
Abstract
CbnR, a LysR-type transcriptional regulator from Cupriavidus necator NH9, activates the transcription of chlorocatechol-degradative enzymes. To activate the transcription, CbnR needs to bind not only to the cbnA promoter but also to the inducer. In this study, the transcriptional activity and DNA-binding activity of twenty-five mutants of CbnR were analyzed. Of the 17 mutants of the DNA-binding domain, 11 mutants lost their ability to activate transcription. While most mutants without transcriptional activation did not show DNA-binding activity, Asn17Ala, Gln29Ala, and Pro30Ala retained DNA-binding activity, suggesting that transcriptional activation by CbnR requires more than its binding to promoter DNA. Of the 8 mutants of the regulatory domain, 6 mutants changed their responses to the inducer, when compared with wild-type CbnR. Interestingly, Arg199Ala and Val246Ala induced constitutive expression of the cbnA promoter without the inducer, suggesting that these mutations brought about a conformational change mimicking that induced by the inducer molecule.
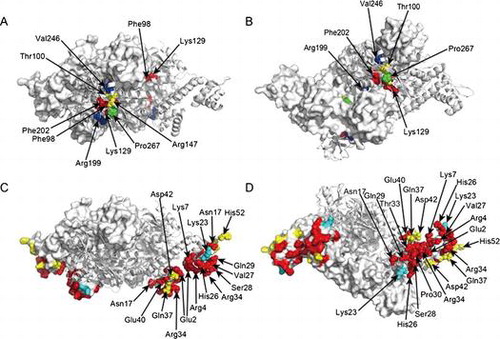
Mutational analysis of a LysR-type transcriptional regulator, CbnR, revealed critical amino acids in regulatory domain (A and B) and DNA-binding domain (C and D).
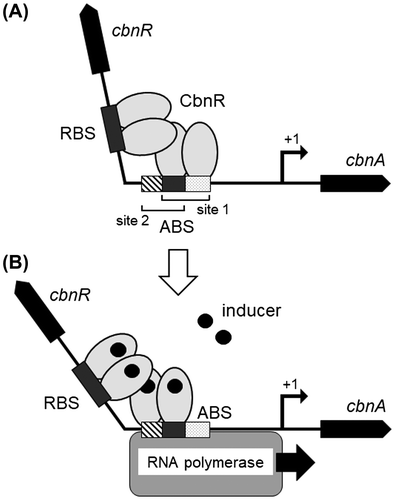
Cupriavidus necator (formerly known as Alcaligenes eutrophus or Ralstonia eutropha) NH9 can degrade 3-chlorobenzoate (3-CB) as the sole source of carbon and energy.Citation1) In C. necator NH9, 3-CB is converted into 3-chlorocatechol, which is further degraded by enzymes encoded in the cbn operon with cbnR-ABCD genes.Citation2,3) Their transcription is regulated by a LysR-type transcriptional regulator (LTTR),Citation4,5) CbnR. CbnR, which is composed of 294 amino acid residues and forms a tetramer,Citation6) specifically binds to recognition-binding and activation-binding sites (RBS and ABS) in the cbnA promoter.Citation2) Electrophoretic mobility shift assay (EMSA) and DNase I protection assay demonstrated that RBS and ABS in the cbnA promoter extend approximately 60 bp with a site of hypersensitivity to DNase I between RBS and ABS. Circular permutation gel shift assay suggested that CbnR bends the DNA by approximately 78°. For the transcriptional activation, CbnR on the promoter needs to bind to an inducer molecule, 2-chloro-cis, cis-muconate or cis, cis-muconate. Upon inducer binding, CbnR is likely to change its conformation and then to alter its binding site in ABS from site 1 to site 2, resulting in a relaxation of the bending angle to approximately 54° (Fig. ).Citation2) These changes seem to allow interactions between LTTR and RNA polymerase to activate the transcription.Citation7,8) Since the transcriptional activation process by CbnR is a typical one for LTTRs, analysis of the transcriptional activation mechanism of CbnR would lead to a general understanding of the transcriptional regulation mechanism of LTTR family proteins.
