis in remodeling tissues and several types of tumors.Citation22–24) For instance, TN-C is highly expressed in glioblastoma multiforme (GBM) and giant cell tumors of the bones.Citation24) In addition, TN-C plays a vital role in cardiac and arterial injury, tumor angiogenesis and metastasis.Citation24–26) Activated HSCs migrate and accumulate at the sites of tissue repair, producing large amounts of ECM especially type I collagen (Col I), and regulating ECM remodeling.Citation26) However, whether TN-C can influence the production of Col I in HSCs remained unknown.
The roles of TN-C in activation and migration of HSCs were explored primarily in this study. Recombinant TN-C protein (rTN-C) and the pTARGET-TN-C overexpression vector were used to treat HSCs. Our results showed that the TN-C level increased with HSC activation. TN-C overexpression promoted Col I production in and migration of HSCs. These promotions could be attenuated by the anti-TGF-β1 or anti-α9β1 integrin antibodies.
Materials and methods
Materials
Adult male Sprague-Dawley (SD) rats were purchased from the Experimental Animal Center of The Fourth Military Medical University. FBS, DMEM/F12, RIPA buffer, PVDF membranes, Tween20, L-glutamine, penicillin/streptomycin, Trypan blue solution, BCA protein assay kit, Triton X-100, RNase inhibitor, pronase-E, collagenase, Dnase I, anti-β-actin antibody, and Transwells were all purchased from Sigma-Aldrich (St Louis, MO, USA). PageRuler Prestained Protein Ladder (Fermentas, St. Leon-Rot, Germany). HRP-conjugated sheep anti-mouse IgG antibodies, HRP-conjugated rabbit anti-goat IgG antibodies and HRP-conjugated sheep anti-rabbit IgG antibodies were purchased from Millipore (Temecula, CA, USA). Enhanced chemiluminescence (ECL) was purchased from Thermo Pierce (Rockford, IL, USA). HEK-293A cells, TRIzol, and LipofectamineTM 2000 were purchased from Invitrogen Life Technologies (Carlsbad, CA, USA). DNase, RT-PCR kit, and Qiagen RNeasy kit were purchased from Qiagen (Hilden, Germany). The SYBR® Green PCR Master Mix was purchased from Applied Biosystems (Foster City, CA, USA). Pyrobest DNA polymerase and avian myeloblastosis virus (AMV) reverse transcriptase were purchased from Takara Biotechnology (Dalian, China). The BrdU cell proliferation assay kit was purchased from Millipore (Billerica, MA, USA). A micro-plate reader was purchased from Bio-Rad (Hercules, CA, USA). Mouse anti-rat α-SMA primary antibody was purchased from Serotec (Kidlington, Oxon, UK). Endotoxin-free recombinant TN-C was prepared by our laboratory. Rabbit anti-rat TN-C antibody, rabbit anti-rat TGF-β1 antibody, rabbit anti-rat α5β1 integrin antibody, goat anti-rat collagen type I α1 chain (Col I α1) antibody and goat anti-rat α9β1, α6β4 or α3β1 integrin antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, Heidelberg, Germany). Moloney Murine Leukemia Virus (M-MLV) reverse transcriptase and pEGFP-N1 was purchased from Clontech (Palo Alto, CA, USA). His-Bind resin (Ni2+-resin) was purchased from Novagen (Darmstadt, Germany).
Animals
All experimental protocols were reviewed and approved by the Institutional Animal Care and Use Committee of Xi’an Jiaotong University. Adult male Sprague-Dawley (SD) rats aged 6–12 months and weighing 400–500 g were housed under conventional conditions in accordance with institutional guidelines. Before experiments, SD rats received 200 IU/100 g Vitamin A via intragastric administration each day for a period of two weeks.
Isolation and Culture of HSCs
HSCs were isolated from rat livers by the pronase-collagenase perfusion technique followed by single-step density gradient centrifugation with Nycodenz. Briefly, SD rats were anesthetized and their livers were operated upon. Subsequently, each liver was rinsed in situ with Ca2+/Mg2+-free Hanks’ balanced salt solution until the liver was free of blood; then, it was gently removed from the body cavity. Following, livers received a series of treatments including pronase-E solution, collagenase solution, and Dnase I solution. Then, the tissue pieces of the liver treated with Dnase I solution were further dissected by rotating on a magnetic stirrer (200 rpm) at 37 °C for several minutes. The obtained cell suspension was subsequently filtered through nylon meshes and centrifuged for 7 min at 450 g. The resulting pellet was resuspended in ice-cold Ca2+/Mg2+-containing HBSS with BSA. Finally, the HSCs were isolated by a density gradient of Nycodenz. The cells were seeded on day 0 and cultured in DMEM/F12 supplemented with 10% FBS, 4 mM L-glutamine and 1 X penicillin/streptomycin at a density of 2.5 × 106 cells per 10-cm Petri dish under a humidified atmosphere of 5% CO2/95% air at 37 °C.
