
Abstract
From investigation of 60 filamentous fungi, we identified Fusarium merismoides var. acetilereum, which uses 4-N-trimethylamino-1-butanol (TMA-butanol) as the sole source of carbon and nitrogen. The fungus produced NAD+-dependent TMA-butanol dehydrogenase (DH) when it was cultivated in medium containing TMA-butanol. The enzyme showed molecular mass of 40 kDa by SDS–PAGE and 160 kDa by gel filtration, suggesting that it is a homotetramer. TMA-butanol DH is stable at pH 7.5–9.0. It exhibits moderate stability with respect to temperature (up to 30 °C). Additionally, it has optimum activity at 45 °C and at pH 9.5. The enzyme has broad specificity to various alkyl alcohols and amino alkyl alcohols, and the carbon chains of which are longer than butanol. Moreover, the activity is strongly inhibited by oxidizing agents, carbonyl and thiol modulators, and chelating agents. This report is the first study examining TMA-butanol DH from eukaryotic microbes.
Graphical abstract
Having broad substrate specificity, 4-N-trimethylamino-1-butanol dehydrogenase (TMA-butanol DH) from Fusarium merismoides var. acetilereum was purified and characterized.
Quaternary ammonium compounds are distributed globally, with more than 100 reported examples such as choline, l-carnitine, and glycine betaine. They are used extensively in consumer applications as disinfectants, fabric softeners, hair conditioners, and emulsifying agents. Although some quaternary ammonium compounds are regarded as pollutants,Citation1) naturally occurring quaternary ammonium compounds are known to have important functions in all living organisms.Citation2–4) For example, choline is a component of neurotransmitter acetylcholine. l-Carnitine functions as a carrier of fatty acids for β-oxidation in mitochondrial inner membrane. Glycine betaine is known to be a compatible solute. Furthermore, the degradation of these compounds by enterobacteria can engender the development of cardiovascular disease through conversion to trimethylamine.Citation4,5) The produced trimethylamine can subsequently contribute to the global carbon and nitrogen cycle after conversion to greenhouse gases by methanogenic bacteria.Citation6) Therefore, the microbial transformations of these compounds and their analogs play important roles in many biological systems.
We have elucidated the aerobic degradation pathway on several quaternary ammonium compounds such as choline,Citation7–9) l-carnitine,Citation10,11) and d-carnitineCitation12) in bacteria, filamentous fungi, and yeast, and we have characterized the related enzymes. We tried to screen bacteria that were able to grow on choline analogs such as homocholine and 4-N-trimethylamino-1-butanol (TMA-butanol). Results show that some bacteria were able to grow on homocholine and TMA-butanol.Citation13,14) It has been elucidated in Pseudomonas sp. that NAD+-dependent TMA-butanol dehydrogenase (TMA-butanol DH) and 4-trimethylaminobutyraldehyde dehydrogenase (TMA-butyraldehyde DH) are involved in the TMA-butanol degradation pathway.Citation14–16) Those two enzymes were characterized, and those genes were cloned.Citation17) However, eukaryotic microbes able to grow on homocholine and TMA-butanol have not been found. It is interesting to compare differences between prokaryotic enzymes and eukaryotic enzymes in the aerobic degradation pathway of quaternary ammonium compounds. Therefore, we tried anew to screen eukaryotic microbes that can grow on TMA-butanol. Results show that some Fusarium strains grew on TMA-butanol. This article describes purification and characterization of NAD+-dependent TMA-butanol DH from F. merismoides var. acetilereum.
Materials and methods
Materials
3-Dimethylamino-1-propanol, 4-dimethylamino-1-butanol, 5-chloro-1-pentanol, and 6-trimethylamino-1-hexanol were purchased from Tokyo Kasei Kogyo Co. Ltd. (Tokyo, Japan). TMA-propanol iodide, TMA-butanol iodide, TMA-pentanol iodide, TMA-hexanol iodide, and TMA-octanol iodides were synthesized using methods described by Hassan et al.Citation14) All other reagents were commercial products of analytical grade.
