
Abstract
Ubiquitous microbial communities in river sediments actively govern organic matter decomposition, nutrient recycling, and remediation of toxic compounds. In this study, prokaryotic diversity in two major rivers in central Thailand, the Chao Phraya (CP) and the Tha Chin (TC) distributary was investigated. Significant differences in sediment physicochemical properties, particularly silt content, were noted between the two rivers. Tagged 16S rRNA sequencing on a 454 platform showed that the sediment microbiomes were dominated by Gammaproteobacteria and sulfur/sulfate reducing Deltaproteobacteria, represented by orders Desulfobacteriales and Desulfluromonadales together with organic degraders Betaproteobacteria (orders Burkholderiales and Rhodocyclales) together with the co-existence of Bacteroidetes predominated by Sphingobacteriales. Enrichment of specific bacterial orders was found in the clayey CP and silt-rich TC sediments, including various genera with known metabolic capability on decomposition of organic matter and xenobiotic compounds. The data represent one of the pioneered works revealing heterogeneity of bacteria in river sediments in the tropics.
Graphical abstract
Analysis of bacterial diversity by tagged 16S rRNA sequencing revealed enrichment of specific taxa in CP and TC rivers.
River sediment is a complex ecological soil niche that results from dynamic accumulation of organic matters, and is inhabited by a wide variety of eukaryotic and prokaryotic microorganisms. Changes in physicochemical characteristics of sediment brought about by urbanization, agriculture, etc., lead to corresponding alteration in microbial community structures.Citation1) Sediment microorganisms are exposed to loads of organic matters and toxic pollutants from land, which has forced them to adapt to the seasonal fluctuation of these substances. Understanding the dynamic nature of river sediment and its microbiome are thus of ecological interest.
The make-up of aquatic microbiomes has direct environmental, ecological, and public health impacts.Citation2,3) Culture-independent analysis of environmental metagenomes by next generation sequencing (NGS) is currently the method of choice for studying community structure and functions of microbial assemblages in various ecosystems. NGS metagenome studies of river sediment in temperate regions have been reportedCitation4); however, to our knowledge an in-depth study of river sediment from the tropics has not been reported. Niches unique to the tropics harbor microbes distinct from temperate regionsCitation5,6); hence, study of microbial diversity in tropical river sediment is thus of interest owing to the expected higher biodiversity and organic matter recycling activity.Citation7,8)
The Chao Phraya (CP) river basin covers a large area of Central Thailand and is a major river system in Southeast Asia. The hydrology of this basin is dynamic owing to seasonal drought and flooding. A previous study of floodwater in the aftermath of the 2011 flood that inundated a large area of the CP river basinCitation9) revealed the transient bacterial and fungal diversity of sediment and water samples from flooded land; however, the microbial communities in established sediments accumulated in this river basin were not studied. Herein, sediments from two major rivers in central Thailand, the CP, flowing through major industrial estates and the Tha Chin (TC), a distributary of CP were investigated. The physicochemical characteristics of sediment samples were measured and the inhabitant prokaryotic communities were analyzed using high-throughput tagged 16S rRNA gene pyrosequencing. Associations between sediment physicochemical characteristics and bacterial assemblages residing in the two rivers were analyzed. The results provide insights into the heterogenous bacterial communities in tropical river sediments and delineate their potential biological functions in this unique freshwater ecosystem.
Materials and methods
Sample collection
Sediment samples were collected from two flood-prone rivers, the CP and TC, using a grab sampler during June–August 2012. Six samples were obtained from each river. The geographic coordinates of each location are provided in Table S1. The CP river is the main river in Thailand, which flows through Bangkok and several major cities in the great central plain to the gulf of Thailand. Sediment samples were obtained from five locations along the CP river (CP1–CP5) close to the delta, and another sample was taken much further upstream (CP6) in Pranakorn Sri Ayutthaya province, at a location close to three industrial estates to the estuary area in Samut Prakarn province (supplementary data Fig. S1(A) and S1(B)). The TC river is a distributary of the CP river. It branches off from the CP in Chainat province and flows westerly through the central plain, until it mouths into the Gulf of Thailand at Samut Sakhon province. The TC river runs through densely populated settlements and agricultural areas in the area, and it is used for water supply, aquaculture, and as a sink for wastewater discharge. Six TC samples (TC1–TC6) were collected along the river, including a sample (TC1) from the delta surrounded by mangrove forest (see supplementary data Fig. S1(C)). The depth of water at each location was <2 m at the time of sampling.