Western blotting
Cultured HSCs were lysed with RIPA buffer. The soluble protein was obtained from the lysate and protein concentration was determined by BCA. Proteins (40 μg/lane) were electrophoresed in 10% (for α-SMA, TN-C, and integrin α9) or 7.5% (for collagen) SDS/polyacrylamide gels and transferred onto PVDF membranes. After blocking with 0.5% skimmed milk powder in 1 × PBS-Tween20, the target proteins were probed with 1:1000 mouse anti-rat α-SMA primary antibodies or anti-β-actin antibodies overnight at 4 °C. Then, the membranes were incubated with HRP-conjugated sheep anti-mouse IgG antibodies (1:4000 dilution) at room temperature for 1 h. The same experiment was performed to detect TN-C. The primary and second antibodies are rabbit anti-rat TN-C antibodies and HRP-conjugated sheep anti-rabbit IgG antibodies, respectively. The reactive bands were detected by enhanced chemiluminescence (ECL) according to the manufacturer’s protocol and the relative levels of each protein to β-actin were analyzed.
qRT-PCR
Total RNA of cultured HSCs (day 0–day 7) was extracted with TRIzol and purified with the Qiagen RNeasy kit. The levels of mRNA were analyzed by one-step quantitative reverse transcriptase-polymerase chain reaction (RT-PCR) using specific primers and the SYBR® Green PCR Master Mix and RT-PCR kit on an ABI Prism 7700 Sequence Detection System. Primers specific for SD rat TN-C, collagen alpha-1(I) chain, and β-actin were synthesized by the Shanghai Sangon Biological Engineering and Technology Service (Shanghai, China). The sequences of primers were 5′- CGG AAT TCA GGG CAG ACA CAA GAG CAA G-3′ (sense-primer) and 5′-AAC TGC AGT TCC TTC GGA GAA CCC ATG G-3′ (anti-sense primer) for TN-C. And, the sequences of primers were 5′- CAC GGC TGT GTG CGA TGA -3′ (sense-primer) and 5′- TCG CCC TCC CGT CTT TG -3′ (anti-sense primer) for collagen alpha-1(I) chain. Briefly, 20 μl reactions containing 50 ng of total RNA, 10 μl of 2 × SYBR Green PCR Master Mix, 6.25 U of AMV reverse transcriptase, 10 U of RNase inhibitor, and 0.1 mM of primers were subjected to one cycle of 95 °C for 10 min and then 40 cycles of 95 °C for 15 s, 56 °C for 30 s, and 72 °C for 45 s. The levels of individual gene mRNA transcripts were firstly normalized to control, β-actin (sense: 5′-CTC CAT CCT GGC CTC GCT GT-3′, anti-sense: 5′-GCT GTC ACC TTC ACC GTT CC-3′). The data were analyzed by the 2−∆∆Ct method and expressed as fold changes.
Expression and purification of endotoxin-free recombinant TN-C
Rat macrophages were incubated for 24 h at 37 °C in 5% CO2. Thereafter, total RNA was extracted with TRIzol reagent and 5 μg total RNA was reverse-transcribed into cDNA using Moloney Murine Leukemia Virus (M-MLV) reverse transcriptase. TN-C gene was amplified by PCR with TN-C forward primer (5′-AGA CAC CTA GCC AAT CCA ACC -3′) and reverse primer (5′- AAA TCC TGT TTT CTC TGG GC -3′), which contained BamH I and Hind III restriction sites, respectively. The DNA fragments of TN-C were obtained and verified by gene sequencing (BGI, Beijing, China). The purified TN-C was then subcloned into pEGFP-N1. Recombinant plasmid pEGFP-N1-TN-C was transfected into HEK-293A cells. Afterwards, the cells were harvested and lysed in PBS containing 0.2% Triton X-100. The complexes were sonicated, and then centrifuged at 15,000 g for 30 min at 4 °C. The supernatant were collected and the fusion proteins were purified using His-Bind resin (Ni2+-resin) according to standard protocols. The purified proteins were analyzed by SDS-PAGE and further verified by Western blot analysis.
