
Abstract
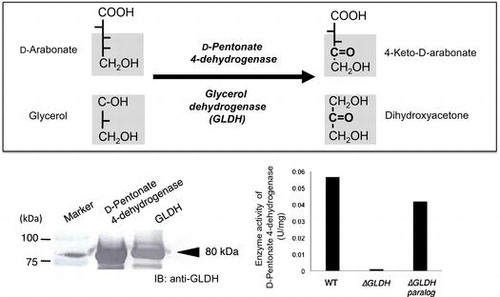
A novel oxidation of D-pentonates to 4-keto-D-pentonates was analyzed with Gluconobacter thailandicus NBRC 3258. D-Pentonate 4-dehydrogenase activity in the membrane fraction was readily inactivated by EDTA and it was reactivated by the addition of PQQ and Ca2+. D-Pentonate 4-dehydrogenase was purified to two different subunits, 80 and 14 kDa. The absorption spectrum of the purified enzyme showed no typical absorbance over the visible regions. The enzyme oxidized D-pentonates to 4-keto-D-pentonates at the optimum pH of 4.0. In addition, the enzyme oxidized D-fructose to 5-keto-D-fructose, D-psicose to 5-keto-D-psicose, including the other polyols such as, glycerol, D-ribitol, D-arabitol, and D-sorbitol. Thus, D-pentonate 4-dehydrogenase was found to be identical with glycerol dehydrogenase (GLDH), a major polyol dehydrogenase in Gluconobacter species. The reaction versatility of quinoprotein GLDH was notified in this study.
The newly isolated Gluconacetobacter liquefaciens (Ga. liquefaciens) RCTMR strains catalyze further oxidation in D-glucose oxidation pathway beyond 2,5-diketo-D-gluconate yielding novel carbonyl sugars.Citation1) Formation of 4-keto-D-pentonate by the oxidative fermentation of acetic acid bacteria was indicated in the previous papers.Citation1–3) There are three different metabolic pathways forming 4-keto-D-arabonate. Enzymatic oxidation of 4-keto-D-arabinose formed from decarboxylation of 2,5-diketo-D-gluconate.Citation1) Alternatively, 4-keto-D-arabinose is formed after oxidation of D-arabinose by a putative D-aldopentose 4-dehydrogenase.Citation2) The third pathway is the direct oxidation of D-arabonate at the C4-postion.Citation3) There are two different metabolic pathways forming 4-keto-D-ribonate, one from D-ribose via 4-keto-D-riboseCitation2) and the direct oxidation at the C4-postion of D-ribonate.Citation3) There was no report about D-pentonate 4-oxidizing enzyme until we reported the existence of the novel enzyme in the membrane of acetic acid bacteria.Citation1) D-Pentonate 4-dehydrogenase is significant because it catalyzes the formation of oxidation products, 4-keto-D-pentonates. It implies that the sequential oxidation pathway of D-glucose by acetic acid bacteria is extending beyond 2,5-diketo-D-gluconate, which had been thought to be the final product in D-glucose oxidation for more than past 50 years. A strong oxidation of D-pentonate was observed in Ga. liquefaciens RCTMR 10Citation1), whereas other strains belonging to Gluconobacter and Gluconacetobacter genera show variable D-pentonate 4-dehydrogenase activity.Citation2)
Interestingly, the same membrane fraction oxidized D-fructose to 5-keto-D-fructose, as preliminarily shown in the previous paper.Citation2) With respect to D-fructose oxidation to 5-keto-D-fructose, a flavoprotein D-fructose dehydrogenase (FDH, EC 1.1.99.11) from Gluconobacter industrius IFO 3260 (presently Gluconobacte japonicus NBRC 3260) is the sole example of FDH so far reported.Citation4) No FDH has been reported in other species over Gluconobacter and Gluconacetobacter. It is also important to solve the question whether a similar enzyme to FDH exists in acetic acid bacteria or an alternative FDH, with a different coenzyme from flavoprotein FDH, catalyzing D-fructose oxidation to 5-keto-D-fructose. Similarly to the case of D-sorbitol oxidation forming L-sorbose, a bound-FAD containing D-sorbitol dehydrogenase (SLDH) from Gluconobacte suboxydans var. α IFO 3254 (presently Gluconobacte thailandicus [G. thailandicus] NBRC 3254) has been characterized.Citation5) The bound flavin-dependent SLDH is the sole example of SLDH, which shows a narrow substrate specificity and oxidizes D-sorbitol exclusively. Except for a few examples, membrane-bound dehydrogenases in acetic acid bacteria have been mainly classified into two categories, PQQ- or FAD-dependent dehydrogenases.Citation6) As an alternative SLDH, PQQ-dependent D-arabitol dehydrogense and SLDH have been purified.Citation7,8) The two PQQ-dependent enzymes show similar and broad substrate specificity to several different polyols. These quinoproteins have been shown to be identical to each other based on their immuno-cross-reactivity and also on gene disruption and finally it was suggested to be the same as the previously isolated glycerol dehydrogenase (GLDH) (EC 1.1.99.22).Citation9)
In this paper, we wish to note that the oxidation of D-pentonate, D-fructose, and D-psicose yielding novel carbonyl sugars is catalyzed by GLDH, a major polyol dehydrogenase, showing novel reaction versatility of the enzyme.
