
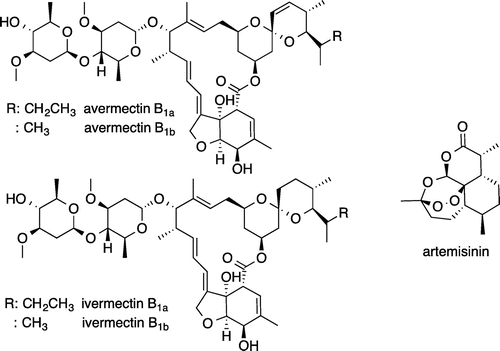
The three major infectious diseases (AIDS, malaria, and tuberculosis) and neglected tropical diseases (NTDs: 17 diseases) unevenly affect more people in developing countries and have been overlooked for research and development of new medicines for a long time. Recently, international organizations, national governments, and private sectors have established support organizations to research and develop innovative drugs for distribution to countries burdened by the three major infectious diseases and NTDs.Footnote1 Thanks to this support, international development of new medicine is progressing, and there are particularly high hopes for drug discovery from natural products. Ōmura and colleagues’ 2015 Nobel Prize in Physiology or Medicine emphasized the importance of natural product drug discovery. IvermectinCitation1) (Fig. ), a chemical derivative of avermectin produced by the actinomycete Streptomyces avermitilis, was discovered by Ōmura et al. and has been developed as a treatment drug for nematode onchocerciasis and other diseases. In addition, Tu and colleagues isolated the malaria treatment drug artemisininCitation2) (Fig. ) from a plant component. Both of these are utilized as highly effective, innovative new drugs. In this manuscript, from the standpoint of natural products, I will introduce the current state of the development of treatment drugs for tuberculosis, one of the three major infectious diseases of modern times.
Fig. 1. Structures of avermectin, ivermectin, and artemisinin.
With over 9 million cases and 1.5 million deaths annually, tuberculosis is the deadliest known infectious disease.Citation3) In the past, tuberculosis was also a severe problem in developed countries, which facilitated the development of many drugs. However, with the decline in the number of new patients, the research and development of new tuberculosis drugs has also diminished. Multidrug-resistant tuberculosis (MDR-TB) and extensively drug-resistant tuberculosis (XDR-TB), against which current antituberculosis medicines are completely ineffective, have become widespread, partially due to the lack of new medicines in the past 40 years.
In addition, coinfection with HIV is serious problem. There are over 35.3 million HIV-positive people worldwide,Citation4) and of the 1.6 million who die of HIV annually, 25% suffer from combined tuberculosis/HIV infection. Tuberculosis and HIV treatments center on chemotherapy, where drug interactions are complex. Even though combination chemotherapy is available for the coinfection, the interaction of drugs for each infection makes the chemotherapy difficult. Thus, overcoming problematic drug interactions is a great challenge for treatment of tuberculosis/HIV coinfection.
Challenges in the development of tuberculosis drugs
Many pharmaceutical companies have been indifferent to serious development of new antituberculosis medicines because they estimate their profits based solely on comparative expenses. However, recently, international organizations such as the Global Alliance for TB Drug Development (TB Alliance) and Stop TB Partnership (WHO) have taken a leading role in initiating efforts to end the tuberculosis epidemic. The main challenges for new antituberculosis drugs are (1) shortening treatment time, (2) simplifying the treatment method, (3) treating MDR/XDR-TB, (4) treating tuberculosis/HIV simultaneously, and (5) ensuring superior safety.Citation5)
The new antituberculosis drugs that display low toxicity, excellent antituberculosis activity, and efficacy against resting-phase bacteria are beneficial for short-course therapy and prevent secondary infections from those already infected. Although long-term combination therapy is currently the basis of tuberculosis treatment, the development of short-term tuberculosis treatments can be safer for children and help prevent MDR/XDR-TB. There is an urgent need for pharmaceutical drugs effective against MDR/XDR-TB that possess novel mechanisms of action and for medicines that are compatible with simultaneous treatment of tuberculosis/HIV or tuberculosis/diabetes.Citation5)
Current status of anti-tuberculosis drug development
Current anti-tuberculosis drugs under development are classified into three groups: (1) first-line drugs that are undergoing re-evaluation to optimize their efficacy, (2) existing drugs developed for other infectious diseases that are modified or applied directly as antituberculosis drugs, and (3) new drugs that possess novel structures and mechanisms of action. In categories 1 and 2, there are many synthesized compounds that can be improved with simple modifications, such as fluoroquinolone compounds (moxifloxacin,Citation6) gatifloxacin,Citation7, 8) and DC-159aCitation9)), oxazolidinone compounds (linezolid,Citation10) sutezolid,Citation11) and AZD-5847Citation12)), and diamine compounds (SQ109Citation13)). Fluoroquinolone compounds and oxazolidinone compounds are effective against a wide range of bacteria in addition to tuberculosis. With compounds like this, care must be taken to avoid cross-tolerance issues.