Physicochemical analysis of sediment samples
Sediment pH was measured using a pH meter. Sediment pH was determined in water (pH H2O) with a sediment to solution ratio of 1:5. The pH in sodium fluoride solution (1 M NaF buffered to pH 8.2) was also measured at a solid to solution ratio of 1:5. The pH measurement in hydrogen peroxide oxidation (pH H2O2) was performed using a solid to solution ratio of 1:2 after sample stirring with a glass rod for 30 min (National Soil Survey Centre, 1996). Electric conductivity (EC) was measured using a conductivity meter. The total carbon content was calculated from the summation of organic carbon (OC) and inorganic carbon (IC). The OC content was determined by the Walkley and Black wet oxidation procedure.Citation10) The IC content was analyzed by gravitational technique after carbonate removal with 3 M HCl.Citation11) Total nitrogen was determined by the Kjeldahl method.Citation12) The concentrations of phosphorus (P), sulfur (S), and other trace elements in sediments were analyzed by a combination of an aqua regia digestion (3:1 HCl: HNO3, 130 °C)Citation13) and a 1 M HCl extraction.Citation14) Concentrations of P and S from the extractions (i.e. pseudo total and reactive elements) were determined by colorimetric procedures.Citation15) It should be noted that the aqua regia digestion method could not completely destroy silicates and other highly resistant sand-sized minerals. The precision and accuracy of analysis for the elements were monitored in a batch system with frequent reference to a reagent blank and the certified international reference stream sediments (STSD1; Canada Center for Mineral and Energy Technology, CANMENT). Reactive elements were extracted by 1 M HCl over 24 h with a solid: solution ratio of 1:20.Citation14) This procedure can dissolve the elements mostly related to micro-crystalline Fe oxides, sulfide and carbonate minerals, elements adsorbed to reactive surfaces of organic constituents and clay minerals.Citation14) Concentrations of polycyclic aromatic hydrocarbons (PAHs) were analyzed for PAH concentration using standard US.EPA. method 8270C.
Next-generation sequencing of 16S rRNA gene amplicons
Metagenomic DNA samples were extracted using a Power Soil DNA extraction kit (MO BIO Laboratories, Inc, Carlsbad, CA). Purified metagenomic DNA was used as a template for amplification of the partial 16S rRNA gene using universal bacterial primers 338F (5′-ACTCCTACGGGAGGCAGCAG-3′)Citation16) and 786R (5′-CTACCAGGGTATCTAATC-3′) encompassing the V3 and V4 hypervariable regions in the prokaryotic 16S rRNA geneCitation17) attached with tagged barcode sequences.Citation18) Polymerase chain reactions were performed using DyNAzyme EXT DNA polymerase (Finnzyme, Espoo, Finland) on a MyCycler thermocycler (Bio-Rad, Hercules, CA) using pre-denaturation at 94 °C for 3 min, and then 30 cycles of denaturation at 94 °C for 1 min, annealing at 55 °C for 1 min, and extension at 72 °C for 1 min 45 s with a final extension at 72 °C for 10 min. The amplicons were gel-purified using QIAGEN Gel Extraction Kit (QIAGEN, Hilden, Germany) and were quantified using a NanoDrop ND-1000 spectrophotometer (Peqlab Biotechnologie GmbH, Erlangen, Germany).
The purified 16S rRNA amplicons were processed for pyrosequencing on a 454-life Science GS-FLX genome sequencer system (Roche, Branford, CT) following the manufacturer’s recommended protocols. The enriched DNA beads were annealed with the sequencing primer and subject to GS FLX sequencing chemistry (GS FLX Titanium sequencing kit). All sequences were deposited in NCBI Sequence Read Archive (SRA) under the accession number SRP057685.
Data cleaning and analysis
The sequencing data-set was initially processed by removing low-quality score reads with cutoff of 20 for Phred quality score. Next, the remaining sequences were de-multiplexed into 12 groups based on their tagged sequences using QIIME (Quantitative Insights Into Microbial Ecology).Citation19) Chimera, hybrid sequences merged from multiple parent sequences from the PCR amplification error, were identified and removed by UCHIMECitation20) using the referenced data-set from the Ribosomal Database Project (RDP).Citation21) Sequences that were 150 bp or longer were selected for further analysis. Taxonomic classification was performed using the composition-based RDP-classifier (version 2.11)Citation22) with bootstrap cutoff of 50%, which allowed ~95% accuracy on identification at the genus level from the V3 and V4 regions to assign taxonomy by the Naïve Bayesian classification method. Operational taxonomic units (OTUs) were determined by the furthest neighbor clustering method in MOTHURCitation23) at a sequence dissimilarity level of 0.03. In order to reduce the bias of unequal numbers of sequences in each sample, normalization was performed by sub-sampling using the read number of the sample with the fewest reads as cutoff. The nonparametric diversity indices including Shannon–Weaver index and the Chao1 richness estimator were calculated at the genetic distance of 0.03 to measure the diversity of bacterial species among the data-set. Good’s coverage (G), an estimator for sampling completeness, was calculated as G = 1 − (n/N), where n is the number of OTUs that have been sampled once and N is the total number of individuals in the sample. The difference of microbial communities between the two rivers was measured using unweighted UniFrac distanceCitation24) and visualized by Principal Coordinates Analysis (PCoA). Microbial and physicochemical profiles were statistically compared using White’s non-parametric t-testCitation25) and Principal Component Analysis (PCA) implemented in STAMP (Statistical Analysis of Metagenomic Profiles).Citation26) The relationship between total OC and microbial profile to the distance from delta was measured using Pearson’s correlation coefficient.