Materials and methods
Chemicals
Potassium D-arabonate was prepared by bromine oxidation of D-arabinose.Citation10) D-Ribonate and D-erythronate were prepared by delactonization of D-ribono-1,4-lactone (Wako, Osaka, Japan) and D-erythronolactone (Tokyo Kasei, Tokyo), respectively. CM-Toyopearl C-650 was purchased from Tosoh Mfg. (Yamaguchi, Japan). n-Dodecyl-β-D-maltoside, n-octyl-β-D-thioglucoside, and n-octyl-β-D-glucoside were purchased from Dojindo (Kumamoto, Japan). Tween 20, Brij 35, Na-cholate, Triton X-100 and sucrose monolarurate were commercially obtained. Mydol 10 and Mydol 12 were kind gift from Kao (Tokyo, Japan). PQQ was prepared and purified as reported previously.Citation11)
Microbial strain and culture medium
G. thailandicus NBRC 3258 was mainly used in this study. A basal medium was composed of 2% Na-gluconate, 0.5% glucose, 0.3% glycerol, 0.3% yeast extract (Oriental Yeast, Co, Tokyo), and 0.2% polypeptone. The bacterial culture (150 mL) in 500 mL of an Erlenmeyer flask was done at 30°C under shaking at 200 rpm.
Preparation of membrane and cytoplasmic fractions
G. thailandicus NBRC 3258 was cultured in 20 L of the basal medium. Cultivation was terminated when the turbidity of the culture medium reached at 350 Klett units, which corresponded to the early stationary phase. A part of cell paste (50 g wet wt.) was suspended in 300 mL of 5 mM acetate buffer, pH 5.0, and disrupted mechanically with a French pressure cell press (Aminco, Silver Spring, MD) at 1000 kg/cm2. To the cell homogenate, phenylmethylsulfonyl fluoride was added to 0.5 mM. After unbroken cells were removed by low-speed centrifugation, 3000 rpm for 5 min, the resulting crude cell-free extract was further centrifuged at 105 ×g for 90 min to separate the membrane fraction from the cytoplasmic fraction.
Enzyme assays
The enzyme activity of D-pentonate 4-dehydrogenase was measured using K-ferricyanide as an electron acceptor, according to the method described by Ameyama.Citation12) The reaction mixture contained 50 μmol of substrate, 100 μmol of acetate buffer, pH 5.0, 10 μmol of K-ferricyanide, and enzyme solution in 1.0 mL. The reaction was started by the addition of K-ferricyanide. The reaction was maintained at 25°C and terminated by adding 0.5 mL of ferric-Dupanol solution. To the reaction mixture, 3.5 ml of water was added and the reaction mixture stood at room temperature for 20 min, before reading the absorbance of the solution at 660 nm. One unit of enzyme activity was defined as the amount of enzyme that catalyzed the oxidation of 1 μmol of D-arabonate per min under the above conditions, which was equivalent to 4.0 of absorbance unit at 660 nm. Alternatively, the enzyme activity was measured with phenazine methosulfate (PMS) and 2,6-dichlorophenol indophenol (DCIP) as the electron acceptors and decrease in absorbance at 600 nm observed at 25°C was recorded by a spectrophotometer. The reaction mixture (1 mL) contained 100 μmol of acetate buffer, pH 5.0, 50 μmol of substrate, 0.3 μmol of PMS, and 0.1 μmol of DCIP. The reaction was started by the addition of PMS and DCIP. In the acetate buffer used, CaCl2 and PQQ were added to 5 mM and 0.5 μM, respectively, to certify the enzyme as holo-enzyme during the assay. One unit of enzyme activity was defined as the enzyme catalyzing oxidation of 1 μmol of substrate per min under the above conditions. The extinction coefficient of DCIP at pH 5.0, of 3.5 mM−1cm−1 was used. Protein concentration measurement was done by reading optical density at 280 nm and 1% protein solution (10 mg/mL) was arbitrarily defined to be 10.0. The specific activity was defined as enzyme activity units/mg protein. Enzyme activity of D-aldopentose 4-dehydrogenase, aldehyde dehydrogenase (ALDH), and alcohol dehydrogenase (ADH) was assayed with K-ferricyanide under the similar conditions described above using D-ribose, acetaldehyde, and ethanol used as substrate, respectively.