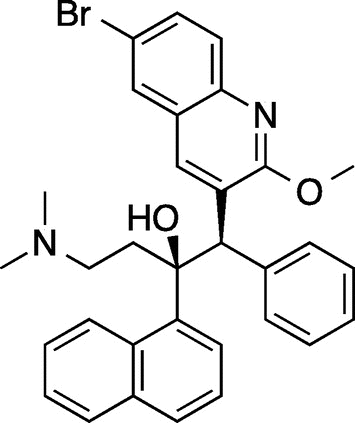
Most of the compounds we are aware of in category 3, compounds with novel structures and mechanisms of action, are natural products. Bedaquiline (TMC207, Fig. ), a type of diarylquinoline, is one of a few recent examples of synthetic compounds.Citation14, 15)
Fig. 2. Structure of bedaquiline.
Antituberculosis drug development from natural products
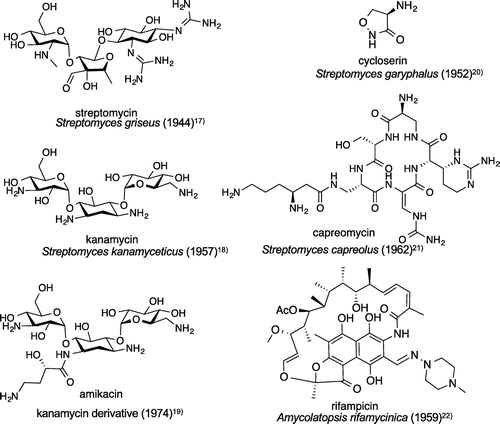
The antituberculosis drugs currently in use are specific for tuberculosis, which is uncommon among antibiotics, and most of them act specifically on Mycobacterium tuberculosis using unique mechanisms. Most antituberculosis drugs of natural-product origin are actinomycete metabolites; hence, actinomycete compounds such as rifamycin, the parent compound of rifampicinCitation21) (Fig. ), are an important starting point for the discovery of antituberculosis substances. Currently, many rifamycin compounds (rifabutin and rifapentine) are being developed.
Fig. 3. Antituberculous drugs of natural product origin, the producing microorganisms, and the respective structures (year discovered).
New drugs possessing new structures and mechanisms of action
Nitroimidazole compounds
Nitroimidazole compounds (Fig. ) include azomycinCitation22) from Streptomyces eurocidicus and its derivatives such as metronidazole, which the French research group Rhône-Poulenc succeeded in developing with reduced toxicity. Previously, compounds with the nitroimidazole structure were used to treat infectious diseases caused by anaerobic bacteria, Helicobacter pylori, and protozoa. However, in recent years, these compounds have been modified as antituberculosis drugs that are activated by an enzyme inside of M. tuberculosis and inhibit mycolic acid synthesis.Citation23) A representative example is shown in Fig. .
Fig. 4. Structures of nitroimidazole compounds.
