
Abstract
Increasing evidence suggests that micro (mi)RNAs play important roles in various biological process. To evaluate the roles of miRNA in the porcine liver, we investigated the dynamic profiles of microRNAomes using liver tissue from pigs during the embryonic period (embryonic day 90), weaning stage (postnatal day 30), and adult stage (7 years old). A total of 186 unique miRNAs were differentially expressed during liver development. We also identified that 17, 13, and 6 miRNAs were specifically abundant at embryonic day 90, postnatal day 30, and at 7 years, respectively. Besides regulating basic cellular roles in development, miRNAs expressed at the three developmental stages also participated in regulating “embryonic liver development,” “early hepatic growth and generating a functioning liver,” and “energy metabolic processes,” respectively. Our study indicates that miRNAs are extensively involved in liver development, and provides a valuable resource for the further elucidation of miRNA regulatory roles during liver development.
Graphical abstract
miRNAs are extensively involved in liver development, especially for these stage-enriched miRNAs.
As one of the largest internal organs in the body, the liver exhibits both endocrine and exocrine properties such as the secretion of several hormones. It also has a range of metabolic functions, including glycogen storage, drug detoxification, the control of metabolism, and the regulation of cholesterol synthesis and transport. Liver development is influenced by inductive signals and genetic regulatory factors that are highly conserved among vertebrates. The regulatory network consists of signaling pathways involving fibroblast growth factors,Citation1) bone morphogenetic proteins,Citation2) Wnt, and multiple transcription factors.Citation3) Analysis of the distinct gene expression pattern of the liver tissue has revealed differences between humans and other primates suggestive of dietary variation.Citation4) Therefore, the liver transcriptome may reflect different dietary or physiological states.
MicroRNAs (miRNAs) are endogenous, non-coding RNAs, ~22 nucleotides (nt) in length that have been demonstrated to have major effects on many aspects of biology by binding to target mRNAs and repressing gene expression. Numerous studies have shown that miRNAs play important roles in liver development, regeneration, and disease.Citation5) For example, miR-122 is specifically expressed in the liver and is essential for liver homeostasis through regulating hepatic networks of genes involved in cell cycle regulation, lipid metabolism, inflammation, and oncogenesis. Loss of miR-122 has been associated with liver fibrosis, inflammation, steatosis, and cancer.Citation6,7) Members of the miR-181 family are highly expressed in the embryonic liver and target the mRNA that encodes GATA6, a transcription factor that regulates liver organogenesis,Citation8) while recent studies concluded that both miRNA-150 and miRNA-194 inhibit hepatic stellate cell (HSC) production.
The pig is an important agricultural animal, but is also emerging as a suitable model for biomedical researchCitation9) and the study of human pathologies.Citation10) Pigs are remarkably similar to humans in terms of their metabolic features, and immune and cardiovascular systems. They have therefore been used as models for research into human skin,Citation11) obesity,Citation12) and lung disease.Citation13) Indeed, recent research studied fumarylacetoacetate hydrolase-deficient pigs as a large animal model for metabolic liver disease.Citation14) This work showed the importance of surveying the specific miRNAome of pigs under diverse physiological states, such as different developmental periods.
The exact role of miRNAs in the developing liver remains unclear, so further study is necessary. The identification of tissue- or stage-specific miRNAs and an increased understanding of the mechanisms involved in miRNA deregulation will improve our knowledge of physiology and development. In the present study, we provide insights into the genome-wide dynamic profiles of miRNA in the developing porcine liver. This comprehensive study extends the miRNA profile and reveals likely roles of liver miRNAs, providing a basis for future research into the development of liver.
Materials and methods
Animal ethics statement
All research involving animals was conducted according to the Regulations for the Administration of Affairs Concerning Experimental Animals (Ministry of Science and Technology, China, revised in June 2004) and was approved by the Institutional Animal Care and Use Committee in the College of Animal Science and Technology, Sichuan Agricultural University, Sichuan, China under permit no. DKY-S20112716.