EDTA treatment of membrane
The membrane suspension (20 mg of protein/mL in 5 mM acetate buffer, pH 5.0) was dialyzed overnight in a cold room against the same buffer containing 20 mM EDTA. The membrane suspension was centrifuged three times to remove excess EDTA at 105 ×g for 60 min. The resulting precipitate was resuspended in 5 mM acetate buffer, pH 5.0, and designated as apo-enzyme. The apo-enzyme was reactivated to the holo-enzyme by the exogenous addition of PQQ and CaCl2 at 5 μM and 5 mM, respectively. To complete the holo-enzyme formation, the membrane suspension was maintained at 20°C for 30 min.
TLC chromatography
Analytical TLC cellulose plate (aluminum sheet 20 × 20 cm, Merck, Darmstadt, Germany) was used. It was done at 25°C with a solvent system of t-butanol:formic acid:water = 4:1:1.5. Detection of reaction products, carbonyl sugars, and carbonyl sugar acids possessing intramolecular ketones was done at room temperature by spraying an alkaline ethanol solution of 2,3,5-triphenyltetrazolium chloride over TLC plate.Citation13)
Purification of D-pentonate 4-dehydrogenase
All operations were done at 4°C. The membrane fraction of G. thailandicus NBRC 3258 was homogenized by a glass homogenizer in 5 mM acetate buffer, pH 4.0 and the protein concentration was adjusted to 20 mg/mL. Before enzyme solubilization, it was brought to holo-enzyme by adding PQQ and CaCl2 to the membrane suspension to 5 μM and 5 mM, respectively. Enzyme solubilization was done by adding 1.0% (v/v) Mydol 10 to the membrane suspension. The membrane suspension was stirred gently overnight and centrifuged at 105 ×g for 60 min. The supernatant was dialyzed for 6 h against 5 mM acetate buffer, pH 4.0 containing 0.2% Mydol 10. Insoluble precipitate was removed by centrifugation before applying the enzyme solution to a CM-Toyopearl C-650 column (1.5 × 15 cm), which was equilibrated with the same acetate buffer. After washing the column with the buffer, the enzyme was eluted by a linear gradient made by 10 to 35 mM CaCl2 in the same acetate buffer. Then, the pooled enzyme fraction was concentrated by a centrifugal filter unit (Millipore Centricon, MW 50,000, Millipore Japan) and applied to a Sephadex G-200 column (1.5 × 120 cm), which had been equilibrated with 5 mM acetate buffer, pH 4.0, containing 5 mM CaCl2 and 0.2% Mydol 10, and fractionated by every 3 mL fraction.
SDS-polyacrylamide gel electrophoresis
SDS-polyacrylamide gel electrophoresis (SDS-PAGE) was done by a slab gel of polyacrylamide (12.5%, w/v) by the method described by Laemmli.Citation14) The dual color standard proteins of molecular masses from 250 kDa to 15 kDa (Bio-Rad, Hercules, CA, U.S.A.) were used as references for measurement of the molecular mass of the enzyme.
Amino acid sequencing of D-pentonate 4-dehydrogenase
D-pentonate 4-dehydrogenase was electrophoresed in SDS-PAGE, and then the protein bands were electroblotted on to a PVDF membrane. After visualization of protein bands with Ponceau S, the 80-kDa subunit was cut out, and then subjected to amino acid sequencing. Amino acid sequencing was done with a Shimadzu model protein sequencer, PPSQ-21A, according to the instructions of the manufacturer.
Preparation of GLDH mutants
GLDH mutants (∆GLDH and ∆GLDH paralog) were prepared from G. thailandicus NBRC 3255. The gene encoding GLDH (NBRC3255_0235) was disrupted by the same method described previously,Citation15) while knock out mutant of GLDH paralog was prepared as follows: for disruption of GLDH paralog gene (NBRC3255_0026), the disruption vector, pSK-∆ParaSldA, was constructed. The 3.1 kb fragment containing NBRC3255_0026 was PCR-amplified with the primer set, 41_ParaSldBA-5′ (5′-AAAGCGGCCGCCGGTGCCACGGGACTTTAT-3′) and 42_ParaSldBA-3′(5′-CCGCTCGAGGACGTGACGCTGGGCTATC-3′) from genomic DNA of G. thailandicus NBRC 3255 with TaKaRa Ex Taq DNA polymerase (Takara Bio Inc., Shiga, Japan). The purified PCR product was digested with NotI/XhoI and ligate to pBluescript SK+ vector (Stratagene, CA, USA). The resultant plasmid, pSK-ParaSldBA, was linearlized with EcoRI digenstion and treated with Calf intestine alkaline phosphatase, then ligated with the EcoRI-digested tetracycline-resistant cassette provided from pKRP12 to yield pSK-∆ParaSldA. To perform homologous recombination in the NBRC3255_0026 locus of genomic DNA, circular pSK-∆ParaSldA was electroporated into G. thailandicus NBRC 3255 at the setting 2.5 kV, 600 Ω, 10 μF, using a 2 mm gap electrocuvette using MicroPluser (Bio-Rad Laboratories, Hercules, California, USA). The tetracycline-resistant colonies confirmed gene disruption by PCR-amplification with same primer set using genome DNA as a template, comparing with wild-type cells (3.1 kb) and ∆GLDH paralog cells (5.1 kb).