Fig. 1. A transcriptional activation model by CbnR. A model of the interaction between the CbnR tetramer and the cbnA promoter DNA before (A) and after (B) the inducer binding. (A) CbnR binds to RBS (−76 to −49) and ABS (−44 to −19) in the cbnA promoter without an inducer. (B) Upon inducer binding, CbnR seems to undergo a conformational change, altering the binding site in ABS from site 1 to site 2, and relaxing DNA bending, leading to the recruitment of RNA polymerase to the promoter region.
Many efforts have been made to understand the molecular mechanism of transcriptional regulation by LTTR.Citation4,5,8–16) In particular, tertiary structural information of LTTRs was found to be critical to understanding the molecular mechanism underlying the transcriptional regulation by LTTR. The first structural information of LTTR was the crystal structure of the regulatory domain (RD) of CysB.Citation17) The RD contains a binding site of the inducer molecule. While tertiary structural information of the tetrameric LTTR had long been unknown due to the difficulties in purification of recombinant LTTRs, we succeeded in purifying a large amount of CbnR and determined the crystal structure of CbnR at 2.2 Å resolution; this is the first crystal structure of the full-length LTTR.Citation6,18) The crystal structure of CbnR revealed that: (i) CbnR forms a tetramer in the crystal, (ii) the tetrameric CbnR contains two different conformers of the CbnR subunit, and (iii) four DNA-binding domains (DBDs) of the tetrameric CbnR are arranged linearly at the V-shaped bottom of the tetramer. A docking study between the CbnR tetramer and B-form DNA showed that the four DBDs in tetrameric CbnR can interact with 60 bp DNA and the interaction bends the DNA due to the V-shaped arrangement of the four DBDs. In addition to the overall structural information, conformational changes upon inducer binding have been demonstrated by structural studies of BenM, DntR, OxyR, and TsaR.Citation10,11,15,16) Based on these earlier studies, a model of transcriptional activation by CbnR has been proposed (Fig. ).Citation2,6) Two DBDs in the CbnR tetramer are thought to bind to RBS. RBS of the cbnA promoter is located at −76 bp to −49 bp of the transcriptional start site of the cbnA gene.Citation2) The two DBDs seem to recognize inverted repeat sequences of about 4 bps in the canonical T-N11-A motif of RBS. The other two DBDs of the CbnR tetramer seem to bind to the ABS, which is located at −44 bp to −19 bp of the cbnA gene.Citation2) The binding of an inducer to RD would evoke a conformational change of RD, leading to a quaternary structural change of the tetrameric CbnR. As a result, the DBD-binding site is shifted from site 1 to site 2 in the ABS to activate transcription of the regulated gene.Citation2) Recently, the crystal structure of the BenM DBD in complex with its target DNA revealed interactions between DBD and DNA.Citation14) While these crystal structures have provided much information about LTTR, the molecular mechanisms of transcriptional activation of LTTR are still elusive. In this study, biochemical analysis of CbnR mutant proteins was performed based on the earlier obtained tertiary structural information. The results revealed important amino acid residues in DBD and RD of CbnR and suggested mechanistic roles of the amino acid residues in the transcriptional activation by CbnR.
Materials and methods
Bacterial strains, plasmids, and media
The bacterial strains and plasmids used in this study are listed in Table . Escherichia coli DH5α was grown in Luria–Bertani (LB) broth at 37 °C.Citation19) Pseudomonas putida PRS4020Citation20) was grown on Pseudomonas Isolation Agar (Difco), TYP medium (16 g of tryptone, 16 g of yeast extract, 5 g of NaCl, 2 g of K2HPO4 per liter), or basal synthetic mediumCitation21) supplemented with 10 mM glucose, 5 mM benzoate (BA), or 5 mM 3-CB at 30 °C as described previously.Citation2,22) Antibiotics were added at the following concentrations for plasmid selection: 50 μg/mL of ampicillin (Ap) for E. coli, 1000 μg/mL of carbenicillin (Cbpc) (or viccillin (Vic)) for P. putida PRS4020. Ten μg/mL gentamycin (Gm) was used for maintenance of P. putida PRS4020. Isopropyl-β-D(-)-thiogalactopyranoside (IPTG) was used for protein expression at a final concentration of 1 mM.