Sample collection and total RNA isolation
Liver tissue was obtained from three female Jinhua pigs at embryonic day 90 (E90), three pigs at postnatal day 30 (30d), and three pigs at postnatal year 7 (7y). All samples were immediately frozen in liquid nitrogen and stored at −80 °C. Total RNA was extracted using the mirVana™ miRNA isolation kit (Ambion, Austin, TX) according to the manufacturer’s protocol. The integrity of total RNA was determined by the Agilent 2100 Bioanalyzer and RNA 6000 Nano LabChip Kit (Agilent Technologies, Santa Clara, CA) with suitable RNA samples having an RNA integrity number >6.0.
Small RNA library construction and sequencing
Liver tissues from each set of three pigs at the three different developmental stages were pooled and prepared according to the Illumina EAS Small RNA Sample Prep Protocol (Illumina, San Diego, CA). Approximately, 15 μg of small RNA-enriched total RNA was prepared for high-throughput sequencing. For each library, small RNA ranging from 14 to 40 nt was purified by polyacrylamide gel electrophoresis and ligated using proprietary adaptors (Illumina). The modified small RNA was then reverse-transcribed and amplified by reverse transcription-PCR. Finally, the enriched cDNA was sequenced on a Genome Analyzer II (GAI, Illumina) according to the manufacturer’s instructions. The small RNA-sequencing data discussed in this publication have been deposited in NCBI’s Gene Expression Omnibus and are accessible through GEO Series accession number GSE81192.
Analysis of high-throughput sequencing data
Raw sequencing reads were processed using a proprietary pipeline script, ACGT101-miR program (Illumina), which used a series of additional filters to remove unmappable reads.Citation15,16) After trimming off the sequencing adapters and junk reads, the resulting reads were successively filtered by read length and sequence component (reads removed due to 3ADT not found and length with <16 and >29 were removed). As shown in Supplemental Fig. S1, They were then compared against NCBI (Sus scrofa genome, version: 10.2), Rfam, and Repbase databases to remove known classes of porcine RNAs (i.e. mRNAs, rRNAs, tRNAs, small nuclear [sn]RNAs, small nucleolar [sno]RNAs, and repeats). The resulting reads that had undergone strict filtering were deemed “clean reads.” The clean reads were mapped to the pig genome (Sscrofa 10.2) and miRbase database with following steps in order: (1) map the clean reads to the known porcine pre-miRNAs and then to the known pre-miRNAs from 74 other mammals in miRBase 19.0 to identify porcine known and conserved miRNAs, respectively; (2) the unmapped sequences in step 1 that can mapped to the pig genome and form hairpin RNA structure from the adjacent 60 nt sequences in either direction from the pig genome using UNAFold were regarded as porcine putative novel miRNA. Because of low-confidence and little reference study about this putative miRNA, so we mainly focus on the porcine known and conserved miRNAs with high confidence in this study.
Differential expression analysis of miRNAs
To identify differentially expressed miRNAs, miRNA expression in each library (E90, 30d, and 7y) was normalized to obtain the expression of transcripts per million using the following formula: normalized expression = (actual miRNA sequencing reads count/total clean reads count) × 1,000,000. If the normalized expression value of a given miRNA was zero, the expression value was modified to 0.01. If the normalized expression of a given miRNA was less than 1 in all libraries, it was removed from future differential expression analysis. The normalized data were used to calculate the expression fold change and p-value (DEGseq, an R package for difference analysis) between two libraries. A unique miRNA was considered to be differentially expressed when |log2 (fold-change)| ≥1 & p-value < 0.001 (Benjamini corrected).
Prediction and functional annotation of miRNA target genes
The potential targets of an miRNA were predicted by PicTarCitation17) and TargetScan (Release 6.2: June 2012)Citation18) programs, and the overlapping results comprised the final predicted targets. The predictions were based on the interactions between human mRNAs and miRNAs because of the absence of porcine miRNA from the current version of the above-mentioned algorithm. Gene ontology (GO) terms and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway terms enriched in predicted target genes were identified using a DAVID Bioinformatics Resources 6.7 (http://david.abcc.ncifcrf.gov/home.jsp).
q-PCR validation
The expression changes of nine randomly selected miRNA from the differentially expressed miRNAs were validated by an SYBR-based High-Specificity miRNA qRT-PCR Detection Kit (TaKaRa, Dalian, China) on the CFX96TM Real-Time PCR Detection System (Bio-Rad, CA, USA). The endogenous control genes (U6 snRNA and 18S rRNA) were used in this assay. The q-PCR validations were carried out on three biological replicates and each sample was run in triplicates. The 2−ΔΔCt method was used to determine the expression level difference between surveyed samples.