Results and discussion
Resolution of D-pentonate 4-dehydrogenase with EDTA
When the membrane fraction of G. thailandicus was incubated with D-arabonate and D-ribonate, 4-keto-D-arabonate and 4-keto-D-ribonate predominated as the sole reaction product, as indicated in the previous paper.Citation2) To characterize the enzyme in the membrane fraction whether it was a quinoprotein or a flavoprotein, the effect of EDTA to the enzyme activity was examined with D-arabonate and D-ribonate. EDTA-treatment of the membrane fraction caused a clear decrease in D-pentonate 4-dehydrogenase activity and the decreased enzyme activity was reactivated to almost the original level by the addition of PQQ and Ca2+ (Fig. ). Other species of divalent metallic ions, such as Mg2+, Sr2+, Ba2+, and several kinds of lantanides, were ineffective and no appreciable reactivation of the apo-enzyme was observed. Thus, D-pentonate 4-dehydrogenase was confirmed tentatively to be a quinoprotein, to which PQQ is attached by the aid of Ca2+. On the contrary, in the case of flavoproteins such as D-gluconate dehydrogenase (GADH),Citation16, 17) D-fructose dehydrogenase,Citation4) SLDH,Citation5), and 2-keto-D-gluconate dehydrogenase,Citation18) enzyme activity was not affected with EDTA treatment at all.
Fig. 1. Effects of EDTA on D-pentonate 4-dehydrogenase.
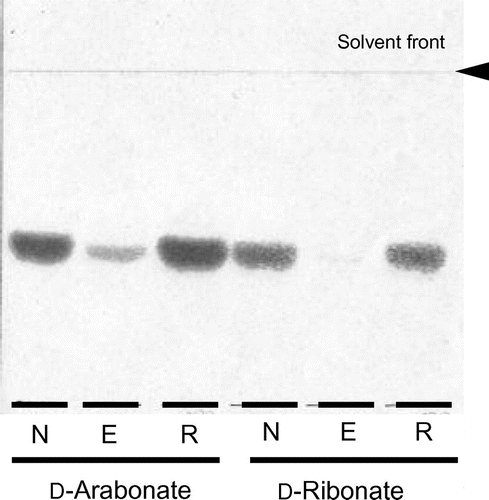
Fig. 2. Effects of EDTA on D-fructose 5-dehydrogenase.
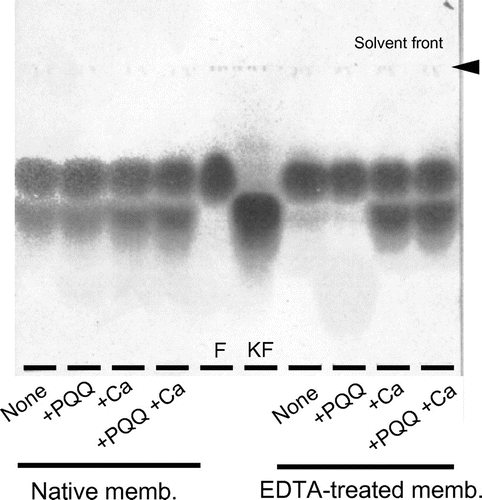
Fig. 3. CM-Toyopearl chromatography of D-pentonate 4-dehydrogenase.
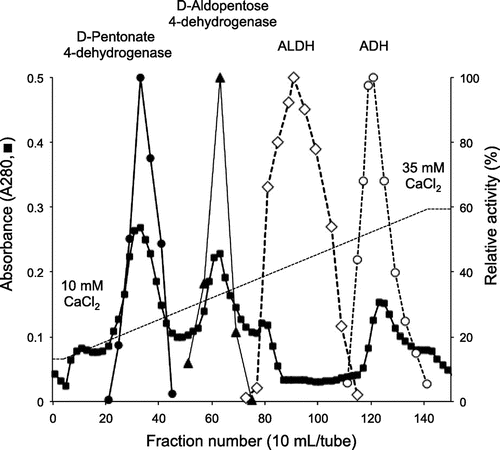
Fig. 4. Gel filtration, absorption spectrum (A) and SDS-PAGE (B) of purified D-pentonate 4-dehydrogenase.