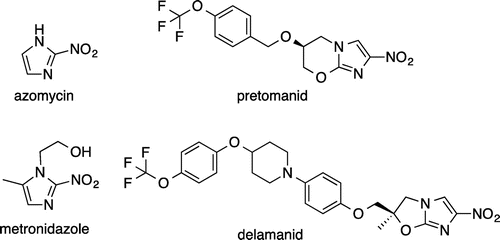
Pretomanid (PA-824, Fig. )
Pretomanid is a novel nitroimidazole compound developed by the U.S. research group Chiron. It possesses two mechanisms of action against M. tuberculosis: inhibition of mycolic acid lipid biosynthesis and inhibition of bacterial protein synthesis. The antimicrobial activity is specific to M. tuberculosis and does not affect general bacteria. This drug shows microbicidal action against M. tuberculosis in the proliferative phase and in the resting phase. Furthermore, it has superior antimicrobial action against MDR-TB.Citation24) According to the TB Alliance, a three-drug combination therapyCitation25) with moxifloxacin and pyrazinamide has entered the Phase III clinical trial development stage.
In addition, in efforts to shorten treatment time, pretomanid has been utilized in three-drug combination therapies with bedaquiline and linezolid or with bedaquiline and pyrazinamide. Both of these combined therapies are progressing through Phase II clinical trials.Citation26)
Delamanid (OPC-67683, Fig. )
Delamanid is a nitroimidazole compound developed by Otsuka Pharmaceutical in Japan that inhibits only methoxymycolic and ketomycolic acid biosynthesis among the mycolic acids constituting the cell wall. Its antimicrobial spectrum, which is specific for the M. tuberculosis complex, and physicochemical properties resemble the aforementioned pretomanid.Citation27) Each drug can result in cross-resistance to the other. However, in terms of antituberculosis activity and safety, delamanid well surpasses pretomanid. In 2014, this drug was approved for the treatment of MDR-TB in Japan and Europe, making it the first new antituberculous drug from Japan in 40 years.
Fig. 5. Structures of CPZEN-45/caprazamycins and liposidomycin.
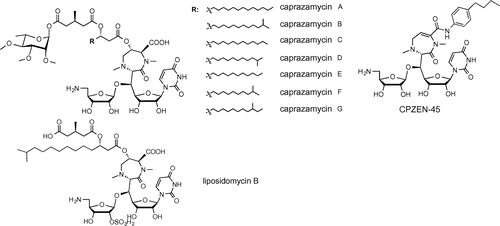
CPZEN-45/caprazamycins (Fig. )Citation28)
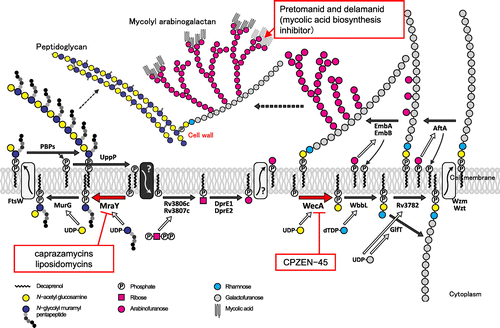
Caprazamycins were discovered from Streptomyces sp. and possess a novel antituberculous mechanism of action. This compound inhibits the phospho-N-acetylmuramyl pentapeptide transferase MraY, which is involved in cell wall synthesis (Fig. ). The compound liposidomycinCitation29) (Fig. ) is a known analog. CPZEN-45Citation30), developed from structure–activity relationship research on caprazamycins, exhibits specificity against M. tuberculosis and a superior antimicrobial spectrum of activity, lacks cross-resistance issues with existing drugs, and is effective against MDR/XDR-TB. The selective anti-M. tuberculosis activity of CPZEN-45 occurs through specific inhibition of the phospho-N-acetylglucosaminyl transferase WecA, which is involved in arabinogalactan biosynthesisCitation31) (Fig. ). In infectious disease animal model testing, CPZEN-45 showed high efficacy against not only sensitive M. tuberculosis but also an XDR-TB strain resistant to 10 drugs. In tests using sensitive M. tuberculosis, CPZEN-45 combination therapy with isoniazid and rifampicin exhibited promising synergistic effects.Citation32)
Fig. 6. Structures of A54556 A, cyclomarin A, and ilamycin A.