Results and discussion
Summary of high-throughput sequencing data
A total of 12.89 million, 8.72 million, and 13.16 million raw reads were generated from E90, 30d, and 7y livers, respectively. After filtering out unmappable reads, 10.68 million, 5.04 million, and 10.78 million reads were deemed “clean reads.” Supplemental Table 1 shows the distribution of small RNAs during filtration. Those reads mapped to known classes of RNA (i.e. mRNA, rRNA, tRNA, snRNA, snoRNA, and repetitive sequence elements) were also eliminated from analysis (Supplemental Table 2).
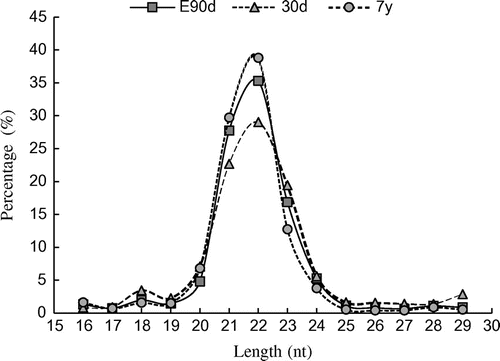
The size distribution of clean reads was similar among the three libraries. In all libraries, the 22-nt small RNA was found to be the most abundant (E90: 35.30%; 30d: 29.03%; and 7y: 38.80%), followed by 21- and 23-nt small RNAs (Fig. ), which is consistent with the typical size of miRNAs obtained from Dicer-derived products and agrees with previous deep sequencing results.Citation19)
Fig. 1. Length distributions of small RNAs.
Identification and characterization of the liver microRNAome at different developmental stages
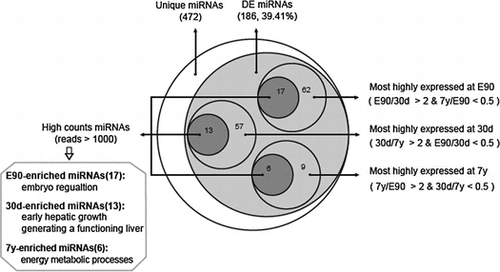
In total, 503 mature miRNAs corresponding to 297 miRNA precursors (pre-miRNAs) were found to be expressed in three stages of porcine liver (Table ). Among them, 393 miRNAs (78.13%) corresponded to previously described porcine miRNAs and 110 orthologous miRNAs (21.87%). As shown in Table , the identified mature miRNAs and their precursors were divided into the following two subgroups in order from high-to-mid confidenceCitation20): (1) Porcine known miRNAs: 393 miRNAs corresponding to 223 known porcine pre-miRNAs (Supplemental Table 3). (2) Porcine conserved miRNAs: 110 miRNAs corresponding to 74 other known mammalian pre-miRNAs in miRBase 19.0, which mapped to the pig genome (Supplemental Table 3). These results agree with previous observations that many miRNAs are conserved among species.Citation21) Because distinct pre-miRNAs and genomic loci exist that express identical mature sequences, these 503 miRNAs sequences correspond to 472 unique miRNA sequences. Compared with human (2588 mature corresponding to 1881 pre-miRNA) and mouse (1915 mature miRNA corresponding to 1193 pre-miRNA), miRNAs identified in porcine is relative few, which also highlight the further study is necessary.
Table 1. Known and conserved miRNA identified in porcine livers (miRNA/pre-miRNA).