Results
Physicochemical properties of sediment samples
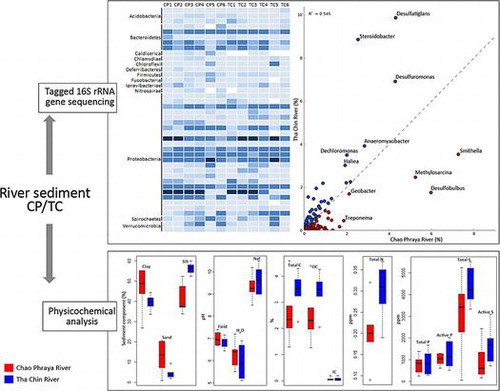
The measured physicochemical properties of sediments sampled from the two rivers are given in Table and Table S1. The CP sediments (CP1–CP3) contained high proportions of clay particles (52–55%), while samples taken from sites upriver (CP4–CP6) had relatively higher proportions of high sand sized particles (19–21%). Field sediment pH measurements were in the neutral range (pH field = 6.7–7.4). The values of pH measured in water varied from slightly acidic to neutral (pH H2O = 5.5–7.2), with the lowest value observed at site CP2. The pH measured in NaF solution ranged from 8.5 to 10.2. This pH value provides an estimation of the amounts of exposed hydroxyl groups (OH–) on the reactive surfaces of organic constituents, aluminosilicate minerals, and sesquioxides (i.e. oxides and oxyhydroxides of Fe and Al). Elevated values of pH in NaF demonstrated that OH– groups were abundant, which provide sites for adsorption of plant nutrients and potentially toxic trace elements. The values of EC for most samples varied in the range of 0.11–2.5 ms cm−1. However, the sediment CP5 had a remarkably high EC value of 1927 ms cm−1. Total OC varied from 1.3 to 4.4% with the mean value of 2.6%. The CP6 sample had the lowest OC content (1.3%), whereas the CP5 sample had the highest OC accumulation (4.6%). Concentrations of IC in all samples were minor (0.010–0.199%). The nitrogen content was low in all samples (0.090–0.37%). Active P varied from 609 to 1298 mg kg−1. Reactive S also differed markedly among the samples (166–2431 mg kg−1). The presence of sulfur in the sediments was also evidenced as a substantially lower pH in H2O2 compared with pH measured in water due to reduction of sulfur to sulfuric acid.
Table 1. Physicochemical properties of sediments of the Chao Phraya and the Tha Chin rivers in Thailand.
Most of the TC samples had a silty clay texture, as reflected by the significantly higher silt content compared with CP samples (p-value < 0.05 according Welch’s t test), while the TC1 sample collected at the estuary of the river had the highest sand content (Table ). The field pH varied from slightly acidic to neutral, with samples taken from upstream locations tending to be less acidic. The EC varied in the range of 1.0–1216 ms cm−1, with the highest value measured for the TC6 sample, reflecting that this sample also contained a large amount of soluble salts. The pH measured in H2O ranged from pH 5.1 and pH 6.9 with the mean value of 5.9. The mean OC content of TC sediment samples was higher than CP sediment samples. A trend of decreasing OC content with sampling distance from delta (2.3–4.3%) was observed for both CP and TC samples (Fig. S2). Sediment samples from TC contained high amounts of active S ranging from 710 to 3097 mg kg−1 and active P from 513 to 2017 mg kg−1.
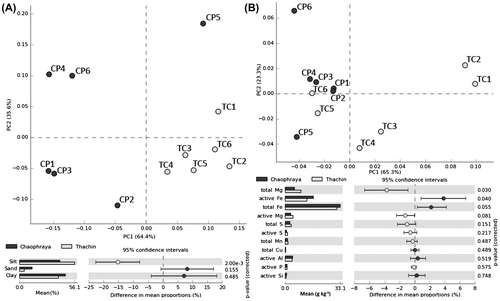
Concentrations of various PAH compounds including naphthalene, acenapthylene, acenaphtene, fluorine, phenanthrene, anthracene, fluoranthene, benzo(a)anthracene, chrysene, benzo(b)fluranthene, benzo(k)fluranthene, benzo(A)pyrene, indeno, dibenzo, and benzo(ghi)perylene in all sediment samples from both rivers were negligible (<0.1 mg/kg). PCA (Fig. (A)) and box plot (Fig. S3) showed that sediment soil compositions in CP samples were significantly different from TC samples. The silt content from TC samples was significantly higher than that from CP samples according to White’s non-parametric t-test (p-value < 0.05). No significant difference was found for the content of trace elements of samples collected from different locations on both rivers except total magnesium (Mg) and active iron (Fe) (Fig. (B)). The content of total OC tended to increase with the distance from the deltas of both rivers, according to Pearson’s correlation coefficients (0.78 for CP and 0.84 for TC) (Fig. S4).
Fig. 1. Principal Component Analysis and White’s non-parametric t-test of (A) sediment component (percent redundancy); (B) total and active trace elements (g kg−1).
Pyrosequencing data-sets and bacterial diversity
Purified environmental DNA was used as a template for amplification of the partial 16S rRNA gene (0.44 kb). The pyrosequencing data-set is summarized in Table . In total, 296,370 filtered reads were obtained. The average sequence length (401–410 bases) and %GC content (53.98–55.37%) were in the same range among all samples. The microbial community richness was in the same range among all samples, which ranged from 1286 to 1966 OTUs. Moreover, all samples showed similar levels of taxonomic diversity, as shown by Chao1 and Shannon diversity indices. The taxa present were sampled to similar extents in each sample and significantly covered the majority of bacterial diversity, as shown by Good’s coverage index which varied from 75.75 to 89.54% at the species level.