Microorganisms and cultures
Various strains of Fusarium were screened for their ability to grow on TMA-butanol as the sole source of carbon and nitrogen. These Fusarium were grown aerobically for 4 days at 30 °C in a culture medium containing 5.0 g of TMA-butanol iodide, 3.0 g of KH2PO4, 3.0 g of K2HPO4, 0.5 g of MgSO4·7H2O, and 0.5 g of yeast extract in 1000 mL of distilled water at pH 7.5. TMA-butanol DH activities were assayed in strains showing good growth (greater than 1.0 turbidity) on the medium. For large-scale cultivation of F. merismoides var. acetilereum (NBRC30040), a medium comprising the following was used: 5.0 g of TMA-butanol iodide, 3.0 g of KH2PO4, 3.0 g of K2HPO4, 0.5 g of MgSO4·7H2O, 1.0 g of yeast extract, 3.0 g of peptone, and 0.8 mL of trace metal mixture [3130 mg/L FeCl3·6H2O, 940 mg/L ZnCl2, 375 mg/L H3BO3, 250 mg/L MnCl2·4H2O, 250 mg/L CuSO4·5H2O, 190 mg/L CoSO4·7H2O, 125 mg/L (NH4) Mo7O24·4H2O] in 1000 mL of distilled water at pH 7.0. The cultivation was conducted at 30 °C for 3 days.
Enzyme assay
TMA-butanol DH activity was measured by the increase in absorbance at 340 nm at 30 °C. The standard reaction mixture (1.5 mL) contained 225 μmol glycine-NaOH buffer (pH 9.5), 4.5 μmol TMA-butanol iodide, and 1.5 μmol NAD+, with an appropriate amount of the enzyme solution. The reaction was started by the addition of NAD+. For the reduction reaction, the reaction mixture (1.5 mL) included 225 μmol potassium phosphate buffer (pH 6.0), 1.5 μmol TMA-butyraldehyde iodide, 0.5 μmol NADH, and an appropriate amount of the enzyme solution. For substrate specificity analysis, trimethylamino alcohols (TMA-alcohols), dimethylamino alcohols (DMA-alcohols), amino alcohols, and alkyl alcohols with different carbon chain lengths were used in the standard assay. The activity was calculated using an extinction coefficient of 6200 M−1 cm−1 for NADH. One unit of enzyme activity was defined as the amount of enzyme that catalyzes the formation of 1 μmol of NADH per 1 min under assay conditions.
Purification of TMA-butanol DH
All operations were performed at 5 °C. About 50 mM triethanolamine-NaOH (TEA-NaOH) buffer (pH 7.0) containing 1 mM dithiothreitol (DTT) was used. Centrifugation was conducted at 12,000 × g for 20 min at 4 °C through purification procedures unless otherwise stated. The fungal cells, harvested from 250 mL culture broth, were disrupted using Bead-Beater (Biospec Products Inc., USA) with 50 mM TEA-NaOH buffer (pH 7.0) and 15 mL of zirconia–silica beads (0.6 mm) for 30 s under cooling with dry ice. After centrifugation at 8600 × g for 20 min at 4 °C, the resultant supernatant was brought to 30% saturation using ammonium sulfate. After centrifugation to remove the precipitate, the resultant supernatant was brought to 50% saturation with ammonium sulfate. The precipitate, which was collected by centrifugation, was dissolved in 50 mM TEA-NaOH buffer (pH 7.0). Then, an equal volume of 2.0 M ammonium sulfate solution in 50 mM TEA-NaOH buffer (pH 7.0) containing 1 mM DTT was added to the sample. The resulting solution was centrifuged. The supernatant was loaded onto a Phenyl-Toyopearl 650 M column (1.6 × 25 cm) equilibrated with 50 mM TEA-NaOH buffer (pH 7.0) containing 1.0 M ammonium sulfate, and 1 mM DTT. After the column had been washed using the same buffer, the enzyme was eluted with a linear gradient of ammonium sulfate (1.0–0 M) at a 1.5 mL/min flow rate. Active fractions were combined and dialyzed against the 50 mM TEA-NaOH buffer (pH 7.0).