The 472 unique miRNAs were then divided into three gradients based on their expression count (normalized expression): high-count miRNAs (counts > 1000), middle-count miRNAs (10 < counts ≤ 1000), and low-count miRNAs (0 < counts ≤ 10). The average counts for low-count miRNAs were only 2.29, whereas the average counts for high-count miRNAs reached >10,000. As shown in Table , the E90 library included 456 unique miRNAs representing 464,203 counts, the 30d library included 428 unique miRNAs representing 389,867 counts, and the 7y library included 366 unique miRNAs representing 483,326 counts. The low-count miRNAs accounted for 56.79% of the total types of miRNA on average, but only for 0.12% of the total miRNA expression. By contrast, the high-count miRNAs accounted for 94.94% of the total miRNA expression on average, but only for 10.05% of the total types of miRNA, suggesting that only a few types of miRNA comprise the majority of sequences and play a leading role in porcine liver tissue.
Table 2. Counts and types of unique known and conserved miRNAs.
miRNAs highly expressed in porcine livers
In each library, miRNA expression exhibited a large dynamic range of counts ranging from three to millions, with the top 20 miRNAs contributing more than 80% of the total miRNA expression (E90: 80.74%; 30d: 81.66%; and 7y: 90.14%) (Fig. ). Interestingly, 14 unique miRNAs simultaneously appear at the top 20 position of all libraries, although the rankings differ (Fig. (D)).
Fig. 2. The 20 most highly expressed miRNAs in E90 (A), 30d (B), and 7y (C).
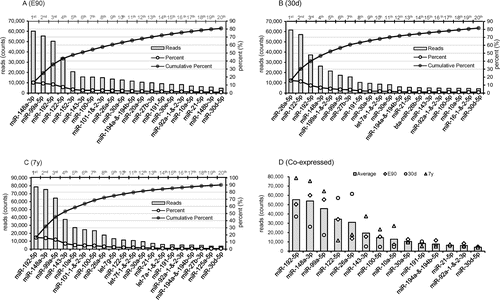
The high abundance of these miRNAs highly expressed in all libraries implies that they may have housekeeping roles in the development and maintenance of liver tissues and basic cellular metabolism.Citation22) For example, miR-192 is involved in the regulation of liver cell deathCitation23) and has the potential to be used as a noninvasive prognostic indicator for cholangiocarcinoma.Citation24) miR-148a increases the proliferation of hepatic cells via inhibiting the expression of Akt and Erk proteins,Citation25) and miRNA-99a directly regulates argonaute-2 through translational repression in hepatocellular carcinoma.Citation26) On a functional level, miR-122 is a key factor and therapeutic target in liver disease,Citation27,28) while miR-26a inhibits the epithelial–mesenchymal transition process in hepatocellular carcinoma by down-regulating the expression of zeste homolog 2.Citation29) miRNA-143 is an integral component of the regulatory network whereby serum response factor controls cytoskeletal remodeling and phenotypic switching of smooth muscle cells during vascular disease.Citation30) miR-194 is specifically expressed in liver parenchymal cells, and prevents liver cancer cell metastasis,Citation31) while miR-30d promotes tumor invasion and metastasis by targeting Galphai2 in hepatocellular carcinoma.Citation32) Together, these findings and other evidence suggest that these miRNAs participate in the regulation of numerous hepatic physiological processes.
miRNAs differentially expressed in porcine liver tissues at different developmental stages
To identify the miRNAs that may be involved in liver development, we carried out comparative miRNA expression analysis across liver samples collected from the three developmental stages. As shown in Fig. , 186 unique miRNAs (out of 472, 39.41%) were differentially expressed among the three libraries (fold change > 2 & p-value < 0.001 (Benjamini corrected), Supplemental Table 4). To validate the sequencing data, nine differentially expressed miRNAs (the primer pairs were available in Supplemental Table 5) were selected randomly to quantify their relative expression levels using qPCR. As shown in Supplemental Fig. S2, our results showed that there was a significant positive correlation between the sequencing and qPCR results (Pearson’s r = 0.905, p < 10−3), which highlighted the high reliability of our sequencing data.
Fig. 3. Characteristics of the differentially expressed miRNAs among porcine livers at different developmental stages.