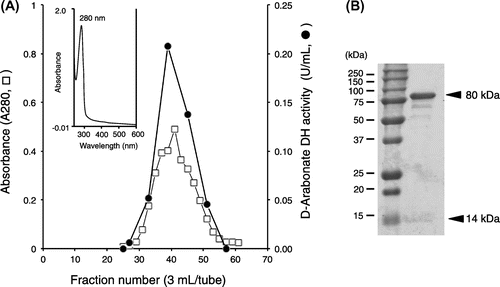
Fig. 5. Reaction versatility of GLDH to D-pentonate, D-fructose, D-psicose, and D-erythronate on the basis of the Bertrand–Hudson’s rule.
Fig. 6. Immunoblotting analysis of D-pentonate 4-dehydrogenase and glycerol dehydrogenase (GLDH).

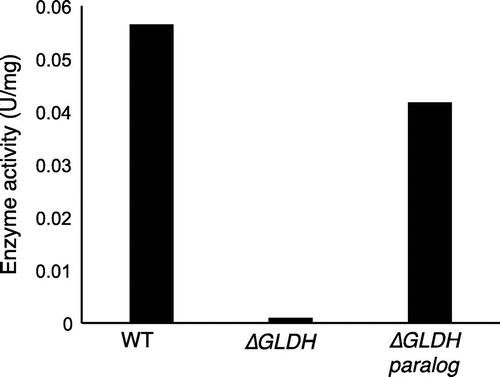
Fig. 7. Effect of gene disruption on D-pentonate 4-dehydrogenase activity.
Resolution of D-fructose 5-dehydrogenase with EDTA
Using the same membrane fraction that had been resolved with EDTA, the profile of D-fructose oxidation was examined under the similar criteria employed above. The native membrane showed a strong D-fructose oxidation irrespective to the presence of PQQ or Ca2+ and 5-keto-D-fructose was obtained as the direct oxidation product (Fig. ). On the other hand, D-fructose oxidation was lost after EDTA treatment. Since the enzyme activity was not recovered only by the addition of PQQ, Ca2+ seemed to be essential. These data demonstrate that Ca2+ plays an important role to link PQQ with PQQ-binding site in the apo-enzyme. D-Fructose 5-dehydrogenase predominated over Gluconobacter and Gluconacetobacter, suggesting its profound physiological roles in D-fructose metabolism. Enzyme activity of the flavin-dependent FDHCitation4) was not resolved with EDTA, while D-fructose 5-dehydrogenase in this study was readily converted to the apo-enzyme. Different from the case of membrane enzyme, once the solubilized D-fructose 5-dehydrogenase was resolved with EDTA, it was difficult to resume the lost enzyme activity to the original level by adding PQQ and Ca2+. Though the data are not shown here, D-psicose was also oxidized to 5-keto-D-psicose. Since a standard 5-keto-D-psicose is not available from commercial sources, the oxidation product was judged putatively to be 5-keto-D-psicose.
Solubilizatioin of D-pentonate 4-dehydrogenase from the membrane fraction
Before solubilization of D-pentonate 4-dehydrogenase, PQQ and CaCl2 were added to the membrane fraction (20 mg of protein/mL in 5 mM of acetate buffer, pH 4.0) to 5 μM and 5 mM, respectively, preventing partial resolution of the target quinoproteins. Solubilization of D-pentonate 4-dehydrogenase was done with 1% of various detergents as shown in Table . n-Octyl-β-D-thioglucoside, n-octyl-β-D-glucoside, Mydol 10, and Mydol 12 were effective detergents to solubilize the enzyme. From this result, we selected Mydol 10 for solubilization of D-pentonate 4-dehydrogenase since we had been successful in purification of various quinoproteins using this detergent.Citation7,15,19) No appreciable enzyme activity remained in the membrane residue precipitated after solubilization. Solubilized enzyme activity of D-pentonate 4-dehydrogenase was lost almost completely when it was dialyzed against acetate buffer, pH 4.0, in the absence of detergent. These observations strongly indicate that D-pentonate 4-dehydrogenase was a typical membrane-bound enzyme.
Table 1. Solubilization of D-pentonate 4-dehydrogenase from the membrane fraction of G. thailandicus NBRC 3258.