Table 2. Summary of pyrosequencing data-set and diversity index of the 16S rRNA gene sequences at 0.03% dissimilarity.
Taxonomic analysis of microbial community from sediment samples
Taxonomic classification of bacteria showed that only 29.84% of the sequences could be assigned to the genus level, while the remainder could only be roughly assigned to higher taxonomic levels. 8.89% of the total sequences could not be classified into any known phyla and are indicated as unclassified bacteria. The bacterial population was assessed across the two river microbiomes in terms of the total number of possible bacterial taxa present. In terms of overall diversity, two major phyla were shared among all samples. Proteobacteria was the predominant phylum in all sediment samples, accounting for 70.7% of the total sequences on average, followed by the closely related phylum Bacteroidetes as the second most abundant taxon (10.3% of the total sequences) (Fig. (A)). Other phyla such as Acidobacteria, Actinobacteria, Chloroflexi, Firmicutes, and Verrucomicrobia were present as minorities, although they are present in all samples and collectively account for an average of 1% of sequences. The fact that these bacterial taxa were present among all samples might indicate that they play significant roles in the ecosystem.
Fig. 2. The relative abundance and taxonomic profiles of (A) Bacteria in Chao Phraya (CP1-6) and Tha Chin (TC1-6) sediment samples; (B) Proteobacteria in Chao Phraya (CP1-6) and Tha Chin (TC1-6) sediment samples; (C) Heatmap representation of the major bacterial orders.

The diversity of Proteobacteria, which constituted 52.80–77.82% of the total among the samples, is shown in more detail in Fig. (B). Diverse groups of Proteobacteria were found in different sediment samples from the two rivers. The majority of bacteria detected in the phylum Proteobacteria belonged to the order Gammaproteobacteria, which accounted for 31.94% of all sediment bacterial genera on average, and consisted of a highly diverse range of bacteria. The second most commonly represented order was Deltaproteobacteria (26.41%). The Gammaproteobacteria in most samples were dominated by unclassified Gammaproteobacteria incertae sedis, followed by orders Chromatiales and Methylococcales, suggesting that unique species may be represented among these samples. The abundance of Gammaproteobacteria incertae sedis was highest in the sediments collected near the deltas of both rivers and tended to decrease with distance from the delta, as reflected from the overall decreasing trend of their abundance from CP1 to CP6 and TC1 to TC6 (Perason’s correlation coefficients between Gammaproteobacteria incertae sedis (% of assigned sequences) and distance from delta are −0.90 and −0.89 for CP and TC, respectively) (Fig. S5). The Deltaproteobacteria was predominated by sulfate-reducing bacteria belonging to orders Desulfobacterales (9.95%), Synthrophobacteriales (4.62%), and Desulfuromonadales (2.93%) with a substantial fraction (1.45–8.25%) of Myxococcales in some samples. The highest abundance of Deltaproteobacteria was found in CP5 and TC5, which were the only two samples not dominated by Gammaproteobacteria. A substantial number of Betaproteobacteria was also found in the sediments with Alphaproteobacteria as the minority. The highest abundance of Betaproteobacteria was found in the samples furthest upriver, i.e. CP6 and TC6 where the microbiomes contained a substantial fraction of Burkhoderriales and Rhodocyclales together with unclassified Betaproteobacteria.
The proportions of the most abundant bacterial orders among all sediment samples are shown in Fig. (C). Several Proteobacteria orders presented as commonly existing taxa in both rivers including the sulfate- and sulfur-reducing bacteria Desulfobacteriales and Desulfluromonadales as well as order Myxococcales and Synthrophobacterales in class Deltaproteobacteria in addition to the Betaproteobacteria orders Burkholderiales and Rhodocyclales. These taxa were the six most abundant Proteobacteria taxa in the microbiomes from all samples, whereas Bacteroidetes in all samples were represented mostly from the order Sphingobacteriales.
Comparative analysis of bacterial diversity in CP and TC
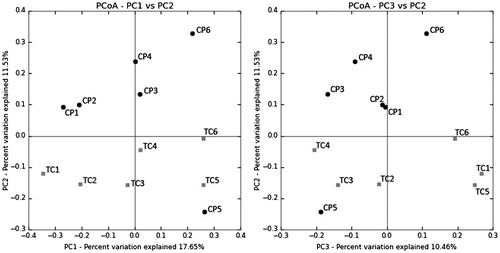
The dissimilarity of bacterial communities between the two rivers was measured using Unifrac method. The PCoA of the unweighted distance metric (Fig. ) shows that the CP samples cluster separately from TC samples. All of TC samples were on the quadrant 3 and 4, meanwhile most of CP samples were on the quadrant 1 and 2 except CP5. This would be related to the highest sand content with the lowest clay among the sample (Table ), which may result in a more unique microbial community in this sample.
Fig. 3. Relationships of microbial communities illustrated by Principal Coordinates Analysis (PCoA) of the unweighted UniFrac distance matrix.