Bromodeoxyuridine (BrdU) assay
HSCs were synchronized for 16 h by serum starvation and plated in 96 wells (3 × 103 cells/well) in DMEM/F12 containing 0.5% FBS, and 10 μL BrdU solution and 10 μL endotoxin-free recombinant TN-C solution (2, 4, 6, 8 μg/μL) were added and incubated for 12 h, 24 h, 36 h, and 48 h, respectively. Then, 100 μL/well of the fixing solution was added and incubated for 15 min. Then 100 μL/well of pre-prepared detection antibody solution was added and incubated for 1 h. After that, 100 μL/well of prepared HRP-conjugated secondary antibody was added and incubated for 30 min. Finally, 100 μL of TMB substrate was added before being incubated for 30 min. The amount of BrdU incorporated into the cells was determined at 450 nm by a micro-plate reader.
Cell migration assay
Migration assay was performed in Transwells (8.0-μm pore size). For the migration assay, 96 h cultured HSCs, approximately 2 × 104 cells per well in serum-free medium were added to the upper wells. Media containing 10% FBS and 10 μL of endotoxin-free rat rTN-C solution at different concentrations (2, 4, 6, and 8 μg/μL) were added to the lower wells, while media containing only 10% FBS was added to the lower wells as control. Cells that migrated through the filter after 12, 24, 36, or 48 h were stained and counted by phase microscopy. Here, platelet-derived growth factor (PDGF) was chosen to be the positive control molecules to compare the migration of HSCs. The time-dependence experiment of PDGF was same as rTN-C, while 2 μg/μL PDGF was used as stimulus dose.
Expression and secretion of collagen I under stimulation of rTN-C
HSCs (2.5 × 105 cells/well) were seeded on six-well plates in DMEM/F12 with 10% FBS. The cells were cultured using DMEM/F12 for 7 days, which was replaced by serum-deprived DMEM/F12 prior to 10 μL endotoxin-free rat rTN-C (8 μg/μL) treatment for 72 h. Heat-denatured rat rTN-C (10 μL, 8 μg/μL) was used for the control. In the following, the expression of collagen I in HSCs total proteins and the medium of HSCs were detected by Western blotting.
The pTARGET-TN-C vector transfection
The full length cDNA of TN-C was sub-cloned with pyrobest DNA polymerase from the HSC RNA of SD rats and inserted into the pTARGET vector. These recombination vectors were transfected into day 7 cultured HSCs using LipofectamineTM 2000, pTARGET blank vector (pTARGET-HSXs) as control. After 5 h of incubation at 37 °C with 5% CO2, the cells were washed and cultured in DMEM/F12 with 10% FBS for experiments at 72 h after transfection.
The effect of TGF-β1of α9β1 integrin on collagen I
HSCs (2.5 × 105 cells/well) were seeded on six-well plates in DMEM/F12 with 10% FBS. The cells were cultured using DMEM/F12 for 7 days, which was replaced by serum-deprived DMEM/F12 prior to 10 μL endotoxin-free rat rTN-C (8 μg/μL) and 10 μL anti-TGF-β1 antibody (5 μg/μL), or 10 μL anti-rat α9β1 integrin antibody (5 μg/μL) treatments for 72 h. HSCs only treated with 10 μL endotoxin-free rTN-C (8 μg/μL) or 10 μL control antibody (5 μg/μL) were used as control.
Silencing α9β1 integrin expression with siRNA
The cells were transfected with siRNA (20 μM, Ambion, #86946) targeted to exon 4 of the α9 integrin using siPort Amine (Ambion) in OptiMem media. Cell media was replaced with full growth media for 72 h after transfection. Then the α9-siRNA transfected HSCs were treated with 10 μL endotoxin-free rat rTN-C (8 μg/μL) for 72 h.
Statistical analysis
Each assay performed on cell proliferation, cell migration, and gene expression detection were run in quadruplicate plates or pores and repeated three times. Data are expressed as mean ± standard error of the mean (SEM) and differences were analyzed by Student’s t-test.