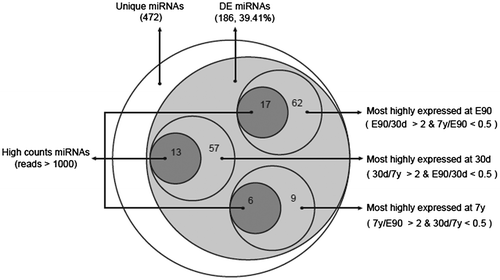
First, we calculated the miRNAs that were highly expressed in just one library. In the E90 library, 62 unique miRNAs (out of 186, 33.33%) were most abundantly expressed (E90/30d >2 and 7y/E90 <0.5), of which 17 unique miRNAs belonged to the high-count miRNAs (counts >1000). Compared with E90 and 7y, 57 unique miRNAs (out of 186, 30.65%) were most highly expressed at 30d (30d/E90 >2 and 7y/30d <0.5), of which 13 unique miRNAs belonged to the high-count miRNAs. A total of 9 unique miRNAs (out of 186, 4.84%) were most highly expressed at 7y (7y/E90 >2 and 30d/7y <0.5), of which six unique miRNAs belonged to the high-count miRNAs (Table ). The remaining 58 unique miRNAs (out of 186, 31.18%) were highly expressed in two of the three libraries or differentially expressed only between two libraries. In this study, we focused on the differentially expressed miRNAs that were specifically expressed in only one library with high reads (reads >1000), which we deemed stage-enriched miRNAs.
Table 3. The stage-enriched miRNAs.
Embryo-enriched miRNAs appear to facilitate the regulation of embryonic liver development. For example, previous studies indicated that miR-106a and miR-17 are expressed at significantly higher levels in human embryonic stem cells (hESCs) than in mature quiescent hepatocytes. They promote hESC self-renewal, which is essential for normal liver development, especially during the period of embryonic development and liver regeneration.Citation33) Additionally, miR-221 promotes hepatocyte proliferation.Citation34)
The 30d-enriched miRNAs appear important in controlling early hepatic growth. However, because nascent piglets suckle during this period, the distinct miRNA expression pattern may reflect the establishment of primary hepatic function. Previous studies indicated that the miR-30 family is required for hepatobiliary development,Citation35) which regulates hepatoblast migration and maturation.Citation36) Moreover, miR-199 shows differential expression among normal human livers, and those affected by hepatitis and hepatocellular carcinoma,Citation37) while miR-27a regulates fat metabolism and cell proliferation during HSC activation.Citation38)
The 7y-enriched miRNAs participate in the regulation of several metabolic processes. For example, miR-143 and let-7 were shown to play essential roles in the metabolism of glucose, high-density lipoprotein, and insulin, so is a potential therapeutic target for diabetes.Citation39,40)
miRNA target gene prediction and functional annotation
To further understand the physiological functions and biological processes associated with these stage-enriched miRNAs, their potential targets were predicted using the public database (PicTar and TargetScan). As shown in Fig. , 1736 and 1498 target genes were identified from 17 embryo-enriched miRNAs and 13 miRNAs enriched in the 30d liver, respectively, while relatively fewer target genes (492) were predicted from the 6 adult-enriched miRNAs (7y).
Fig. 4. Characteristics of the target genes predicted from stage-enriched miRNAs.