Table 1. Bacterial strains and plasmids used in this study.
DNA manipulation
Standard methods were used to manipulate plasmids and DNA fragments.Citation19) QIAprep Spin Miniprep Kit and Plasmid Midi Kit (QIAGEN) were used for plasmid extraction. Restriction endonucleases were purchased from Nippon Gene, TaKaRa, or TOYOBO. Polymerase chain reaction (PCR) amplification was performed using KOD DNA polymerase (TOYOBO). A QIAquick Gel Extraction Kit or QIAEX II Gel Extraction Kit (QIAGEN) was used for DNA extraction from the agarose gel after electrophoresis. In construction of plasmids, vector and insert DNA were ligated using a DNA Ligation Kit Version 2 or a DNA Ligation Kit Mighty Mix (TaKaRa). All enzymes were used according to the manufacturer’s recommendations. Plasmids pQcbnR-AB’ and pQcbnRmutant-AB’ were transformed into P. putida PRS4020 by electroporation as described previously.Citation2,22) Plasmids pTrccbnR and pTrccbnRmutant were transformed into E. coli DH5α by the heat shock method.Citation23)
Site-directed mutagenesis and sequencing
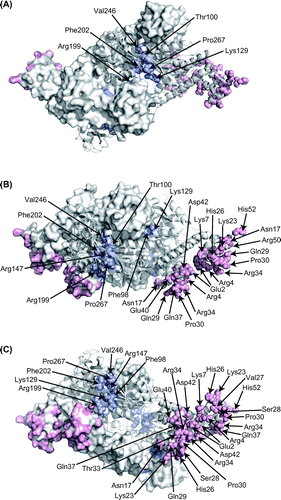
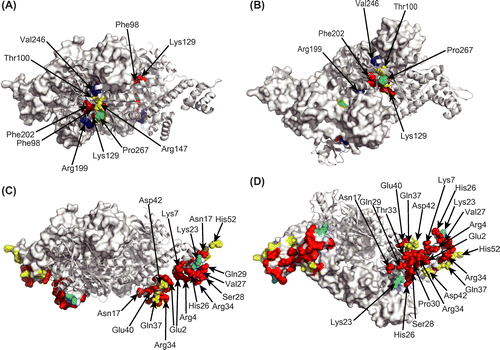
CbnR mutants with an introduced point mutation were constructed by site-directed mutagenesis based on the overlap extension method using PCR reaction with mutagenic primers.Citation24) Two sets of primers were used for site-directed mutagenesis. The first one consisted of vector sequence-specific primers located outside of the CbnRHis coding region, and the second one was forward and reverse mutagenic primers encompassing the site of mutation in the CbnRHis gene. The vector sequence-specific primers were 5′-TGTAAAACGACGGCCAGT-3′ (-21M13) and 5′-CAGGAAACAGCTATGACC-3′ (M13Rev). The nucleotide sequences of the (forward and reverse) mutagenic primers are complimentary to each other and their nucleotide sequences match those of the mutation region of the cbnR gene except the substituted codons. The nucleotide sequences of the mutant cbnR genes cloned in pBluescript SKNco(-) were confirmed. The -21M13, M13Rev, and cbnR_SmaIRev (5′-AGGTCTTGCCGAACTTCC-3′) primers were used for the sequence analysis. The primers used for the site-directed mutagenesis in this study are listed in Supplementary Table 1. In this study, we did not check the conformation of mutant proteins using physicochemical methods such as circular dichroism (CD) spectra analysis. Since most of the mutations were introduced on surface residues (Fig. ), it is unlikely that the proteins lost their native structures due to the mutations. However, we cannot exclude the possibility that some mutant proteins lack the native tertiary structure and lose transcriptional activity.