Results
TN-C expression was elevated during activation of HSCs
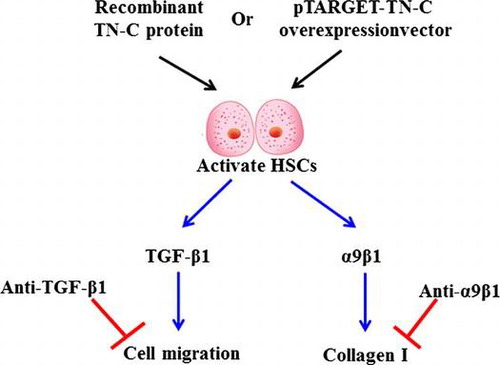
The data on Western blotting and showed that expression of α-SMA was being increased, which indicated that activated HSCs emerged gradually from day 2 (Fig. (A)). TN-C expression was detected by qRT-PCR and Western blotting during HSC activation. As shown in Fig. (B and C), expression of TN-C mRNA and protein gradually increased. These results suggested that TN-C might be associated with activation of HSCs.
Fig. 1. TN-C level was being increased in activated HSCs.
TN-C facilitated HSC migration
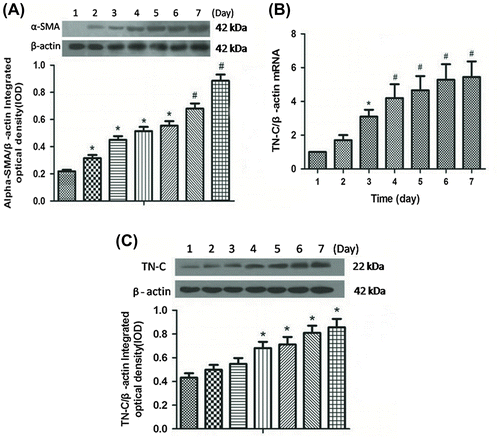
To explore the role of TN-C in activation of HSCs, the cells were incubated with various concentration of rTN-C ranging from 2 to 8 μg/μL, and then cell proliferation and migration were detected with the BrdU and transwell migration methods, respectively. The data showed that there were no significant changes in HSC proliferation under treatment with different doses of rTN-C (Fig. (A)). While, the number of migrated HSCs were increased with time and increasing rTN-C doses (Fig. (B)).
Fig. 2. Migration of HSCs was enhanced by the rTN-C treatment.
Collagen I expression in HSCs was promoted by the treatment of rTN-C
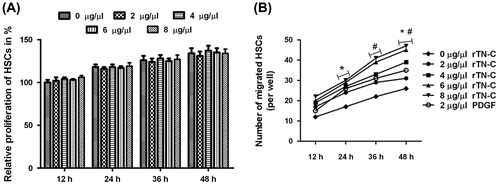
Then Collagen I mRNA and protein (Col I α1) levels in rTN-C treated HSCs and the media were detected. Col I α1 was up-regulated in both in HSCs and the media by WT rTN-C treatment but not by the denatured rTN-C (Fig. (A and B)). Moreover, a pTARGET-TN-C vector containing TN-C cDNA was constructed and transfected into HSCs (mimicking the elevation of TN-C autocrine). Western blotting showed that TN-C expression in these HSCs was significantly elevated (Fig. (C and D)) to detect the production of Col I. From Fig. (E and F), it can be seen that THE production of Col I is up-regulated significantly in TN-C-overexpressing HSCs compared with HSCs (p < 0.05). Consistent with the results of rTN-C treatment, the Collagen I mRNA and protein levels of were both markedly increased response to TN-C overexpression (Fig. (E–G)). These results demonstrate that TN-C contributed to Col I production in activated HSCs.
Fig. 3. Expression of Collagen I was induced by the rTN-C protein treatment in HSCs.
TGF-β1 was required in Col I synthesis in HSCs
Commonly, TGF-β1 is stored as an inactivated protein. When activated, TGF-β1 interacted downstream Smad proteins and induced collagen production. In order to explore whether the effect of TGF-β1 on Col I production in HSCs can be influenced by TN-C, anti-TGF-β1 Ab was added into rTN-C pre-treated HSCs, while rTN-C pre-treated HSCs added IgG was regarded as a control. As shown in Fig. (A and B), Col I α1 was down-regulated in the anti-TGF-β1 Ab treated group compared with the control, indicating that TGF-β1 was required during TN-C-induced Col I production.
Fig. 4. TGF-β1 and α9β1 were essential in the induction of collagen I by TN-C.