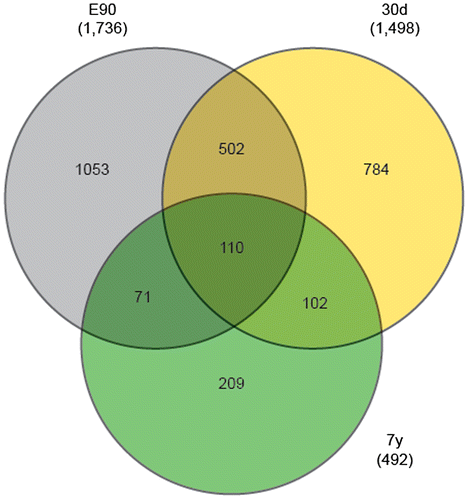
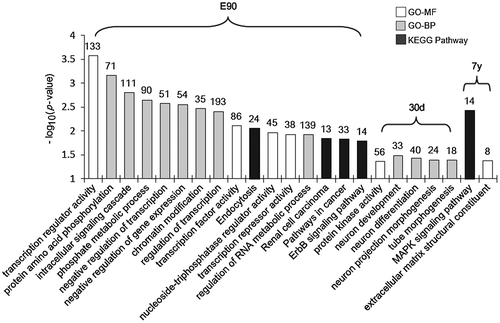
To further explore the characteristics of stage-enriched miRNAs, we focused on the target genes predicted from a single development stage. In total, 209, 784, and 1053 genes were uniquely predicted from 7y, 30d, and E90, respectively. As shown in Fig. , GO and KEGG analysis revealed that the E90-specific genes were significantly enriched in “transcription regulation activity,”, “phosphate metabolic process,” “transcription factor activity,” and the “ErbB signaling pathway,” suggesting they facilitated the regulation of embryonic liver development.Citation41) The 30d-specific genes were significantly enriched in “neuron development and differentiation” and “tube morphogenesis,” which are important in controlling early hepatic growth and generating a functioning liver. The 7y-specific genes were most significantly enriched in the “MAPK signaling pathway,” which participates in controlling a number of cell regulation processes, including nuclear transport, nucleosome assembly, cytoskeletal regulation, and energy metabolism.Citation42)
Fig. 5. KEGG pathway and gene ontology categories enriched in the stage-specific target genes.
Conclusion
This study effectively extends the miRNA profile expressed in the porcine liver by comparing the microRNAome across three different stages of development. Besides regulating basic cellular roles in liver development and functional maintenance, miRNAs expressed at E90, 30d, and 7y also participated in the regulation of “embryonic liver development,” “early hepatic growth and generating a functioning liver,” and “energy metabolic processes,” respectively. Our results provide a valuable resource for the further elucidation of miRNA regulatory roles in liver development, and could be used to develop molecular markers for liver development.
Author contributions
Yihui Liu, Long Jin, and Mingzhou Li conceived and designed the experiments. Yihui Liu performed the experiments. Yihui Liu and Pengbo Lou analyzed the data. Yiren Gu, Mingzhou li, and Xuewei Li contributed reagents/materials/analysis tools. Yihui Liu and Long Jin wrote the paper.
Disclosure statement
No potential conflict of interest was reported by the authors.
Funding
This work was supported by grants from the National Special Foundation for Transgenic Species of China [grant number 2014ZX0800950B], [grant number 2014ZX08006-003]; the National Natural Science Foundation of China [grant number 31530073], [grant number 31522055], [grant number 31301942]; the Program for Innovative Research Team of Sichuan Province [grant number 2015TD0012].
Supplemental material
The supplemental material for this paper is available at http://dx.doi.org/10.1080/09168451.2016.1240602.
TBBB_1240602_Supplemental_Material.zip
Download Zip (409.6 KB)Notes
Abbreviations: BP, biological processes; DE, differentially expressed; GO, gene ontology; hESCs, human embryonic stem cells; HSC, hepatic stellate cell; NE, normalized expression.
Related Research Data
References
- Calmont A, Wandzioch E, Tremblay KD, et al. An FGF response pathway that mediates hepatic gene induction in embryonic endoderm cells. Dev. Cell. 2006;11:339–348.10.1016/j.devcel.2006.06.015
- Agarwal S, Holton KL, Lanza R. Efficient differentiation of functional hepatocytes from human embryonic stem cells. Stem Cells. 2008;26:1117–1127.10.1634/stemcells.2007-1102
- Yin C, Evason KJ, Asahina K, et al. Hepatic stellate cells in liver development, regeneration, and cancer. J. Clin. Invest. 2013;123:1902–1910.10.1172/JCI66369
- Brawand D, Soumillon M, Necsulea A, et al. The evolution of gene expression levels in mammalian organs. Nature. 2011;478:343–348.10.1038/nature10532
- Tzur G, Israel A, Levy A, et al. Comprehensive gene and microRNA expression profiling reveals a role for micrornas in human liver development. PLoS One. 2009;4:e7511.10.1371/journal.pone.0007511
- Tsai W-C, Hsu S-D, Hsu C-S, et al. MicroRNA-122 plays a critical role in liver homeostasis and hepatocarcinogenesis. J. Clin. Invest. 2012;122:2884–2897.10.1172/JCI63455
- Yin S, Fan Y, Zhang H, et al. Differential TGFβ pathway targeting by miR-122 in humans and mice affects liver cancer metastasis. Nat. Commun. 2016;7:11012.10.1038/ncomms11012
- Zaret KS. Regulatory phases of early liver development: paradigms of organogenesis. Nat. Rev. Genet. 2002;3:499–512.10.1038/nrg837
- Gün G, Kues WA. Current progress of genetically engineered pig models for biomedical research. Biores. Open Access. 2014;3:255–264.