Fig. 2. Amino acid residues for mutations. (A) Top view, (B) side view, (C) and bottom view of the tetrameric CbnR. One pair of compact- and extended-form subunits is shown in the cartoon model, and the other pair is shown in surface representations. Light blue and pink residues are mutated residues in RD and DBD, respectively. Mutated residues on the cartoon models are shown with sphere models.
Construction of plasmids
Plasmids used in this study were constructed as shown in Supplementary Fig. . Briefly, a NcoI-XbaI fragment or a NcoI-SmaI fragment of a mutant cbnR gene was used to replace the corresponding fragment of the wild-type cbnR gene in pBSKNco(-)cbnRncoHis, yielding pBLcbnRmutant. Protein expression vectors were prepared based on plasmid pTrc99A.Citation25) The NcoI-XbaI fragment of the wild-type (or a mutant) cbnR gene was transferred from pBSKNco(-)cbnRncoHis (or pBLcbnRmutant) to pTrc99A. Vectors for the reporter assay were constructed as follows: first, pBSKNco(-)cbnAB’ was constructed by inserting a 1,715 bp NcoI-HindIII fragment containing a complete intergenic region of cbnR and cbnA with the cbnA promoter, the full length of the cbnA gene, and a part of the cbnB gene into NcoI-HindIII sites of pBluescript SKNco(-). A NcoI-XbaI fragment containing the wild-type (or a mutant) cbnR gene was inserted into the NcoI-XbaI sites of pBSKNco(-)cbnAB′ in divergent orientation to the cbnA promoter and the cbnAB′ genes, yielding pBLcbnR-AB′ (or pBLcbnRmutant-AB′). Then, the 2,620 bp XbaI-HindIII fragment containing the cbnR (or its mutant) gene and the cbnAB′ genes from pBLcbnRmutant-AB′ was inserted into the XbaI-HindIII sites of the pQF50 vector,Citation26) yielding reporter plasmid pQcbnR-AB’ (or pQcbnRmutant-AB′).
β-galactosidase assay
The transcriptional activity of the cbnA promoter by the wild-type CbnR or its mutants was measured quantitatively by β-galactosidase assay as described previously.Citation2,27) P. putida PRS4020 strains carrying pQcbnR-AB′ or pQcbnRmutant-AB’ were grown at 30 °C in 5 mL of LB medium with Cbpc (or Vic) and Gm overnight. One hundred microliters of the preculture medium were inoculated into 10 mL of basal synthetic medium supplemented with 10 mM glucose, Cbpc (or Vic), and Gm, with or without 5 mM BA or 3-CB, and incubated at 30 °C for a fixed time between 16 and 19 h for a series of assays using the wild-type CbnR as a control for normalization, as described below. Our data showed that β-galactosidase activities increased only slightly from 16 to 19 h of incubation time, probably due to saturation. After incubation, the cells were harvested and washed with 5 mL of LacZ buffer (8.5 g Na2HPO4, 6.22 g NaH2PO4·2H2O, 0.75 g KCl, 0.246 g MgSO4·7H2O, 2.7 mL β-mercaptoethanol per liter).Citation27) The cells were resuspended in 1 mL of LacZ buffer. The crude extract of the bacterial cells was prepared either by chemical lysis (for all Gln29 mutants except Gln29Ala and all RD mutants) or by sonication (the other mutants). In chemical lysis, 400–800 μL of harvested cells were washed with the LacZ buffer and the resuspended cells were lysed with 200 μL of CelLytic B Plus Working solution (Sigma–Aldrich) for 8 min. In sonication, 1 mL of cell suspension was disrupted by sonication using US-150E (Nissei) (tip 3 mm; output level 6.6; two times of 20 s sonication with an interval of 20 s on ice) or using VP-15s (TAITEC) (tip 3 mm; output level 4; three cycles of 20 s sonication and 20 s interval on ice). After centrifugation, the supernatant was used as a crude extract for the β-galactosidase activity assay. Protein concentrations were measured using a Bradford protein assay kit (Bio-Rad) with bovine serum albumin as the standard. One milliliter of the reaction mixture