The Institute of Microbial Chemistry and Lilly TB Drug Discovery Initiative are proceeding with joint development of CPZEN-45 as an XDR-TB treatment drug, which has reached the preclinical stage.
Disruption of Clp proteases
A serine protease that exists universally in bacteria, Clp (caseinolytic protease, Fig. ) degrades abnormal proteins with incorrect folds or translation errors. Clp is an enzyme complex with a similar function to the eukaryotic proteasomes; however, it is not essential in most bacteria. The acyldepsipeptide antibiotic A54556 A (Fig. ) from Streptomyces hawaiiensis is a new type of antibiotic discovered by the German company Bayer. A54556 binds to the Bacillus subtilis ClpP subunit, deregulating its function and inducing cell death.Citation33) Because ClpP is essential for the viability of M. tuberculosis unlike most bacteria, the possibility of using this type of antibiotic as an antituberculous drug is being examined.Citation34) Currently, exploratory research for compounds that target this enzyme complex is propelled by the TB Alliance.
Fig. 7. Bacterial Clp protease and the substances that disrupt it.
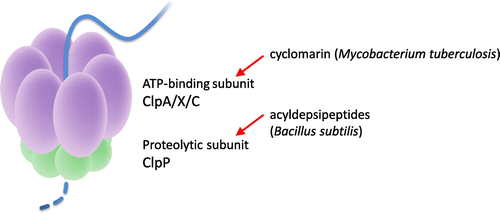
Fig. 8. Mycobacterium tuberculosis cell wall biosynthesis pathway and the natural product inhibitor.
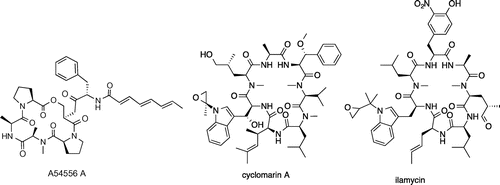
Cyclomarin (Fig. )
Cyclomarin is a heptapeptide derived from the marine actinomycete Streptomyces sp.Citation35) It acts upon the ClpC1 subunit of the M. tuberculosis Clp, causing deregulated protein degradation and subsequent cell deathCitation36) (Fig. ). A similar compound, ilamycin, is produced by Streptomyces sp.Citation37) (Fig. ) and, like cyclomarin, acts specifically on acid-fast bacilli.
Concluding remarks
Historically, discoveries of key pharmaceutical drugs such as streptomycin, rifampicin, and pyrazinamide have had large clinical effects. Among these medicinal compounds, natural products have indirectly and directly played a momentous role. It is believed that in the future, natural products, especially actinomycete metabolites, will continue to lead to the appearance of novel antituberculous drugs.
Development of novel antituberculous drugs will shorten and simplify treatment and increase the cure rate. With the arrival of new drugs possessing novel mechanisms of action, it is now possible to establish treatment methods for MDR/XDR-TB and tuberculosis/HIV superinfections. These pharmaceutical drugs could also have hidden potential as treatments for nontuberculosis mycobacterial infections, particularly NTDs such as leprosy and Buruli ulcers.
Around the time, I started researching actinomycete metabolites, I had the honor of listening to the Academy Prize commemorative speeches of Prof. Hata and Prof. Ōmura. I remember being deeply impressed by their discoveries of new medicines from microorganisms. Twenty-five years later, Prof. Ōmura’s Nobel Prize has reaffirmed the importance of drug discovery from natural products and has greatly encouraged all of us who pursue research on natural product drug discovery.
Disclosure statement
No potential conflict of interest was reported by the author.
Acknowledgments
I would like to express my gratitude to Dr. Yoshimasa Ishizaki of the Microbial Chemistry Research Foundation who assisted with proofreading this manuscript and creating figures.
Notes
† This work is a translation of an original work in Japanese in the Japan Society for Bioscience, Biotechnology, and Agrochemistry http://doi.10.1271/kagakutoseibutsu.54.37.
