
Abstract
The synthesis of aurachin B, an antibiotic that features a C3-oxygen-substituted quinoline N-oxide nucleus bearing a farnesyl side chain at C4, was accomplished in 60% overall yield from o-nitrotoluene by a concise five-step sequence. An enantioselective synthesis of aurachin H was also achieved for the first time in only two steps from an optically active epoxy iodide.
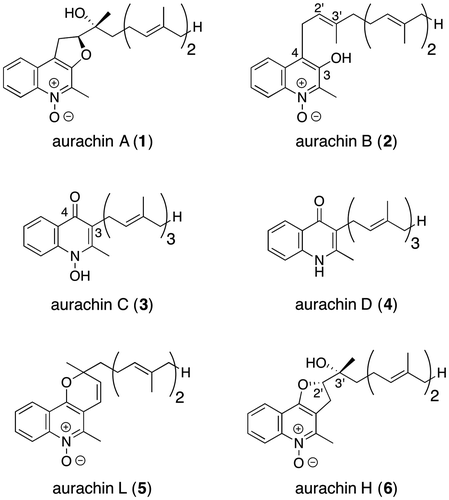
Aurachins are a family of quinoline alkaloids first isolated from the culture broth of Stigmatella sp. by Höfle et al. and later from Rhodococcus sp. by Kitagawa et al. as well as by Fiedler et al.Citation1–6) Aurachins A–D (1–4) are known to show various pharmacologically important biological properties such as antibacterial, antifungal, and antiplasmodial activities (Fig. ).Citation1, 4–6) In particular, aurachins A (1), C (3), and D (4) were found to be extremely potent inhibitors of the quinol oxidation sites of bacterial cytochromes bo and bd, and thus aurachins are regarded as promising lead compounds for therapeutic antibiotic agents.Citation7, 8) These intriguing biological profiles as well as the unique molecular architectures featuring a 4-quinolone or quinoline N-oxide nucleus prompted synthetic efforts toward this family of natural products, which culminated in the total synthesis of aurachin D (4) through the Conrad–Limpach cyclization by two research groups.Citation9, 10) We also succeeded in the total synthesis of aurachins C (3), D (4), and L (5) by utilizing reductive cyclization of δ-nitro ketone intermediates with zinc or iron as the key step.Citation11) All of the previous synthetic studies on aurachins have thus been directed toward those with an oxygen-functionality at C4 and a farnesyl chain at C3 (see 3–5) or their analogs.Citation8–13) However, synthesis of aurachin B (2) or its analogs with another substitution pattern, namely, an oxygen functionality at C3 and a farnesyl chain at C4, has never been reported so far, which probably means the difficulty to realize the aurachin B-type substitution pattern. As for the synthesis of aurachins A (1) and H (6), the absolute configurations of which are still unknown, or their analogs that are characterized by the presence of a dihydrofuran ring unit, only one report on the synthesis of analogs of aurachin H has been disclosed by Müller and Nay’s group.Citation9) In their synthesis, however, treatment of quinoline intermediates possessing a geranyl or farnesyl side chain with m-chloroperbenzoic acid to oxidize the quinoline nitrogen as well as to epoxidize the Δ2’ double bond on the side chains was accompanied by unfavorable overepoxidation of a distal double bond,Citation9) which means that the oxidative approach to the quinoline N-oxide nucleus is not suitable not only for the synthesis of 6 but also for that of 1 and 2. As part of our ongoing efforts toward the total synthesis of aurachins,Citation11) we describe herein the first total synthesis of aurachin B (2) and (2’S,3’R)-aurachin H (6) exploiting our reductive approach to the quinoline N-oxide structural motif.
Fig. 1. Structures of aurachins A–D (1–4), L (5) and H (6).