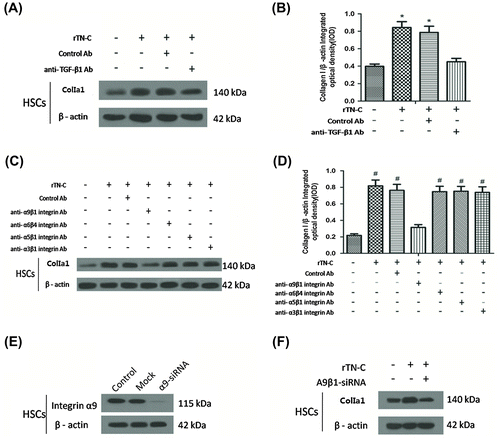
Anti-α9β1 integrin Ab specifically blocks the TN-C-induced Col I production
We next evaluated the role of extracellular TN-C on induction of Col I production in HSC (mimicking the elevation of TN-C paracrine). The anti-α9β1 integrin antibody incubation blocked the rTN-C-driven Col I production (Fig. (C and D)). Neutralization of other integrins (i.e. α6β4, α5β1, and α3β1) failed to prevent induction of Col I production by rTN-C (Fig. (C and D)). To verify that α9β1 integrin is indispensable in rTN-C-driven Col I production, α9β1siRNA was transfected into HSCs and then Col I α1 expression was detected by Western blotting. The protein of α9β1 was sharply decreased by the siRNA transfection (Fig. (E)). Col I α1 expression induced by rTN-C was resisted by α9β1 antibody (Fig. (F)), revealing that α9β1 integrin is indispensable in rTN-C-driven Col I production.
Discussion
In the normal liver, HSCs are quiescent and contain numerous vitamin A lipid droplets, constituting the largest reservoir of vitamin A in the body.Citation27) They participated in ECM homeostasis, drug detoxification, immunotolerance, and possibly the preservation of hepatocyte mass through the secretion of mitogens, including hepatocyte growth factor.Citation28) During liver injury caused by hepatic toxins, viral infection, or other factors, HSCs were activated and differentiated into alpha smooth muscle actin-expressing contractile myofibroblasts, which greatly contributed to the hepatic fibrogenesis process.Citation29) HSCs play an important role in the response to multiple etiologies of liver injury, including accumulation of the ECM, mitogen-mediated proliferation, amplified inflammation, and immunoregulation, as well as altered matrix degradation.Citation2)
Tenascin-C (TN-C) is an ECM glycoprotein that is expressed during wound healing in various tissues. Previous studies reported that TN-C was up-regulated during liver fibrotic processes, particularly at parenchymal-mesenchymal interfaces in fibrotic areas.Citation30) Furthermore, double immunohistochemistry in a rat model of liver fibrosis showed the accumulation of desmin-positive cells with TN-C deposition, and an immunoelectron microscopic study suggested that TN-C also existed in HSCs.Citation30) However, the significance of TN-C in HSCs was still unclear. Here, we reported that TN-C played a positive role in HSC activation and migration. We found that the expression of TN-C mRNA was being increased in cultured SD rat HSCs from day 1 to day 7 cultured HSCs, suggesting that TN-C was accumulated with the activation of HSCs. Then the effect of TN-C on proliferation and migration of HSCs was investigated. TN-C enhanced the migration of HSCs in a dose- and time-dependent manner, but cannot influence the proliferation. This is consistent with a recent study which concluded that tenascin-C knock-down in the human LN229 GBM cell line had no significant effect on cell proliferation in vitro but abolished cell migration by promoting focal adhesion disassembly signals.
HSCs played a significant role in Col I deposition when hepatocellular injury is concentrated within the liver lobule and sinusoids.Citation31) In addition, activated HSCs migrate and accumulate at the sites of tissue repair, secreting large amounts of ECM, mostly Col I, and regulating ECM remodeling.Citation11) In this study, we also reported that TN-C promoted the production of Col I by HSCs. Overexpression of TN-C by rTN-C and pTARGET-TN-C construct both induced Col I production. These data implied that TN-C may be a therapeutic target in hepatic fibrosis for its promoting effect on Collagen I expression. Hence, we speculated that inhibition of TN-C may decrease the Collagen I expression and moderate hepatic fibrosis, which needs further research.