1. The Global Alliance for TB Drug Development (TB Alliance) and the Medicine for Malaria Venture (MMV) are known to be a major resource. In Japan, the Global Health Innovative Technology Fund (GHIT) was founded.
Related Research Data
References
- Burg RW, Miller BM, Baker EE, et al. Avermectins, new family of potentanthelmintic agents: producing organism and fermentation. J Antimicrob Chemother. 1979;15:361.10.1128/AAC.15.3.361
- Tu YY, Ni MY, Zhong YR, et al. Studies on the constituents of Artemisia annua L. Yao Xue Xue Bao. 1981;16:366.
- Global TB control WHO report 2014.
- Global report: UNAIDS report on the global AIDS epidemic 2013.
- Igarashi M, Takahashi Y. Developmental status of new antituberculous drugs. Nihon Rinsho. 2011;69:1482–1488.
- Nuermberger EL, Yoshimatsu T, Tyagi S, et al. Moxifloxacin-containing regimen greatly reduces time to culture conversion in murine tuberculosis. Am J Respir Crit Care Med. 2004;169:421–426.10.1164/rccm.200310-1380OC
- Rustomjee R, Lienhardt C, Kanyok T, et al. A Phase II study of the sterilising activities of ofloxacin, gatifloxacin and moxifloxacin in pulmonary tuberculosis. Int J Tuberc Lung Dis. 2008;12:128–138.
- Cynamon M, Sklaney MR, Shoen C. Gatifloxacin in combination with rifampicin in a murine tuberculosis model. J Antimicrob Chemother. 2007;60:429–432.10.1093/jac/dkm200
- Disratthakit A, Doi N. In vitro activities of DC-159a, a novel fluoroquinolone, against Mycobacterium species. Angew Chem Int Ed. 2010;54:2684–2686.10.1128/AAC.01545-09
- Alcalá L, Ruiz-Serrano MJ, Pérez-Fernández Turégano C, et al. In vitro activities of linezolid against clinical isolates of Mycobacterium tuberculosis that are susceptible or resistant to first-line antituberculous drugs. Antimicrob Agents Chemother. 2003;47:416–417.10.1128/AAC.47.1.416-417.2003
- Williams KN, Stover CK, Zhu T, et al. Promising antituberculosis activity of the oxazolidinone PNU-100480 relative to that of linezolid in a murine model. Antimicrob Agents Chemother. 2009;53:1314–1319.10.1128/AAC.01182-08
- Balasubramanian V, Solapure S, Iyer H, et al. Bactericidal activity and mechanism of action of AZD5847, a novel oxazolidinone for treatment of tuberculosis. Antimicrob Agents Chemother. 2014;58:495–502.10.1128/AAC.01903-13
- Nikonenko BV, Protopopova M, Samala R, et al. Drug therapy of experimental tuberculosis (TB): improved outcome by combining SQ109, a new diamine antibiotic, with existing TB drugs. Antimicrob Agents Chemother. 2007;51:1563–1565.10.1128/AAC.01326-06
- Andries K, Verhasselt P, Guillemont J, et al. A diarylquinoline drug active on the ATP synthase of Mycobacterium tuberculosis. Science. 2005;307:223–227.10.1126/science.1106753
- Diacon AH, Pym A, Grobusch M, et al. The diarylquinoline TMC207 for multidrugresistant tuberculosis. N Eng J Med. 2009;360:2397–2405.10.1056/NEJMoa0808427
- Jones D, Metzger HJ, Schatz A, et al. Control of Gram-negative bacteria in experimental animals by streptomycin. Science. 1944;100:103–105.
- Umezawa H, Ueda M, Maeda K, et al. Production and isolation of a new antibiotic: kanamycin. J Antibiot. 1957;10:181–188.
- Price KE, Pursiano TA, DeFuria MD. Activity of BB-K8 (amikacin) against clinical isolates resistant to one or more aminoglycoside antibiotics. Antimicrob Agents Chemother. 1974;5:143–152.10.1128/AAC.5.2.143
- Kurosawa H. The isolation of an antibiotic produced by a strain of streptomyces K-300. Yokohama Med Bull. 1952;3:386–399.