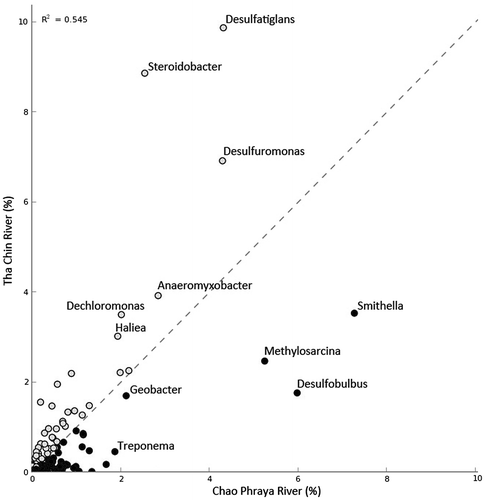
The bacterial distribution was further studied at the genus level as shown in Fig. . Geobacter, Methylosarcina, Smithella, Desulfobulbus, and Treponema were more abundant in the CP samples compared with the TC samples. In contrast, Desulfatiglans, Steroidobacter, Desulfuromonas, Anaeromyxobacter, Dechloromonas, and Haliea were more abundant in TC samples. The results clearly revealed heterogeneity of bacteria between the two river sediment samples.
Fig. 4. Scatter plot showing relative abundances of bacterial genera from Chao Phraya with Tha Chin rivers.
Discussion
The properties of river sediment vary considerably, depending on the nature of diffused minerals and dissolved organic matter. Physicochemical analysis of sediments obtained in this study revealed significant variation in OC content and particle size distribution between CP and TC samples. This difference is expected to contribute to variation in groups of inhabiting microorganisms with different metabolic capabilities which interactively play roles in. complex biochemical processes directly related to water quality.
Using the culture-independent approach, various groups of bacteria were identified in the river sediments in this study, including those with previously reported inherent capabilities as primary producers, saprophytic microbes, and sulfate reducers. Several bacterial phyla are common in sediments from both CP and TC rivers. Proteobacteria, the largest bacterial phylum with versatile metabolic capabilities, is the predominant bacterial group in sediments from both rivers. The Proteobacteria comprise mainly Gamma- and Deltaproteobacteria. Gammaproteobacteria are predominant in sediment from estuary and coastal areas,Citation27) while Deltaproteobacteria are common in sediments from freshwater and marine environments.Citation7) The majority of the Gamma- and Deltaproteobacteria in the CP and TC sediments are sulfur reducers. The complex organic matters in these river sedimentsCitation28) can also support other primary consumers and saprophytic microbes including Bacteroidetes, Firmicutes, and Actinobacteria, which collectively constitute a complex ecological niche in the sediment ecosystem under conditions with varying oxygen availability.
Enrichment of Gammaproteobacteria has been reported in marine sediment and intertidal wetland, while Deltaproteobacteria are present in sediments from freshwater and marine environments,Citation7) and Gamma-/deltaproteobacterial orders were found to be predominant in flood water.Citation9) The majority of these bacteria found in aquatic sediments are sulfur reducers. The Deltaproteobacteria comprise a branch of predominantly aerobic genera, which contains most of the known sulfate- and sulfur-reducing bacteria alongside several other anaerobic bacteria. The presence of various sulfate- and sulfur reducing Deltaproteobacteria in the sediment samples in this study was related to the substantial amount of sulfur in the environmental samples, particularly in TC2 where the highest enrichment of Desulfobacterales were found with the highest active sulfur content in this environmental sample.
Besides the predominant Gamma- and Deltaproteobacteria, minor populations of Alpha- and Betaproteobacteria were identified in the CP and TC sediments. The most prominent order of Betaproteobacteria is Burkholderiales, which comprises the families Burkholderiaceae, Oxalobacteraceae, Alcaligenaceae, and Comamonadaceae. These bacterial taxa are strictly aerobic and facultative anaerobic chemoorganotrophs, obligate, and facultative chemolithotrophs, nitrogen-fixing organisms, as well as plant, animal, and human pathogens.Citation29) Some of them contain genes encoding dioxygenase protein to degrade a vast array of aromatic compounds, including several priority pollutants. Although Burkholderiales were found as a minority in the CP and TC sediments, these bacteria were found as a major taxon in the Mississippi river in a recent study using high throughput sequencing of 16S rRNA gene diversity on an Illumina platform. Environmental factors including temperature, rainfall, and nutrient concentrations were shown to have significant influences on seasonal dynamic changes in the bacterial community structures in this habitat.Citation30) Influences of organic matter properties as well as pH on shaping the diversity of sediment bacteria have also been documented for other river systems.Citation31,32) The diversity of CP and TC river sediment communities found in this study is different from that reported in the Pearl river, China, where Acidobacteria, Nitrospora, Verrucomicobia, and Alpha- and Beta-proteobacteria are in abundance.Citation7)