Activation of HSCs is a complex process that is regulated by multiple factors, among which TGF-β1 functions essentially. Hence, we blocked TGF-β1 signaling by its specific antibody in rTN-C pre-treated HSCs and then detected change of Collagen I expression. The results revealed that TN-C could enhance the Col I production and migration of HSC through TGF-β1. Next, we evaluated the role of extracellular TN-C-mediated signaling (paracrine role) in collagen I expression in HSC. Incubation with the anti-α9β1 integrin Ab blocked the rTN-C-driven Collagen I expression in rat HSCs. These results indicated that TGF-β1 and α9β1 integrin were essential in Tenascin-C induced Collagen I expression in HSCs.
Conclusions
TN-C was accumulated with the activation of HSCs, and rTN-C can enhance the collagen I expression and HSC migration through TGF-β1 and α9β1 integrin signaling pathways. Our results provide a potential target for the therapy of hepatic fibrosis.
Author contributions
Jian-Cang Ma and Xin Huang performed the majority of the experiments and drafted the manuscript; Ya-Wei Shen, Chen Zheng, Qing-Hua Su, and Jin-Kai Xu supplemented a small part of the experiments and revised the manuscript; Jun Zhao designed the study and provided the platform and fundings (the following two grants mentioned in the Acknowledgments were led by Jun Zhao) for the study.
Disclosure statement
No potential conflict of interest was reported by the authors.
Ethical approval
All procedures performed in this study involving human participants were in accordance with the ethical standards of the Ethical Committee of Xi’an Jiao Tong University and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. Informed consent was obtained from all individual participants included in the study.
All procedures performed in this study involving animals were reviewed and approved by the institutional animal care and use committee of Xi’an Jiao Tong University.
Supplemental materials
The supplemental material for this paper is available at http://dx.doi.org/10.1080/09168451.2016.1165600.
TBBB_1165600_Figure_S1.jpg
Download JPEG Image (921.9 KB){kind=link}
Additional information
Funding
Notes
Abbreviations: TN-C ,Tenascin-C; HSCs, hepatic stellate cells; Col I, type I collagen; ECM, extracellular matrix; MMP, matrix metalloproteases; HCC, hepatocellular carcinoma; GBM, glioblastoma multiforme; SD, Sprague-Dawley; ECL, Enhanced chemiluminescence; AMV, avian myeloblastosis virus.
Related Research Data
References
- Bataller R, Brenner DA. Hepatic stellate cells as a target for the treatment of liver fibrosis. Semin. Liver Dis. 2001;21:437–451.
- Hellerbrand C. Hepatic stellate cells-the pericytes in the liver. Pflügers Archiv-Eur. J. Physiol. 2013;465:775–778.10.1007/s00424-012-1209-5
- Puche JE, Lee YA, Jiao J, et al. A novel murine model to deplete hepatic stellate cells uncovers their role in amplifying liver damage in mice. Hepatology. 2013;57:339–350.10.1002/hep.26053
- Li YC, Wang JH, Asahina KJ. Mesothelial cells give rise to hepatic stellate cells and myofibroblasts via mesothelial-mesenchymal transition in liver injury. Proc. Nat. Acad. Sci. 2013;110:2324–2329.10.1073/pnas.1214136110
- Williams MJ, Clouston AD, Forbes SJ. Links between hepatic fibrosis, ductular reaction, and progenitor cell expansion. Gastroenterology. 2014;146:349–356.10.1053/j.gastro.2013.11.034
- Raj A, Holtmann G, Bhat P, et al. Hepatic fibrosis is associated with small intestinal permeability in chronic liver disease due to hepatitis C, hepatitis B and non-alcoholic fatty liver disease, in subjects without ascites. J. Gastroenterol. Hepatol. 2014;29:200.
- Iwaisako K, Jiang C, Zhang M, et al. Origin of myofibroblasts in the fibrotic liver in mice. Proc. Nat. Acad. Sci. 2014;111:E3297–E3305.10.1073/pnas.1400062111
- Depito DH, Schwabe RG. Hepatic Stellate Cells and Liver Cancer. Stellate Cells in Health and Disease. 2015;145.