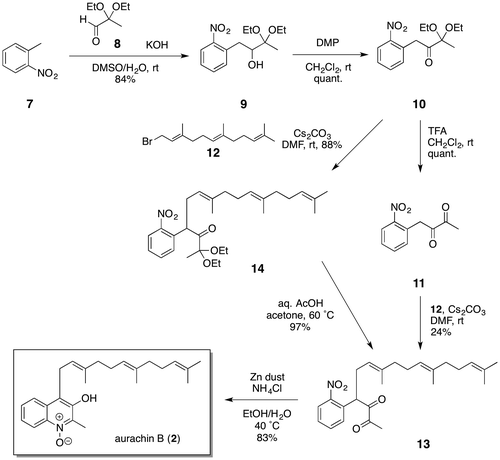
As shown in Scheme , our synthesis of aurachin B (2) began with the nucleophilic addition of a benzylic anion generated from o-nitrotoluene (7) to known aldehyde 8, which in turn was readily prepared in two steps from ethyl pyruvate via diethyl acetal formation and DIBAL reduction.Citation10, 14) Although the nucleophilic addition was low-yielding (31%) when sodium hydride was used as the base in DMF, treatment of 7 with potassium hydroxide and 8 in DMSO/H2O significantly improved the process, giving 9 in 84% yield. The oxidation of the alcohol 9 with DMP followed by removal of the acetal protecting group with TFA proceeded quantitatively to afford α-diketone 11. The alkylation of 11 with farnesyl bromide (12) and cesium carbonate, however, gave a complex mixture, from which desired product 13 was isolated in only 24% yield; this may be ascribable to the self-aldol reaction of the α-diketone 11. We therefore changed the order of reactions and conducted, at first, the alkylation of the α,α-diethoxyketone 10 with 12 under the same conditions as used for 11, furnishing 14 in a good yield of 88%. Subjection of 14 to acidic hydrolysis conditions then afforded the desired diketone 13 in 97% yield. Finally, reductive cyclization of 13 using zinc dust at 40 °C gave aurachin B (2) in 83% yield, the 1H and 13C NMR spectra of which showed good agreement with those reported for natural aurachin B.
Scheme 1. Synthesis of aurachin B (2).
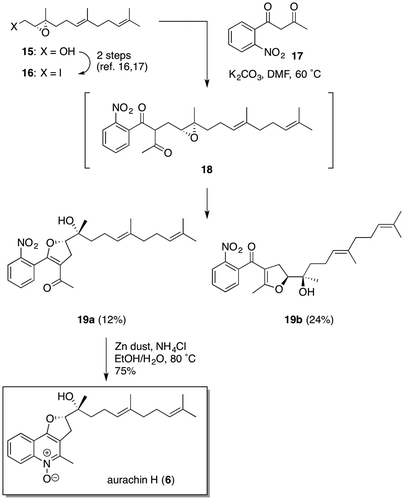
Having completed the efficient total synthesis of aurachin B (2), we then turned our attention to the synthesis of aurachin H (6) (Scheme ). Based on a biosynthetic pathway proposed by Höfle et al. that the dihydrofuran moiety of 6 would be constructed via the intramolecular epoxide-opening reaction of the corresponding N-hydroxy quinolone precursor bearing an epoxide ring in the side chain,Citation2) we envisaged a three-stepwise sequence for the synthesis of 6 that comprises: (1) alkylation of known β-diketone 17 with epoxy iodide 16Footnote1,Citation15); (2) reduction of the resulting product 18 with zinc generating the N-hydroxy quinolone ring;Citation11) and (3) intramolecular dihydrofuran ring formation. Contrary to the initial plan, treatment of 17 with 16, which was obtained in 86% enantiomeric excess by a known procedure,Citation16, 17) in DMF in the presence of K2CO3 afforded 19a and its isomer 19b in isolated yields of 12 and 24%, respectively, which means that the alkylation product 18 directly cyclized into the two isomers via the respective enolate forms. All attempts to improve the yield and selectivity of 19a by conducting the reaction under various conditions (KOt-Bu/t-BuOH at rt, NaH/HMPA/THF at reflux, Cs2CO3/DMF at 60 °C, K2CO3/DMF at 100 °C, etc.) were unsuccessful. Finally, the minor product 19a was reduced under the same conditions as used for 13 to furnish aurachin H (6) in 75% yield [[α]20D + 14 (c = 0.30, CHCl3)]. The 1H NMR spectrum of 6 exhibited good agreement with that of natural aurachin H. Although the efficiency of the transformation of 16 into 19a was far from satisfactory, it would be worth noting that the two processes (alkylation and dihydrofuran ring formation) in the initially planned three-step sequence took place in one pot.
Scheme 2. Synthesis of aurachin H (6).
In conclusion, the synthesis of aurachin B (2) was accomplished in 60% overall yield from o-nitrotoluene (7) by the concise five-step sequence featuring the alkylation of α,α-diethoxyketone 10 with farnesyl bromide 12 to install the C4 substituent and the reductive cyclization of 13 to construct the quinoline N-oxide nucleus. The efficient approach to aurachin B (2) would surely be applicable to the synthesis of other aurachin B-type natural products and their analogs as well. The enantioselective synthesis of aurachin H (6) was also achieved for the first time in only two steps from optically active epoxy iodide 16, which would help determine the absolute configuration of aurachin H, although the specific rotation value of the natural product is not known at present.