At the genus level, Geobacter, Methylosarcina, Smithella, Desulfobulbus, and Treponema were among the most enriched bacterial taxa in CP, possibly reflecting the clayey nature of this sediment, whereas Sulfurovum, Hydrogenophaga, Haliea, Anaeromyxobacter, Desulfuromonas, and Dechloromonas predominate the silt-rich TC sediment. This community structure suggested complex activities involving aerobic and anaerobic metabolism on the decomposition and mineralization of organic matter and xenobiotic compounds in the sediments. Recent metagenomic study reports discovery of various oxidoreductases and hydrolases degrading pollutants especially poly-aromatic, halogenated and poly-ester molecules from complex ecosystems such as river sediments, sludge, and polluted water.Citation33) Many of the bacterial taxa identified in this study using the metagenomic approach have been previously isolated from aquatic or marine environments and their capabilities on bioremediation under aerobic and anaerobic conditions were shown.Citation34–36) These include various Deltaproteobacteria, which are capable of sulfate reduction (Desulfobulbus) and sulfur reduction (Desulfuromonas). Geobacter is a member of Deltaproteobacteria, which is recognized as a powerful microbe in bioremediation under anaerobic conditions. They are capable of oxidizing organic compounds and metals, including iron and petroleum compounds into carbon dioxide by using iron or other metals as electron acceptors.Citation37,38) Anaeromyxobacter is a metabolically versatile Deltaproteobacteria with a bioremediation capability.Citation39,40) This group of bacteria can utilize a wide variety of electron acceptors for growth under anoxic conditions, including ortho-substituted halophenols, nitrate, oxygen, fumarate, and iron(III). Betaproteobacteria play important roles in decomposition of organic matter, as exemplified by the genus Hydrogenophaga, which constitutes a group of versatile anaerobic bacteria shown to decompose organic matter in wastewater in a biofilm bioreactor designed for chemical oxygen demand removal.Citation41) The metabolic capability of Hydrogenophaga on degrading polluted hydrocarbons including benzene, methyl-tert-butyl ether, and polycyclic aromatic hydrocarbons (PAHs) has been reported.Citation42,43) Dechloromonas, another Betaproteobacteria found in the river sediment microbiomes, was also reported as a versatile degrader of aromatic compounds.Citation44,45) Methylosarcina is an aerobic methanotroph that utilizes methane and methanol as carbon sources, and ammonia, nitrate, and various organic compounds as sources of nitrogen. The role of Methylosarcina on consuming methane generated from the sediments or anaerobic zones in lakes has been reported.Citation46,47) According to the revealed diversity profiles of the overall microbial communities in both rivers and their inherent metabolic capabilities, this study suggested predominance of mixed anaerobic and aerobic metabolism related to complex biochemical activities from decomposition of organic matter, mineralization of nutrients, and degradation of xenobiotic compounds in the river sediments, which can potentially be related to environmental quality of the ecosystem.
Conclusion
In conclusion, our study of bacterial diversity reveals diverse bacterial taxa reflecting the biochemical activities and microbial processes in tropical river sediment. Similarity and differences in bacterial taxa distribution in the sediments from both rivers indicated shared and different dominated biochemical activities and microbial processes in the two rivers. This diversity survey provides a foundation for more detailed studies on how microbial communities in river sediments can adapt to changes in physicochemical conditions resulting from natural processes and human activities.
Author contributions
HT collected sediments and processed DNA samples for analysis. WM was responsible for bioinformatics analysis of the data-set and prepared the manuscript. WW analyzed physicochemical properties of sediment samples. OP, AY, NF, and LE provide information and comments for the manuscript. VC was a project co-principal investigator and prepared the manuscript.
Disclosure statement
No potential conflict of interest was reported by the authors.
Funding
This project was financially supported by the National Science and Technology Development Agency [grant number P-12-01060] under the J-RAPID program.
Supplemental material
The supplemental material for this paper is available at http://dx.doi.org/10.1080/09168451.2016.1234927.
TBBB_1234927_Supplemental_Material.docx
Download MS Word (1.8 MB)Acknowledgments
Any opinions, findings, and conclusions or recommendations expressed in this material are those of the author(s) and do not necessarily reflect the views of the NSTDA and National Science Foundation.
Related Research Data
References
- Acosta-Martínez V, Dowd S, Sun Y, et al. Tag-encoded pyrosequencing analysis of bacterial diversity in a single soil type as affected by management and land use. Soil Biol. Biochem. 2008;40:2762–2770.10.1016/j.soilbio.2008.07.022
- Alongi DM The role of bacteria in nutrient recycling in tropical mangrove and other coastal benthic ecosystems. In: Sasekumar A, Marshall N, Macintosh DJ, editors. Ecology and conservation of southeast Asian marine and freshwater environments including wetlands. Developments in Hydrobiology. 98. Dordrecht: Springer; 1994. p. 19–32.10.1007/978-94-011-0958-1
- Cabral JP. Water microbiology. Bacterial pathogens and water. Int. J. Environ. Res. Publ. Health. 2010;7:3657–3703. Epub 2010 Dec 09.10.3390/ijerph7103657
- Xia X, Xia N, Lai Y, et al. Response of PAH-degrading genes to PAH bioavailability in the overlying water, suspended sediment, and deposited sediment of the Yangtze River. Chemosphere. 2015;128:236–244. Epub 2015 Feb 28.10.1016/j.chemosphere.2015.02.011
- Kanokratana P, Uengwetwanit T, Rattanachomsri U, et al. Insights into the phylogeny and metabolic potential of a primary tropical peat swamp forest microbial community by metagenomic analysis. Microb. Ecol. 2011;61:518–528. Epub 2010 Nov 09.10.1007/s00248-010-9766-7
- Somboonna N, Assawamakin A, Wilantho A, et al. Metagenomic profiles of free-living archaea, bacteria and small eukaryotes in coastal areas of Sichang island, Thailand. BMC Genomics. 2012;13:S29.