- Seki E, Schwabe RF. Hepatic inflammation and fibrosis: functional links and key pathways. Hepatology. 2015;61:1066–1079.10.1002/hep.v61.3
- Eda H, Kulig KM, Steiner TA, et al. A nanofiber membrane maintains the quiescent phenotype of hepatic stellate cells. Dig. Dis. Sci. 2012;57:1152–1162.10.1007/s10620-012-2084-9
- Troeger JS, Mederacke I, Gwak GY, et al. Deactivation of hepatic stellate cells during liver fibrosis resolution in mice. Gastroenterology. 2012;143:1073–1083.10.1053/j.gastro.2012.06.036
- Hernández-Gea V, Ghiassi-Nejad Z, Rozenfeld R, et al. Autophagy releases lipid that promotes fibrogenesis by activated hepatic stellate cells in mice and in human tissues. Gastroenterology. 2012;142:938–946.10.1053/j.gastro.2011.12.044
- Tacke F, Weiskirchen R. Update on hepatic stellate cells: pathogenic role in liver fibrosis and novel isolation techniques. Expert. Rev. Gastroenterol. Hepatol. 2012;6:67–80.10.1016/j.jhep.2015.02.039
- Mederacke I, Dapito DH, Affò S, et al. High-yield and high-purity isolation of hepatic stellate cells from normal and fibrotic mouse livers. Nat. Protoc. 2015;10:305–315.10.1038/nprot.2015.017
- Mikuriya Y, Tashiro H, Kuroda S, et al. Fatty liver creates a pro-metastatic microenvironment for hepatocellular carcinoma through activation of hepatic stellate cells. Int. J. Cancer. 2015;136:E3–E13.10.1002/ijc.29096
- Thompson AI, Conroy KP, Henderson NC. Hepatic stellate cells: central modulators of hepatic carcinogenesis. BMC gastroenterol. 2015;15:63.
- Tan W, Li Y, Lim SG, Tan TM. miR-106b-25/miR-17-92 clusters: polycistrons with oncogenic roles in hepatocellular carcinoma. World. J. Gastroenterol. 2014;20:5962–5972.
- Huang L, Xu AM, Liu S, et al. Cancer-associated fibroblasts in digestive tumors. World. J. Gastroenterol. 2014;20:17804–17818.
- Adams JC, Chiquet-Ehrismann R, Tucker RP. The evolution of tenascins and fibronectin. Cell Adh. Migr. 2015;9:22–33.
- Chiquet-Ehrismann R, Tucker RP. Tenascins and the importance of adhesion modulation. Cold. Spring. Harb. Perspect. Biol. 2011;3:a004960.
- Kasprzycka M, Hammarström C, Haraldsen G. Tenascins in fibrotic disorders—from bench to bedside. Cell Adh. Migr. 2015;9:83–89.
- Nie S, Gurrea M, Zhu J, et al. Tenascin-C: a novel candidate marker for cancer stem cells in glioblastoma identified by tissue microarrays. J. Proteome Res. 2014;14:814–822.
- Berndt A, Richter P, Kosmehl H, et al. Tenascin-C and carcinoma cell invasion in oral and urinary bladder cancer. Cell Adh. Migr. 2015;9:105–111.
- Brösicke N, van Landeghem FK, Scheffler B, et al. Tenascin-C is expressed by human glioma in vivo and shows a strong association with tumor blood vessels. Cell Tissue Res. 2013;354:409–430.10.1007/s00441-013-1704-9
- Spenlé C, Saupe F, Midwood K, et al. Exploitation and collateral damage in cancer management. Cell Adh. Migr. 2015;9:141–153.
- Midwood KS, Hussenet T, Langlois B, et al. Advances in tenascin-C biology. Cell. Mol. Life Sci. 2011;68:3175–3199.10.1007/s00018-011-0783-6
- Rojkind M, Reyes-gordillo K. Hepatic stellate cells. In: Irwin M. Arias, editor. The liver: biology and pathobiology. 5th ed. Hoboken, NJ: John Wiley & Sons;2009. p. 407–432.10.1002/9780470747919
- Geerts A. History, heterogeneity, developmental biology, and functions of quiescent hepatic stellate cells. Semin. Liver Dis. 2001;21:311–336.10.1055/s-2001-17550
- Kumar P, Smith T, Rahman K, et al. Adiponectin modulates focal adhesion disassembly in activated hepatic stellate cells: implication for reversing hepatic fibrosis. FASEB J. 2014;28:5172–5183.10.1096/fj.14-253229
- El-Karef A, Kaito M, Tanaka H, et al. Expression of large tenascin-C splice variants by hepatic stellate cells/myofibroblasts in chronic hepatitis C. J. Hepatol. 2007;46:664–673.10.1016/j.jhep.2006.10.011
- Moreira RK. Hepatic stellate cells and liver fibrosis. Arch. Pathol. Lab. Med. 2007;131:1728.