The dialysate was next loaded onto a DEAE-Sepharose column (1.6 × 13.5 cm) equilibrated with 50 mM TEA-NaOH buffer (pH 7.0). After the column had been washed with the same buffer, the enzyme was eluted with a linear gradient of KCl (0–1.0 M) at a 1.5 mL/min flow rate. Active fractions were combined and concentrated using a Millipore Ultrafree 4 centrifugal filter device (MW: 10,000). The concentrated enzyme solution was loaded onto a Sephacryl S-200 column (1.6 × 90 cm) equilibrated with 50 mM TEA-NaOH buffer (pH 7.0) containing 0.1 M KCl and 1 mM DTT. The enzyme was eluted with the 50 mM TEA-NaOH buffer (pH 7.0) containing 0.1 M KCl and 1 mM DTT at a 1.0 mL/min flow rate. Active fractions were combined and concentrated using a centrifugal filter device (MW, 10,000; Millipore Ultra free 4; Nihon Millipore Ltd.). The concentrated enzyme solution was loaded onto a Mono-Q 5/50 GL column equilibrated with 20 mM TEA-NaOH buffer (pH 7.0) containing 1 mM DTT. After washing the column with the same buffer, the enzyme was eluted with a linear gradient of KCl (0–1.0 M) at a 0.5 mL/min flow rate. Active fractions were pooled and stored at −20 °C. Protein was measured using the method described by Lowry et al.Citation18) using bovine serum albumin as standard protein, or by absorbance at 280 nm.
Polyacrylamide gel electrophoresis and N-terminal amino acid sequencing
Polyacrylamide gel electrophoresis (Native-PAGE) was done using 7.5% gel according to the method described by Williams and Reisfeld.Citation19) The gels were either stained directly with CBB R-250 or used for activity staining to detect the active band. The enzyme activity in the gel was detected by incubating the gels in the reaction mixture of 150 mM glycine-NaOH buffer (pH 9.5), 1.98 mM NAD+, 1.13 mM TMA-butanol iodide, 0.24 mM nitroblue tetrazolium, and 64 μM 1-methoxy phenazine methosulfate. SDS–PAGE was done using 12.5% gel following the method of LaemmliCitation20) and stained with CBB. For N-terminal amino acid sequence analysis, the protein was electroblotted onto a PVDF membrane at a constant current of 126 mA using semidry blotting apparatus (AE-6677P/S/N; ATTO Bioscience). The PVDF membrane was stained with CBB. The protein band was excised from the gel and was used for Edman degradation with a sequencing system (491 PROCISE; Applied Biosystems, Foster City, CA, USA).
Results
Screening and purification of TMA-butanol DH from Fusarium merismoides var. acetilereum
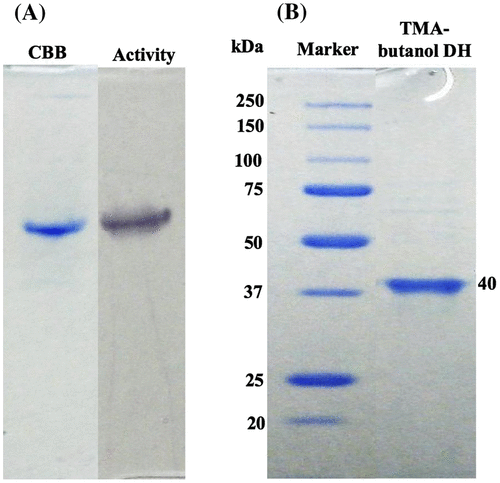
Initially, we screened various eukaryotic microbes including filamentous fungi and yeasts on TMA-butanol as the sole source of carbon and nitrogen. In growth tests, several Fusarium strains showed good growth. Furthermore, 60 strains of the genus Fusarium were screened for their ability to grow on TMA-butanol, using it as the sole source of carbon and nitrogen. F. merismoides var. acetilereum (NBRC30040), which showed high growth and the highest enzyme activity on the medium, was selected (see Table S1 in Supplementary materials). TMA-butanol DH activity increased with cell growth, reaching maximum activity at OD600 2–3 at 3 days of growth. The enzyme was purified homogeneously through six purification steps (see Table S2 in Supplementary materials). Activity staining showed a single band at the same position of the band detected on the CBB-stained gel (Fig. (A)).
Fig. 1. Assessment of homogeneity (A and B) of TMA-butanol DH. Enzyme purity was checked using Native-PAGE (A) and SDS–PAGE (B).
Characterization of the purified enzyme
The molecular mass of the native enzyme was estimated as 160 kDa using size exclusion chromatography on a TSK-gel G3000SW column (0.78 × 30 cm) equilibrated with 50 mM TEA-NaOH buffer (pH 7.0) containing 1 mM DTT and 0.1 M KCl.