- Wang Y, Sheng HF, He Y, et al. Comparison of the levels of bacterial diversity in freshwater, intertidal wetland, and marine sediments by using millions of illumina tags. Appl. Environ. Microbiol. 2012;78:8264–8271. Epub 2012 Sep 25.10.1128/AEM.01821-12
- Costa PS, Reis MP, Ávila MP, et al. Metagenome of a microbial community inhabiting a metal-rich tropical stream sediment. PLoS ONE. 2015;10:e0119465. Epub 2015 Mar 06.10.1371/journal.pone.0119465
- Mhuantong W, Wongwilaiwalin S, Laothanachareon T, et al. Survey of microbial diversity in flood areas during Thailand 2011 flood crisis using high-throughput tagged amplicon pyrosequencing. PLoS ONE. 2015;10:e0128043. Epub 2015 May 29.10.1371/journal.pone.0128043
- Nelson DW, Sommers LE. Total carbon, organic carbon, and organic matter. In: Sparks DL, Page AL, Helmke PA, Loeppert RH, editors. Methods of soil analysis: part 3-chemical methods. SSSA Book Series No. 5. Madison, WI: Soil Science Society of America, American Society of Agronomy; 1996. p. 961–1010.
- Allison LE, Moodie CD. Carbonate. In: Norman AG, editor. Methods of soil analysis part 2 chemical and microbiological properties. Agronomy monograph. Madison, WI: American Society of Agronomy, Soil Science Society of America; 1965. p. 1379–1396.
- Bremner JM, Sparks DL, Page AL, et al. Nitrogen-total. Methods of soil analysis: part 3-chemical methods. Madison, WI: Soil Science Society of America, American Society of Agronomy; 1996.
- Lynch J. Additional provisional elemental values for LKSD-1, LKSD-2, LKSD-3, LKSD-4, STSD-1, STSD-2, STSD-3 and STSD-4. Geostandard. Newslett. 1999;23:251–260.10.1111/ggr.1999.23.issue-2
- Wallmann K, Hennies K, König I, et al. New procedure for determining reactive Fe(III) and Fe(II) minerals in sediments. Limnol. Oceanogr. 1993;38:1803–1812.10.4319/lo.1993.38.8.1803
- Sparks DL, Page AL, Helmke PA, et al. Methods of soil analysis part 3 – chemical methods. Madison, WI: Soil Science Society of America, American Society of Agronomy; 1996.
- Humblot C, Guyot JP. Pyrosequencing of tagged 16S rRNA gene amplicons for rapid deciphering of the microbiomes of fermented foods such as pearl millet slurries. Appl. Environ. Microbiol. 2009;75:4354–4361. Epub 2009 May 05.10.1128/AEM.00451-09
- Baker GC, Smith JJ, Cowan DA. Review and re-analysis of domain-specific 16S primers. J. Microbiol. Methods. 2003;55:541–555.10.1016/j.mimet.2003.08.009
- Meyer M, Stenzel U, Hofreiter M. Parallel tagged sequencing on the 454 platform. Nat. Protocols. 2008;3:267–278.10.1038/nprot.2007.520
- Caporaso JG, Kuczynski J, Stombaugh J, et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods. 2010;7:335–336. Epub 2010 Apr 13.10.1038/nmeth.f.303
- Edgar RC, Haas BJ, Clemente JC, et al. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics. 2011;27:2194–2200. Epub 2011 Jun 28.10.1093/bioinformatics/btr381
- Cole JR, Wang Q, Fish JA, et al. Ribosomal database project: data and tools for high throughput rRNA analysis. Nucleic Acids Res. 2014;42:D633–D642. Epub 2013 Nov 30.10.1093/nar/gkt1244
- Wang Q, Garrity GM, Tiedje JM, et al. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007;73:5261–5267. Epub 2007 Jun 26.10.1128/AEM.00062-07
- Schloss PD, Westcott SL, Ryabin T, et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009;75:7537–7541. Epub 2009 Oct 06.10.1128/AEM.01541-09
- Lozupone C, Knight R. UniFrac: a new phylogenetic method for comparing microbial communities. Appl. Environ. Microbiol. 2005;71:8228–8235. Epub 2005 Dec 08.10.1128/AEM.71.12.8228-8235.2005
- White JR, Nagarajan N, Pop M. Statistical methods for detecting differentially abundant features in clinical metagenomic samples. PLoS Comput. Biol. 2009;5:e1000352.10.1371/journal.pcbi.1000352
- Parks DH, Tyson GW, Hugenholtz P, et al. STAMP: statistical analysis of taxonomic and functional profiles. Bioinformatics. 2014;30:3123–3124. Epub 2014 Jul 26.10.1093/bioinformatics/btu494
- Feng BW, Li XR, Wang JH, et al. Bacterial diversity of water and sediment in the Changjiang estuary and coastal area of the East China Sea. FEMS Microbiol. Ecol. 2009;70:80–92. Epub 2009 Sep 29.
- Ehrhardt M, Wattayakorn G, Dawson R. GC/MS based analyses of individual organic constituents of chao phraya river water and estimated discharge rates into the upper gulf of Thailand. Estuar. Coast. Shelf Sci. 1990;30:439–451.10.1016/0272-7714(90)90066-Z
- Garrity GM, Bell JA, Lilburn T. Order I. Burkholderiales ord. nov. In: Garrity GM, Brenner DJ, Krieg NR, Staley JT, editors. Bergey’s Manual® of Systematic Bacteriology. 2nd ed., Vol. 2, Part C. New York: Springer; 2005. p. 575.