- Herr EB, Redstone MO. Chemical and physical characterization of capreomycin. Ann. N. Y. Acad. Sci. 1966;135:940–946.
- Sensi P, Margalith P, Timbal MT. Rifomycin, a new antibiotic; preliminary report. Farmaco Sci. 1959;14:146–147.
- Maeda K, Osato T, Umezawa H. A new antibiotic, azomycin. J Antibiot. 1953;6:182.
- Singh R, Manjunatha U, Boshoff HI, et al. PA-824 kills nonreplicating Mycobacterium tuberculosis by intracellular NO release. Science. 2008;322:1392–1395.10.1126/science.1164571
- Stover CK, Warrener P, VanDevanter DR, et al. A small-moleculenitroimidazopyran drug candidate for the treatment of tuberculosis. Nature. 2000;405:962–966.10.1038/35016103
- Dawson R, Diacon AH, Everitt D, et al. Efficiency and safety of the combination of moxifloxacin, pretomanid (PA-824), and pyrazinamide during the first 8 weeks of antituberculosis treatment: a phase 2b, open-label, partly randomised trial in patients with drug-susceptible or drug-resistant pulmonary tuberculosis. Lancet. 2015;385:1738–1747.10.1016/S0140-6736(14)62002-X
- http://www.tballiance.org/pipeline/pipeline.php.
- Matsumoto M, Hashizume H, Tomishige T, et al. OPC-67683, a nitro-dihydroimidazooxazole derivative with promising action against tuberculosis in vitro and in mice. PLoS Med. 2006;3:e446.
- Igarashi M, Nakagawa N, Doi N, et al. Caprazamycin B, a novel anti-tuberculosis antibiotic, from Streptomyces sp. J Antibiot. 2003;56:580–583.10.7164/antibiotics.56.580
- Isono K, Uramoto M, Kusakabe H, et al. Liposidomycins: novel nucleoside antibiotics which inhibit bacterial peptidoglycan synthesis. J Antibiot. 1985;38:1617–1621.10.7164/antibiotics.38.1617
- Takahashi Y, Igarashi M, Miyake T, et al. Novel semisynthetic antibiotics from caprazamycins A-G: caprazene derivatives and their antibacterial activity. J Antibiot. 2003;66:171–178.
- Ishizaki Y, Hayashi C, Inoue K, et al. Inhibition of the first step in synthesis of the mycobacterial cell wall core, catalyzed by the GlcNAc-1-phosphate transferase WecA, by the novel caprazamycin derivative CPZEN-45. J Biol Chem. 2013;288:30309–30319.10.1074/jbc.M113.492173
- Miyake T, Takahashi, Y, Igarashi M. Synthesis and activity of CPZEN-45, a new antituberculous drug candidate. 235th ACS National Meet: CARB. 2008;104.
- Brötz-Oesterhelt H, Beyer D, Kroll HP, et al. Dysregulation of bacterial proteolytic machinery by a new class of antibiotics. Nat Med. 2005;11:1082–1087.10.1038/nm1306
- Raju RM, Unnikrishnan M, Rubin DH, et al. Mycobacterium tuberculosis ClpP1 and ClpP2 function together in protein degradation and are required for viability in vitro and during infection. PLoS Pathog. 2012;8:e1002511.10.1371/journal.ppat.1002511
- Renner MK, Shen YC, Cheng XC, et al. Cyclomarins A-C, new antiinflammatory cyclic peptides produced by a marine bacterium (Streptomyces sp.). J Am Chem Soc. 1999;121:11273–11276.10.1021/ja992482o
- Schmitt EK, Riwanto M, Sambandamurthy V, et al. The natural product cyclomarin kills Mycobacterium tuberculosis by targeting the ClpC1 subunit of the caseinolytic protease. Angew Chem Int Ed Engl. 2011;50:5889–5891.10.1002/anie.v50.26
- Kaku R. Studies on the antitubercular activity of a new antibiotic, ilamycin. 1. Antitubercular activity of water-insoluble ilamycin. J Antibiot B. 1963;16:93–98.