The molecular mass of the denatured enzyme SDS–PAGE gel was 40 kDa (Fig. (B)). These results suggest that the purified enzyme is homotetramer having four identical subunits. The N-terminal amino acid was not able to determine using Edman degradation. Therefore, the N-terminal amino acid of the enzyme is regarded as modified.
The effect of pH and temperature on the activity and stability of the purified enzyme are shown in Figure (A)–(D). The optimal pH for the oxidation reaction was found to be 9.5 (Fig. (A)). That of the reduction reaction was 7.5 (Fig. (B)). The pH stability test showed that the enzyme is stable at pH 8.0 (Fig. (C)). At pH 9.5, it lost more than 55% of its activity, whereas the pH is optimal for enzyme reactions. The optimal temperature of the enzyme reaction was 45 °C. The relative activity between 35 and 50 °C was greater than 80% (Fig. (D)). Thermal stability tests showed that the activity was completely stable up to 30 °C, but at 45 °C, the enzyme lost about 60% of its activity (Fig. (D)).
Fig. 2. Effects of pH and temperature on the catalytic activity and stability of TMA-butanol DH.
Investigation of the effect of inhibitors on the enzyme activity revealed that the purified enzyme is strongly inhibited by ammonium persulfate, hydrogen peroxide, N-ethylmaleimide, 4-(chloromercuri) benzoic acid, and 1,10-phenanthroline, with remaining activity of less than 10% (Table ).
Table 1. TMA-butanol DH from Fusarium merismoides var. acetilereum and Pseudomonas sp. 13CM.
Substrate specificity and enzyme kinetics
Substrate specificity of the enzyme was tested using TMA-alcohols, DMA-alcohols, amino alcohols, and alkyl alcohols. Clearly, the enzyme had broad substrate specificity (Table ). However, the enzyme did not react to choline (TMA-ethanol), homocholine (TMA-propanol), DMA-propanol, methanol, 1-nonanol, or 1-decanol. Furthermore, kinetic parameters were calculated (Table ). Among TMA-alcohols (C4–C8), the enzyme recognized all of them as substrate, displaying higher affinity for TMA-pentanol (Km: 0.13 mM) and TMA-octanol (Km: 0.60 mM), than for TMA-butanol (Km: 2.2 mM). In terms of DMA-alcohols (C4–C8), the enzyme recognized all of them as substrate, displaying higher affinity for DMA-pentanol (Km: 0.36 mM), DMA-hexanol (Km: 0.18 mM), and DMA-octanol (Km: 0.25 mM), than that for DMA- butanol (Km: 2.9 mM). On the other hand, the longer the alkyl-carbon chain becomes in amino alcohol group and alkyl alcohol group, the higher the affinity for substrate becomes. Finally, 8-amino-1-octanol showed the lowest Km value (0.061 mM) and the highest Kcat/Km value (2140) among all tested substrates. And the enzyme did not recognize NADP+.
Table 2. Kinetic parameters between of Fusarium enzyme and Pseudomonas enzyme.
Discussion
This study newly revealed that several fungi of genus Fusarium were able to grow sufficiently on TMA-butanol as a carbon and nitrogen source. And we found that F. merismoides var. acetilereum inductively produced a first enzyme, NAD+-dependent TMA-butanol DH, in TMA-butanol degradation pathway. We detected the activity of NAD+-dependent TMA-butyraldehyde DH in the cell extract of F. merismoides var. acetilereum grown on TMA-butanol (22 mU/mg). This enzyme activity was detected in Neurospora crassa as l-carnitine biosynthetic enzyme.Citation21) Additionally, we newly detected the activity of NAD+-dependent l-carnitine DH (11 mU/mg). l-Carnitine DH activity was found in several bacteria,Citation10,11,22) but this enzyme activity has not been found in eukaryotic microbes. Therefore, the TMA-butanol degradation pathway of F. merismoides var. acetilereum is presumably the same as that of Pseudomonas sp. 13CM: TMA-butanol → TMA-butyraldehyde → γ-butyrobetaine → l-carnitine → 3-dehydrocarnitine.