- Bassols A, Costa C, Eckersall PD, et al. The pig as an animal model for human pathologies: a proteomics perspective. Proteomics Clin. Appl. 2014;8:715–731.10.1002/prca.201300099
- Summerfield A, Meurens F, Ricklin ME. The immunology of the porcine skin and its value as a model for human skin. Mol. Immunol. 2015;66:14–21.10.1016/j.molimm.2014.10.023
- Spurlock ME, Gabler NK. The development of porcine models of obesity and the metabolic syndrome. J. Nutr. 2008;138:397–402.
- van der Merwe M, Ewing H, Caminita F, et al. The preterm pig as a model for acute lung disease after preterm birth (668.3). FASEB J. 2014;28: 668.3.
- Hickey RD, Mao SA, Glorioso J, et al. Fumarylacetoacetate hydrolase deficient pigs are a novel large animal model of metabolic liver disease. Stem Cell Res. 2014;13:144–153.10.1016/j.scr.2014.05.003
- Bigham A, Bauchet M, Pinto D, et al. Identifying signatures of natural selection in Tibetan and Andean populations using dense genome scan data. PLoS Genet. 2010;6:e1001116.10.1371/journal.pgen.1001116
- Li M, Liu Y, Wang T, et al. Repertoire of porcine microRNAs in adult ovary and testis by deep sequencing. Int. J. Biol. Sci. 2011;7:1045–1055.10.7150/ijbs.7.1045
- Krek A, Grün D, Poy MN, et al. Combinatorial microRNA target predictions. Nat. Genet. 2005;37:495–500.10.1038/ng1536
- Lewis BP, Shih I-h, Jones-Rhoades MW, et al. Prediction of mammalian MicroRNA targets. Cell. 2003;115:787–798.10.1016/S0092-8674(03)01018-3
- Ma J, Yu S, Wang F, et al. MicroRNA transcriptomes relate intermuscular adipose tissue to metabolic risk. Int. J. Mol. Sci. 2013;14:8611–8624.10.3390/ijms14048611
- Ambros V, Bartel B, Bartel DP, et al. A uniform system for microRNA annotation. RNA. 2003;9:277–279.10.1261/rna.2183803
- Lagos-Quintana M, Rauhut R, Lendeckel W, et al. Identification of novel genes coding for small expressed RNAs. Science. 2001;294:853–858.10.1126/science.1064921
- Copley MR, Babovic S, Benz C, et al. The Lin28b–let-7–Hmga2 axis determines the higher self-renewal potential of fetal haematopoietic stem cells. Nat. Cell Biol. 2013;15:916–925.10.1038/ncb2783
- Roy S, Benz F, Alder J, et al. Down-regulation of miR-192-5p protects from oxidative-stress induced-acute liver injury. Clin. Sci. 2016;130:1197–1207.