Spectral data of aurachins B (2) & H (6). Aurachin B (2): mp 186–187 °C; IR νmax: 2924 (s), 1377 (w), 1121 (s), 1095 (m); 1H NMR (400 MHz, CDCl3) δ: 1.58 (3H, s), 1.60 (3H, s), 1.66 (3H, s), 1.90 (3H, s), 1.95–1.99 (2H, m), 2.02–2.09 (2H, m), 2.14–2.18 (4H, m), 2.70 (3H, s), 3.76 (2H, d, J = 7.2 Hz), 5.04–5.10 (2H, m), 5.30 (1H, t, J = 6.8 Hz), 5.98 (1H, s), 7.56–7.62 (2H, m), 7.89–7.92 (1H, m), 8.77–8.79 (1H, m); 13C NMR (100 MHz, CDCl3) δ: 12.6, 16.2, 16.7, 17.8, 24.7, 25.8, 26.5, 26.8, 39.7, 39.8, 119.9, 120.8, 123.2, 123.7, 123.9, 124.4, 127.5, 127.6, 128.0, 131.4, 135.6, 137.0, 138.7, 142.3, 147.5; HRMS (ESI) m/z: calcd. for C25H33NO2Na ([M + Na]+) 402.2409, found 402.2415.
Aurachin H (6): [α]20D = + 14 (c = 0.30, CHCl3); mp 190–191 °C; IR νmax: 3236 (m), 2926 (s), 1227 (m), 1071 (m); 1H NMR (400 MHz, CDCl3) δ: 1.40 (3H, s), 1.56–1.67 (1H, m), 1.60 (3H, s), 1.65 (3H, s), 1.67 (3H, s), 1.71–1.82 (1H, m), 2.00 (2H, t, J = 7.2 Hz), 2.05–2.26 (4H, m), 2.65 (3H, s), 3.29 (1H, dd, J = 15.6, 9.6 Hz), 3.45 (1H, dd, J = 15.6, 8.8 Hz), 4.98 (1H, dd, J = 9.6, 8.8 Hz), 5.09 (1H, t, J = 7.2 Hz), 5.16 (1H, t, J = 7.2 Hz), 7.54 (1H, dd, J = 7.6, 7.2 Hz), 7.71 (1H, dd, J = 8.0, 7.2 Hz), 7.91 (1H, d, J = 8.0 Hz), 8.71 (1H, d, J = 7.6 Hz); 13C NMR (100 MHz, CDCl3) δ: 16.2, 16.2, 17.9, 22.0, 22.8, 25.8, 26.7, 29.7, 37.6, 39.8, 73.7, 91.3, 116.0, 117.3, 120.4, 122.0, 123.8, 124.3, 127.1, 130.5, 131.7, 136.3, 141.1, 144.4, 153.6; HRMS (ESI) m/z: calcd. for C25H33NO3Na ([M + Na]+) 418.2353, found 418.2360.
Authors’ contribution
M.E. and S.K. designed the synthetic route and wrote the manuscript. K.T. conducted the synthetic experiments with the aid of M.E.
Funding
This work was financially supported by JSPS KAKENHI [grant number 16K07708].
Supplemental materials
Spectral data and experimental procedures for 2, 6, 9–11, 13–14, 19a, and 19b are available at http://10.1080/09168451.2017.1325711.
Disclosure statement
No potential conflict of interest was reported by the authors.
TBBB_1325711_Supplemental_Material.docx
Download MS Word (1.9 MB)Acknowledgments
We thank Prof Yamashita, Dr Cho and Ms Taguchi (Tohoku University) for their help in measuring NMR and MS spectra.
After our manuscript was sent to the journal, a synthesis of aurachins A and B was published (See, Ref 19).