Table presents some properties of Fusarium enzyme and Pseudomonas enzyme. Fusarium enzyme is a homotetramer having molecular mass of 160 kDa, whereas Pseudomonas enzyme is a monomer with molecular mass of 45 kDa. Both TMA-butanol DHs from Fusarium and Pseudomonas are strongly inhibited by 1,10-phenanthroline, which is known as a chelating agent for Fe2+ ions. Alcohol dehydrogenases are classified into three groups: zinc-dependent enzyme, group I; zinc-independent enzyme, group II; and iron containing or activated enzyme, group III.Citation23,24) Group III alcohol dehydrogenases are structurally diverse, having subunits with approximately 385 amino acid residues,Citation24,25) which is equivalent to the approximate molecular mass (40–45 kDa) of the subunit of Fusarium enzyme and of Pseudomonas enzyme. Consequently, both TMA-butanol DHs are regarded as members of NAD(P)+-dependent alcohol dehydrogenase superfamily group III.
Fusarium enzyme has broad substrate specificity, recognizing TMA-alcohols, DMA-alcohols, amino alcohols, and alkyl alcohols while Pseudomonas enzyme reacts to TMA-alcohols and DMA-alcohols (Table ). In the TMA-alcohol group, Fusarium enzyme displays the highest affinity for TMA-pentanol while Pseudomonas enzyme displays the highest affinity for TMA-butanol. Furthermore, in the DMA-alcohol group, Fusarium enzyme displays the highest affinity for DMA-hexanol while Pseudomonas enzyme displays the highest affinity for DMA-pentanol.
About substrate recognition mechanism, trimethylated amino group of choline is crucially important for binding at the active site of choline oxidase.Citation26,27) As with choline oxidase, Hassan et al. inferred that substrate binding site of Pseudomonas enzyme consists of an anionic site consisted of negatively charged or aromatic amino acid residue and a catalytic site. The anionic site recognizes the trimethylated amino moiety of the substrate. The catalytic site recognizes the hydroxyl moiety of the substrate.Citation14,16) In contrast, Fusarium enzyme recognizes alcohols of many kinds. Therefore, results suggest that Fusarium TMA-butanol DH has a unique substrate recognition mechanism that is distinct from that of Pseudomonas TMA-butanol DH.
Author contributions
Yuko, T analyzed the enzyme substrate specificity. Sae, T analyzed inhibitors effects and researched several preceding studies. Hiroshi F, Isam AMA, Jiro A, and Nobuhiro M wrote the article.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplemental material
The supplemental material for this paper is available at http://dx.doi.org/10.1080/09168451.2016.1177443.
TBBB_1177443_Supplementary_Material.doc
Download MS Word (90.5 KB)Acknowledgment
We are grateful to Professor Motoichiro Kodama, Tottori University, for providing various Fusarium strains.
Notes
Abbreviations: TMA, trimethylamino; DMA, dimethylamino; TMA-butanol, 4-N-trimethylamino-1-butanol; DH, dehydrogenase; NAD+, nicotinamide adenine dinucleotide; TEA, triethanolamine; DTT, dithiothreitol; SDS, sodium dodecyl sulfate; PAGE, polyacrylamide gel electrophoresis; CBB, Coomassie brilliant blue R-250; PVDF, polyvinylidene difluoride.
Related Research Data
References
- Grabinska-Sota E. Evaluation of impact of quaternary ammonium chlorides on water environment. Academic edition. Gliwice: Politechnika Śląska; 2004.
- Ueland PM. Choline and betaine in health and disease. J. Inherited Metab. Dis. 2011;34:3–15.10.1007/s10545-010-9088-4
- Vaz FM, Wanders RJA. Carnitine biosynthesis in mammals. Biochem. J. 2002;361:417–429.10.1042/bj3610417
- Yancey PH. Organic osmolytes as compatible, metabolic and counteracting cytoprotectants in high osmolarity and other stresses. J. Exp. Biol. 2005;208:2819–2830.10.1242/jeb.01730
- Tang WHW, Hazen SL. The contributory role of gut microbiota in cardiovascular disease. J. Clin. Invest. 2014;124:4204–4211.10.1172/JCI72331