- Staley C, Gould TJ, Wang P, et al. Species sorting and seasonal dynamics primarily shape bacterial communities in the Upper Mississippi River. Sci. Total Environ. 2015;505:435–445. Epub 2014 Dec 03.10.1016/j.scitotenv.2014.10.012
- Fagervold SK, Bourgeois S, Pruski AM, et al. River organic matter shapes microbial communities in the sediment of the Rhone prodelta. ISME J. 2014;8:2327–2338. Epub 2014 May 27.10.1038/ismej.2014.86
- Liu S, Ren H, Shen L, et al. pH levels drive bacterial community structure in sediments of the Qiantang River as determined by 454 pyrosequencing. Front. microbiol. 2015;6:285. Epub 2015 May 06.
- Ufarté L, Laville É, Duquesne S, et al. Metagenomics for the discovery of pollutant degrading enzymes. Biotechnol. Adv. 2015;33:1845–1854. Epub 2015 Nov 04.10.1016/j.biotechadv.2015.10.009
- Marziah Z, Mahdzir A, Musa MN, et al. Abundance of sulfur-degrading bacteria in a benthic bacterial community of shallow sea sediment in the off-Terengganu coast of the South China Sea. Microbiologyopen. 2016. Epub 2016 Jun 04.
- Purdy KJ, Nedwell DB, Martin Embley T, et al. Use of 16S rRNA-targeted oligonucleotide probes to investigate the distribution of sulphate-reducing bacteria in estuarine sediments. FEMS Microbiol. Ecol. 2001;36:165–168.10.1111/fem.2001.36.issue-2-3
- Coby AJ, Picardal F, Shelobolina E, et al. Repeated anaerobic microbial redox cycling of iron. Appl. Environ. Microbiol. 2011;77:6036–6042.10.1128/AEM.00276-11
- Lovley DR, Giovannoni SJ, White DC, et al. Geobacter metallireducens gen. nov. sp. nov., a microorganism capable of coupling the complete oxidation of organic compounds to the reduction of iron and other metals. Arch. Microbiol. 1993;159:336–344.10.1007/BF00290916
- Lovley DR, Ueki T, Zhang T, et al. Geobacter: the microbe electric’s physiology, ecology, and practical applications. In: Robert KP, editor. Advances in microbial physiology. Vol. 59. Amsterdam: Academic Press; 2011. p. 1–100.
- Sanford RA, Cole JR, Tiedje JM. Characterization and description of Anaeromyxobacter dehalogenans gen. nov., sp. nov., an aryl-halorespiring facultative anaerobic myxobacterium. Appl. Environ. Microbiol. 2002;68:893–900.10.1128/AEM.68.2.893-900.2002
- Thomas SH, Wagner RD, Arakaki AK, et al. The mosaic genome of Anaeromyxobacter dehalogenans strain 2cp-c suggests an aerobic common ancestor to the delta-proteobacteria. PLoS ONE. 2008;3:e2103. Epub 2008 May 08.10.1371/journal.pone.0002103
- Zhao HP, Van Ginkel S, Tang Y, et al. Interactions between perchlorate and nitrate reductions in the biofilm of a hydrogen-based membrane biofilm reactor. Environ. Sci. Technol. 2011;45:10155–10162. Epub 2011 Oct 25.10.1021/es202569b
- Fu B, Liao X, Liang R, et al. COD removal from expanded granular sludge bed effluent using a moving bed biofilm reactor and their microbial community analysis. World J. Microbiol. Biotechnol. 2010;27:915–923.
- Bastida F, Rosell M, Franchini AG, et al. Elucidating MTBE degradation in a mixed consortium using a multidisciplinary approach. FEMS Microbiol. Ecol. 2010;73:370–384. Epub 2010 May 25.
- Coates JD, Chakraborty R, Lack JG, et al. Anaerobic benzene oxidation coupled to nitrate reduction in pure culture by two strains of Dechloromonas. Nature. 2001;411:1039–1043.10.1038/35082545
- Chakraborty R, O’Connor SM, Chan E, et al. Anaerobic degradation of benzene, toluene, ethylbenzene, and xylene compounds by Dechloromonas strain RCB. Appl. Environ. Microbiol. 2005;71:8649–8655. Epub 2005 Dec 08.10.1128/AEM.71.12.8649-8655.2005
- Wise MG, McArthur JV, Shimkets LJ. Methylosarcina fibrata gen. nov., sp. nov. and Methylosarcina quisquiliarum sp.nov., novel type 1 methanotrophs. Int. J. Syst. Evol. Microbiol. 2001;51:611–621.10.1099/00207713-51-2-611
- Kalyuzhnaya MG, Stolyar SM, Auman AJ, et al. Methylosarcina lacus sp. nov., a methanotroph from Lake Washington, Seattle, USA, and emended description of the genus Methylosarcina. Int. J. Syst. Evol. Microbiol. 2005;55:2345–2350. Epub 2005 Nov 11.10.1099/ijs.0.63405-0