Notes
Abbreviations: DIBAL, diisobutylaluminium hydride; DMF, N,N-dimethylformamide; DMSO, dimethyl sulfoxide; DMP, Dess-Martin Periodinane; TFA, trifluoroacetic acid; rt, room temperature; HMPA, hexamethylphosphoric triamide; THF, tetrahydrofuran
1. The enantiomeric excess of 15 was determined by comparing its specific rotation value with the reported datumCitation16) and confirmed by a 1H NMR analysis of the corresponding camphanate derivative.Citation18)
References
- Kunze B, Höfle G, Reichenbach H. The aurachins, new quinoline antibiotics from myxobacteria: production, physico-chemical and biological properties. J Antibiot. 1987;40:258–265.10.7164/antibiotics.40.258
- Höfle G, Kunze B. Biosynthesis of aurachins A−L in Stigmatella aurantiaca: a feeding study. J Nat Prod. 2008;71:1843–1849.10.1021/np8003084
- Höfle G, Irschik H. Isolation and biosynthesis of aurachin P and 5-nitroresorcinol from Stigmatella erecta. J Nat Prod. 2008;71:1946–1948.10.1021/np800325z
- Höfle G, Böhlendorf B, Fecker T, et al. Semisynthesis and antiplasmodial activity of the quinoline alkaloid aurachin E. J Nat Prod. 2008;71:1967–1969.10.1021/np8004612
- Kitagawa W, Tamura T. A quinoline antibiotic from Rhodococcus erythropolis JCM 6824. J Antibiot. 2008;61:680–682.10.1038/ja.2008.96
- Nachtigall J, Schneider K, Nicholson G, et al. Two new aurachins from Rhodococcus sp. Acta 2259. J Antibiot. 2010;63:567–569.10.1038/ja.2010.79
- Meunier B, Madgwick SA, Reil E, et al. New inhibitors of the quinol oxidation sites of bacterial cytochromes bo and bd. Biochemistry. 1995;34:1076–1083.10.1021/bi00003a044
- Miyoshi H, Takegami K, Sakamoto K, et al. Characterization of the ubiquinol oxidation sites in cytochromes bo and bd from Escherichia coli using aurachin C analogues. J Biochem. 1999;125:138–142.10.1093/oxfordjournals.jbchem.a022250
- Li XW, Herrmann J, Zang Y, et al. Synthesis and biological activities of the respiratory chain inhibitor aurachin D and new ring versus chain analogues. Beilstein J Org Chem. 2013;9:1551–1558.10.3762/bjoc.9.176
- Dejon L, Speicher A. Synthesis of aurachin D and isoprenoid analogues from the myxobacterium Stigmatella aurantiaca. Tetrahedron Lett. 2013;54:6700–6702.10.1016/j.tetlet.2013.09.085
- Enomoto M, Kitagawa W, Yasutake Y, et al. Total synthesis of aurachins C, D, and L, and a structurally simplified analog of aurachin C. Biosci Biotechnol Biochem. 2014;78:1324–1327.10.1080/09168451.2014.918494
- Reil E, Soll M, Masson K, et al. Synthesis of quinolones and acridones and their inhibitory activity in NADH-dehydrogenases and cytochrome b/cl-complexes. Biochem Soc Trans. 1994;22:62S.10.1042/bst022062s
- Rich PR. Electron transfer reactions between quinols and quinones in aqueous and aprotic media. Biochim Biophys Acta. 1981;637:28–33.10.1016/0005-2728(81)90206-1
- Trost BM, Fettes A, Shireman BT. Direct catalytic asymmetric aldol additions of methyl ynones. Spontaneous reversal in the sense of enantioinduction. J Am Chem Soc. 2004;126:2660–2661.10.1021/ja038666r
- Brie M, Silberg LA. Derivatives of pyrazoles. I. Synthesis and IR-spectra of some new 3-methyl-5-aryl-pyrazoles. Revue Roumaine de Chimie [Roumanian J Chem.]. 1989;34:733–737.
- Marshall JA, Hann RK. A cascade cyclization route to adjacent bistetrahydrofurans from chiral triepoxyfarnesyl bromides. J Org Chem. 2008;73:6753–6757.10.1021/jo801188w
- Kigoshi H, Ojika M, Shizuri Y, et al. Isolation of (10R,11R)-(+)-squalene-10,11-epoxide from the red alga laurencia okamurai and its enantioselective synthesis. Tetrahedron. 1986;42:3789–3792.10.1016/S0040-4020(01)87533-5
- Koohang A, Bailey JL, Coates RM, et al. Enantioselective inhibition of squalene synthase by aziridine analogues of presqualene diphosphate. J Org Chem. 2010;75:4769–4777.10.1021/jo100718z
- Hattori H, Yokoshoma S, Fukuyama T. Angew. Chemie. Int. Ed. DOI:10.1002/anie.201702204