Purification of D-pentonate 4-dehydrogenase
D-Pentonate 4-dehydrogenase was eluted from CM-Toyopearl C-650 at the initial stage of linear gradient of CaCl2 and the enzyme activity appeared as a sharp peak as shown in Fig. . As judged from SDS-PAGE preliminarily done at this stage, D-pentonate 4-dehydrogenase appeared to be highly purified from the purification procedures employed in this study. At this stage of enzyme purification, enzyme activity assayed with K-ferricyanide was lost rapidly. It was caused by the release of co-existed heme c components in the solubilized enzyme solution during the CM-Toyopearl chromatography. Therefore, the enzyme activity of D-pentonate 4-dehydrogenase was no longer assayed with K-ferricyanide and, instead of that, it was assayed with PMS-DCIP as the electron acceptors hereafter, as similarly seen with other examples of membrane-bound quinoproteins.Citation9, 19, 20) On the contrary, D-aldopentose 4-dehydrogenase, ALDH, and ADH were assayed with K-ferricyanide. To certify the purity of enzyme preparation, active enzyme fractions from CM-Toyopearl chromatography were pooled and concentrated by a Centricon and applied to Sephadex G-200 gel filtration. As shown in Fig. , elution of protein by the absorbance at 280 nm coincided with enzyme activity. The enzyme activities oxidizing glycerol, D-sorbitol, D-arabitol, and ribitol were purified for about 50 times from the membrane fraction with 50–60% recovery as summarized in Table . The purified enzyme was recovered with a high yield after the simple enzyme purification method employed. The purification and recovery of the minor enzyme activities oxidizing D-pentonates and D-fructose were deduced based on the purification yields against polyol oxidation. Since enzyme activity ratio of oxidizing glycerol to D-arabonate or glycerol to D-fructose was constant to be about 10 through the purification steps from the membrane fraction to the purified enzyme, the data strongly suggest that a same single enzyme catalyzed oxidation of all these substrates. It should be noted that D-pentonate 4-dehydrogenase was distinct from GADH of oxidative bacteria, such as Serratia marcescens,Citation16) Pseudomonas aeruginosa,Citation17) and acetic acid bacteria.Citation21) D-Pentonate 4-dehydrogenase contained PQQ as the primary coenzyme, while GADH was a flavoprotein containing a covalently bound flavin.Citation22)
Table 2. Summary of purification of D-pentonate 4-dehydrogenase from G. thailandicus NBRC 3258.
Physicochemical and catalytic properties of D-pentonate 4-dehydrogenase
Absorption spectrum of the purified D-pentonate 4-dehydrogenase did not show any appreciable absorption peak over the visible regions (Fig. (A)) and it was alike to that of quinoprotein D-glucose dehydrogenase.Citation23,24) When examined the molecular size of the enzyme with SDS-PAGE, the enzyme was composed of two different subunits of 80 and 14 kDa (Fig. (B)). In addition to D-arabonate, D-ribonate, and D-erythronate, D-fructose and D-psicose were oxidized with D-pentonate 4-dehydrogenase. The optimum pH of D-pentonate oxidation was found at 4.0 and apparent km was determined to be 10.5 mM for D-arabonate and 9.7 mM for D-ribonate. D-Erythronate was also oxidized with the enzyme giving 3-keto-D-erythronate as a putative reaction product, since the purity of the substrate from a commercial source was not guaranteed. Surprisingly, when reactivity of D-pentonate 4-dehydrogenase was examined with different polyols, a higher enzyme activity was observed with glycerol, D-sorbitol, D-arabitol, and ribitol, as suggested above (Table ). D-Gluconate was also oxidized at the similar velocity with that of D-arabonate (Data not shown). On the basis of these results from substrate specificity, it was suggested that D-pentonate 4-dehydrogenase was identical to GLDH, a major polyol dehydrogenase, in acetic acid bacteria.Citation15) D-Pentonate 4-dehydrogenase catalyzed oxidation of a wide variety of substrates which have an erythro-configuration to the bottom primary alcohol residue basing on the Bertrand–Hudson’s rule.Citation6) D-Pentonate 4-dehydrogenase is different from the membrane-bound SLDH, which is composed of three different subunit structure involving heme c subunit.Citation8)
Genomic characterization of D-pentonate dehydrogenase
We prepared the disruptant of pqq1 gene (GOX1857) from G. oxydans 621H and GLDH-like gene (NBRC3255_0026) from G. thailandicus NBRC 3255 and their activities for D-aldopentoses were compared with those of the wild-type strain. Both of the wild-type strains did not show the enzyme activity for D-fructose, indicating that these strains do not have D-fructose 5-dehydrogenase. On the contrary, enzyme activities for D-ribose and D-arabinose were detected in these strains and they are enhanced by the addition of PQQ and CaCl2. However, the loss of enzyme activity in the mutant was not observed, suggesting that GOX1857 and NBRC3255_0026 were not responsible for D-aldopentose 4-dehydrogenase. From our recent investigations about the substrate specificity, D-aldopentose 4-dehydrogenase was different from GDH. GDH oxidized the C1-position of D-glucose giving D-gluconate but did not oxidize the C4-position of D-aldopentose, while D-aldopentose 4-dehydrogenase oxidized the C4-position of D-aldopentose but not C1-position. As shown in Fig. , D-pentonate 4-dehydrogenase was separated clearly from D-aldopentose 4-dehydrogenase in CM-Toyopearl chromatography. Purification and characterization of D-aldopentose 4-dehydrogenase would be the subject in our forthcoming study.