- Silakit R, Loilome W, Yongvanit P, et al. Circulating miR-192 in liver fluke-associated cholangiocarcinoma patients: a prospective prognostic indicator. J. Hepatobiliary Pancreat. Sci. 2014;21:864–872.10.1002/jhbp.2014.21.issue-12
- Damania P, Sen B, Dar SB, et al. Hepatitis B virus induces cell proliferation via HBx-induced microRNA-21 in hepatocellular carcinoma by targeting programmed cell death protein4 (PDCD4) and phosphatase and tensin homologue (PTEN). PLoS One. 2014;9:e91745.10.1371/journal.pone.0091745
- Zhang J, Jin H, Liu H, et al. MiRNA-99a directly regulates AGO2 through translational repression in hepatocellular carcinoma. Oncogenesis. 2014;3:e97.10.1038/oncsis.2014.11
- Bandiera S, Pfeffer S, Baumert TF, et al. miR-122 – a key factor and therapeutic target in liver disease. J. Hepatol. 2015;62:448–457.10.1016/j.jhep.2014.10.004
- Waring J, Dumas E, Abel S, et al. Serum miR-122 may serve as a biomarker for response to direct acting antivirals: effect of paritaprevir/R with dasabuvir or ombitasvir on miR-122 in HCV-infected subjects. J. Viral Hepat. 2016;23:96–104.10.1111/jvh.12470
- Ma D-N, Chai Z-T, Zhu X-D, et al. MicroRNA-26a suppresses epithelial-mesenchymal transition in human hepatocellular carcinoma by repressing enhancer of zeste homolog 2. J. Hematol. Oncol. 2016;9:1–10.10.1186/s13045-015-0229-y
- Xin M, Small EM, Sutherland LB, et al. MicroRNAs miR-143 and miR-145 modulate cytoskeletal dynamics and responsiveness of smooth muscle cells to injury. Genes Dev. 2009;23:2166–2178.10.1101/gad.1842409
- Meng Z, Fu X, Chen X, et al. miR-194 is a marker of hepatic epithelial cells and suppresses metastasis of liver cancer cells in mice. Hepatology. 2010;52:2148–2157.10.1002/hep.23915
- Yao J, Liang L, Huang S, et al. MicroRNA-30d promotes tumor invasion and metastasis by targeting Galphai2 in hepatocellular carcinoma. Hepatology. 2010;51:846–856.
- Jung CJ, Iyengar S, Blahnik KR, et al. Human ESC self-renewal promoting microRNAs induce epithelial-mesenchymal transition in hepatocytes by controlling the PTEN and TGFβ tumor suppressor signaling pathways. Mol. Cancer Res. 2012;10:979–991.10.1158/1541-7786.MCR-11-0421
- Yuan Q, Loya K, Rani B, et al. MicroRNA-221 overexpression accelerates hepatocyte proliferation during liver regeneration. Hepatology. 2013;57:299–310.10.1002/hep.25984
- Hand NJ, Master ZR, Eauclaire SF, et al. The microRNA-30 family is required for vertebrate hepatobiliary development. Gastroenterology. 2009;136:1081–1090.10.1053/j.gastro.2008.12.006
- Zhang J, Zhang H, Liu J, et al. miR-30 inhibits TGF-β1-induced epithelial-to-mesenchymal transition in hepatocyte by targeting Snail1. Biochem. Biophys. Res. Commun. 2012;417:1100–1105.10.1016/j.bbrc.2011.12.121
- Hou J, Lin L, Zhou W, et al. Identification of miRNomes in human liver and hepatocellular carcinoma reveals miR-199a/b-3p as therapeutic target for hepatocellular carcinoma. Cancer Cell. 2011;19:232–243.10.1016/j.ccr.2011.01.001
- Ji J, Zhang J, Huang G, et al. Over-expressed microRNA-27a and 27b influence fat accumulation and cell proliferation during rat hepatic stellate cell activation. FEBS Lett. 2009;583:759–766.10.1016/j.febslet.2009.01.034
- Jordan SD, Krüger M, Willmes DM, et al. Obesity-induced overexpression of miRNA-143 inhibits insulin-stimulated AKT activation and impairs glucose metabolism. Nat. Cell Biol. 2011;13:434–446.10.1038/ncb2211
- Zhu H, Shyh-Chang N, Segrè AV, et al. The Lin28/let-7 axis regulates glucose metabolism. Cell. 2011;147:81–94.10.1016/j.cell.2011.08.033
- Carver RS, Stevenson MC, Scheving LA, et al. Diverse expression of ErbB receptor proteins during rat liver development and regeneration. Gastroenterology. 2002;123:2017–2027.10.1053/gast.2002.37060
- Chang L, Karin M. Mammalian MAP kinase signalling cascades. Nature. 2001;410:37–40.10.1038/35065000