- Thibodeaux CJ, van der Donk WA. Converging on a mechanism for choline degradation. Proc. Nat. Acad. Sci. U.S.A. 2012;109:21184–21185.10.1073/pnas.1219534110
- Nagasawa T, Mori N, Tani Y, et al. Characterization of choline dehydrogenase from Pseudomonas aeruginosa A-16. Agric. Biol. Chem. 1976;40:2077–2084.10.1271/bbb1961.40.2077
- Yamada H, Mori N, Tani Y. Properties of choline oxidase of Cylindrocarpon didymum M-1. Agric. Biol. Chem. 1979;43:2173–2177.10.1271/bbb1961.43.2173
- Mori N, Shirakawa K, Uzura K, et al. Formation of ethylene glycol and trimethylamine from choline by Candida tropicalis. FEMS Microbiol. Lett. 1988;51:41–44.10.1111/fml.1988.51.issue-1
- Mori N, Kasugai T, Kitamoto Y, et al. Purification and some properties of carnitine dehydrogenase from Xanthomonas translucens. Agric. Biol. Chem. 1988;52:249–250.10.1271/bbb1961.52.249
- Mori N, Mitsuzumi H, Kitamoto Y. Purification and properties of carnitine dehydrogenase from Pseudomonas sp. YS-240. J. Ferment. Bioeng. 1994;78:337–340.10.1016/0922-338X(94)90276-3
- Setyahadi S, Ueyama T, Arimoto T, et al. Purification and properties of a new enzyme, d-carnitine dehydrogenase, from Agrobacterium sp. 525a. Biosci. Biotechnol. Biochem. 1997;61:1055–1058.10.1271/bbb.61.1055
- Ahmed IAM, Arima J, Ichiyanagi T, et al. Isolation and characterization of homocholine-degrading Pseudomonas sp. strains A9 and B9b. World J. Microbiol. Biotechnol. 2010;26:1455–1464.10.1007/s11274-010-0320-z
- Hassan M, Morimoto S, Murakami H, et al. Purification and characterization of 4-N-Trimethylamino-1-butanol Dehydrogenase of Pseudomonas sp. 13CM. Biosci. Biotechnol. Biochem. 2007;71:1439–1446.10.1271/bbb.60510
- Hassan M, Okada M, Ichiyanagi T, et al. 4-N-Trimethylaminobutyraldehyde Dehydrogenase: purification and characterization of an enzyme from Pseudomonas sp. 13CM. Biosci. Biotechnol. Biochem. 2008;72:155–162.10.1271/bbb.70514
- Hassan M. Enzymatic studies on the degradation of 4-N-Trimethylamino-1-butanol by Pseudomonas sp. 13CM [ PhD thesis]. Tottori, Japan: Tottori University; 2008.
- Bari MdR, Akai N, Arima J, et al. Evaluation of genes encoding 4-N-trimethylaminobutyraldehyde dehydrogenase and 4-N-trimethylamino-1-butanol dehydrogenase from Pseudomonas sp. 13CM. Int. J. Agric. Biol. 2013;15:238–244.
- Lowry OH, Rosebrough NJ, Farr AL, et al. Protein measurement with the folin phenol reagent. J. Biol. Chem. 1951;193:265–275.
- Williams DE, Reisfeld RA. Disk electrophoresis in polyacrylamide gels: extension to new conditions of pH and buffer. Ann. N.Y. Acad. Sci. 1964;121:373–381.
- Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685.10.1038/227680a0
- Kaufman R, Broquist HP. Biosynthesis of carnitine in Neurospora crassa. J. Biol. Chem. 1977;252:7437–7439.
- Hanschmann H, Ehricht R, Kleber HP. Purification and properties of l(−)-carnitine dehydrogenase from Agrobacterium sp. Biochem. Biophys. Acta. 1996;1290:177–183.10.1016/0304-4165(96)00020-7
- Elleuche S, Fodor K, von der Heyde A, et al. Group III alcohol dehydrogenase from Pectobacterium atrosepticum: insights into enzymatic activity and organization of the metal ion-containing region. Appl. Microbiol. Biotechnol. 2014;98:4041–4051.10.1007/s00253-013-5374-z
- Reid MF, Fewson CA. Molecular Characterization of Microbial Alcohol Dehydrogenases. Crit. Rev. Microbiol. 1994;20:13–56.10.3109/10408419409113545
- Elleuche S, Antranikian G. Bacterial group III alcohol dehydrogenase – function, evolution and biotechnological applications. OA Alcohol. 2013;3:1–6.
- Gadda G, Powell NLN, Menon P. The trimethylammonium headgroup of choline is a major determinant for substrate binding and specificity in choline oxidase. Arch. Biochem. Biophys. 2004;430:264–273.10.1016/j.abb.2004.07.011
- Enokibara S. Purification and characterization of an alkaliphilic choline oxidase of Fusarium oxysporum. Biosci. Biotechnol. Biochem. 2012;76:2219–2224.10.1271/bbb.120513