D-Pentonate 4-dehydrogenase oxidized a variety of polyols in accordance with Bertrand–Hudson’s rule and D-pentonate was also included among the oxidizable substrates. In addition, SDS-PAGE showed that D-Pentonate 4-dehydrogenase composed of 80- and 14-kDa subunit. Membrane-bound quinoprotein consisting of two subunits is limited in Gluconobacter species.
Thus, D-pentonate 4-dehydrogenase could be similar to the major polyol dehydrogenase in acetic acid bacteria such as GLDH (Fig. ). Alternative confirmation showing D-pentonate 4-dehydrogenase is identical to GLDH was conducted with the following criteria: immuno-blotting analysis of the purified enzyme with GLDH and gene disruption of D-pentonate 4-dehydrogenase activity. To determine the identity of D-pentonate 4-dehydrogenase with GLDH, immuno-blotting analysis was carried out by the method described previously (Fig. ).Citation15) The purified D-pentonate 4-dehydrogenase showed immune-cross-reactivity to anti-GLDH antibody with the same level. Moreover, to investigate whether GLDH is involved in D-pentonate 4-dehydrogenation, the enzyme activity was compared with GLDH mutants of G. thailandicus NBRC 3255 (Fig. ). Two sets of GLDH genes (GLDH, NBRC3255_0235; sldA and GLDH paralog, NBRC3255_0026) are known in G. thailandicus NBRC 3255. The enzyme activity was lost completely in GLDH disruptant (∆GLDH), whereas the mutant of GLDH paralog (∆GLDH paralog) did not affect the enzyme activity. These data indicate GLDH catalyzes D-pentonate 4-oxidation. The N-terminal amino acid sequencing of 80-kDa subunit of D-pentonate 4-dehydrogenase, GAGGE, accorded with the sequence encoded by GLDH gene (NBRC3255_0235; sldA). Provided D-pentonate 4-dehydrogenase is identical to the quinoprotein GLDH, this study has given a complete purification of GLDH for the first time. Because it was not realized in the past 40 years after GLDH purification that became a significant research subject in the research of membrane-bound dehydrogenases.
Author contributions
Osao Adachi conceived and designed the experiments. Yoshitaka Ano and Osao Adachi performed the experiments and prepared the manuscript. Roque A. Hours, Yoshihiko Akakabe, and Naoya Kataoka helped in interpreting data. Toshiharu Yakushi and Kazunobu Matsushita participated in critical revision of the manuscript.
Funding
This work was supported by Japan Society for the Promotion of Science (JSPS KAKENHI) [grant number 24780074] (to YA), [26450094] (to OA).
Acknowledgments
The authors wish to thank Dr. N. Nontapong, Ms. A. Kazui, Ms. A. Nishibara and Ms. M. Fujii, for their technical assistances.
Disclosure statement
No potential conflict of interest was reported by the authors.
Notes
A part of this work was presented at the 2016 annual meeting of JSBBA, Sapporo, as 3D014, on March 29 2016.
References
- Adachi O, Hours RA, Akakabe Y, et al. Production of 4-keto-D-arabonate by oxidative fermentation with newly isolated gluconacetobacter liquefaciens. Biosci Biotechnol Biochem. 2010;74:2555–2558.10.1271/bbb.100698
- Adachi O, Hours RA, Shinagawa E, et al. Formation of 4-keto-D-aldopentoses and 4-Pentulosonates (4-Keto-D-pentonates) with unidentified membrane-bound enzymes from acetic acid bacteria. Biosci Biotechnol Biochem. 2011;75:1801–1806.10.1271/bbb.110339
- Adachi O, Hours RA, Shinagawa E, et al. Enzymatic synthesis of 4-pentulosonate (4-keto-D-pentonate) from D-aldopentose and D-pentonate by two different pathways using membrane enzymes of acetic acid bacteria. Biosci Biotechnol Biochem. 2011;75:2418–2420.10.1271/bbb.110575
- Ameyama M, Shinagawa E, Matsushita K, et al. D-Fructose dehydrogenase of Gluconobacter industrius: Purification, characterization and application to enzymatic micro-determination of D-fructose. J Bacteriol. 1981;145:814–823.
- Shinagawa E, Matsushita K, Adachi O, et al. Purification and characterization of D-sorbitol dehydrogenase from membrane of Gluconobacter suboxydans var. α. Agric Biol Chem. 1982;46:135–141.
- Adachi O, Ano Y, Toyama H, et al. Biooxidation with PQQ- and FAD-dependent dehydrogenase. In Schmid RD, Urlacher VB, editors. Modern biooxidation. Enzymes, reactions and applications. Weinheim: Wiley-VCH; 2007. p. 1–41.
- Adachi O, Fujii Y, Ghaly MF, et al. Membrane-bound quinoprotein D-arabitol dehydrogenase of gluconobacter suboxydans IFO 3257: a versatile enzyme for the oxidative fermentation of various ketoses. Biosci Biotechnol Biochem. 2001;65:2755–2762.10.1271/bbb.65.2755
- Choi ES, Lee EH, Rhee SK. Purification of a membrane-bound sorbitol dehydrogenase from Gluconobacter suboxydans. FEMS Microbiol Lett. 1995;125:45–49.10.1111/fml.1995.125.issue-1
- Ameyama M, Shinagawa E, Matsushita K, et al. Solubilization, purification and properties of membrane-bound glycerol dehydrogenase from Gluconobacter industrius. Agric Biol Chem. 1985;49:1001–1010.
- Isbell HS. Oxidation of aldoses with bromine. J Res Nat Bur Stand. 1962;66A:233–239.10.6028/jres.066A.023
- Ameyama M, Hayashi M, Matsushita K, et al. Microbial production of pyrroloquinoline quinone. Agric Biol Chem. 1984;48:561–565.
- Ameyama M. Enzymatic microdetermination of D-glucose, D-fructose, D-gluconate, 2-keto-D-gluconate, aldehyde, and alcohol with membrane-bound dehydrogenases. Meth Enzymol. 1982;89:20–29.10.1016/S0076-6879(82)89006-X
- Lisboa BP. Thin-layer chromatography of steroids, sterols, and related compounds. Meth Enzymol. 1969;15:38–43.
- Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature (London). 1970;227:680–685.10.1038/227680a0
- Matsushita K, Fujii Y, Ano Y, et al. 5-keto-D-gluconate production is catalyzed by a quinoprotein glycerol dehydrogenase, major polyol dehydrogenase, in gluconobacter species. Appl Environ Microbiol. 2003;69:1959–1966.10.1128/AEM.69.4.1959-1966.2003
- Shinagawa E, Matsushita K, Adachi O, et al. Membrane-bound D-gluconate dehydrogenase of Serratia marcescens. Purification and properties. Agric Biol Chem. 1978;42:2355–236.
- Matsushita K, Shinagawa E, Adachi O, et al. Membrane-bound D-gluconate dehydrogenase from Pseudomonas aeruginosa. Purification and structure of cytochrome-binding form. J Biochem. 9179;85:1173–1181.
- Shinagawa E, Matsushita K, Adachi O, et al. D-Gluconate dehydrogenase, 2-keto-D-gluconate yielding, from Gluconobacter dioxyacetonicus. Purification and characterization. Agric Biol Chem. 1984;48:1517–1522.
- Moonmangmee D, Fujii Y, Toyama H, et al. Purification and characterization of membrane-bound cyclic alcohol dehydrogenase from Gluconobacter frateurii CHM 9. Biosci Biotechnol Biochem. 2001;65:2763–2772.10.1271/bbb.65.2763
- Moonmangmee D, Adachi O, Shinagawa E, et al. L-erythrulose production by oxidative fermentation is catalyzed by PQQ-containing membrane-bound dehydrogenase. Biosci Biotechnol Biochem. 2002;66:307–318.10.1271/bbb.66.307
- Shinagawa E, Matsushita K, Adachi O, et al. D-Gluconate dehydrogenase, 2-keto-D-gluconate yielding, from Gluconobacter dioxyacetonicus. Purification and characterization. Agric Biol Chem. 1984;48:1517–1522.
- McIntire W, Singer TP, Ameyama M, et al. Identification of the covalently bound flavins of D-gluconate dehydrogenase from Pseudomonas aeruginosa and Pseudomonas fluorescens and of 2-keto-D-gluconate dehydrogenase from Gluconobacter melanogenus. Biochem J. 1985;231:651–654.10.1042/bj2310651
- Matsushita K, Ohno Y, Shinagawa E, et al. Membrane-bound D-glucose dehydrogenase from Pseudomonas sp.: solubilization, purification and characterization. Agric Biol Chem. 1980;44:1505–1512.
- Ameyama M, Shinagawa E, Matsushita K, et al. D-glucose dehydrogenase of Gluconobacter suboxydans. Solubilization, purification and characterization. Agric Biol Chem. 1981;45:851–861.
- Terada O, Tomizawa K, Suzuki S, et al. Formation of 5-dehydrofructose by members of Acetobacter species. Part II. Studies on the fermentation, purification and properties of crystal. J Agr Chem Soc Jpn. 1961;35:131–134.
- Adachi O, Matsushita K, Shinagawa E, et al. Purification and properties of methanol dehydrogenase and aldehyde dehydrogenase from Methylobacillus glycogens. Agric Biol Chem. 1990;54:3123–3129.