contained 100 or 30 μg of the crude extract in the LacZ buffer. The reaction mixture (1 mL) was pre-incubated at 28 °C, and 200 μL of 4 mg/mL of o-nitrophenyl-β-D-galactopyranoside (ONPG) was added to start the incubation for β-galactosidase assay at 28 °C. When the color of the reaction solution reached an appropriate range of yellow, the reaction was stopped by adding 500 μL of Na2CO3 (1 M). The absorbance of o-nitrophenol at 420 nm was measured. β-galactosidase activity was calculated in Miller units (nmol/min/mg protein).Citation27) Measurements of the β-galactosidase activity were performed in triplicate for one crude extract. The crude extract was also prepared in triplicate from three independent cultures. Thus, nine repetitions for each biological section were derived from the combination of plasmid construct and culture medium. It should be noted that the β-galactosidase activities of the wild-type CbnR were comparable between the sonicated and chemical lysis samples. A reporter assay of mutant CbnR was conducted together with that of wild-type CbnR. The results of mutant CbnR were expressed as relative values against the value of Miller units obtained with the wild-type CbnR in the presence of BA as 100%.
Purification of CbnRHis and its mutants
CbnRHis or its mutants was overexpressed and purified from E. coli DH5α harboring pTrccbnR or pTrccbnRmutant. E. coli cells harboring the plasmids were grown at 37 °C in 5 or 10 mL of LB medium with Ap overnight. The preculture medium was diluted to 50- or 100-fold in 160 to 400 mL of LB medium with Ap and incubated at 37 °C. When the O.D.600 reached 0.4–0.6, expression of CbnRHis or its mutants was induced by adding IPTG at 1 mM, followed by incubation for an additional 2.5 h under the same conditions. After incubation, the cells were harvested by centrifugation for 8 min at 5,000 rpm and 4 °C. Purification of CbnRHis or its mutants was performed using His Bind Quick 900 Cartridges (Novagen) or a Ni-NTA Fast Start Kit (QIAGEN). In the case of using the former kit, the cells harvested from 160-mL culture medium were disrupted by sonication on ice using US-150E (Nissei) with an output level of 6.6, six cycles of 20 s sonication, and 20 s intervals. After the centrifugation, the resultant supernatant was subjected to purification using His Bind Quick 900 Cartridges (Novagen) following the protocol supplied by the manufacturer. The proteins bound to the column were washed with 4 mL of a washing buffer (100 mM imidazole, 300 mM NaCl, 50 mM sodium phosphate buffer, pH 8.0) and then eluted with 4 mL of an elution buffer (400 mM imidazole, 300 mM NaCl, 50 mM sodium phosphate buffer, pH 8.0). The purified CbnRHis or its mutants was concentrated to about 1 mg/mL using CENTRICON YM-10 Centrifugal Filter Devices (MW cut-off 10,000) (Millipore) at 3,000 rpm. In the case of Ni-NTA Fast Start Kit, the cells were harvested from 400-mL culture medium and lysed with lysis buffer supplied by the kit. The supernatant after centrifugation was purified using manufacturer’s protocol with a modification. The eluate obtained by the elution buffer supplied by the kit still contained proteins other than CbnRHis or its mutants abundantly, and thus was discarded (Supplementary Fig. 2, panel B, lane E). Then, CbnRHis or its mutants was further eluted from the column using 1 mL of the elution buffer containing 400 mM imidazole (see above) (Supplementary Fig. 2, panel B, lane P). The purity of the eluted fraction was analyzed by separation on 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) (Supplementary Fig. 2). While most of the mutant proteins of CbnR were obtained by this method, we could not obtain the mutant proteins of Glu2Ala, His26Ala, Gln29Arg, and Gln29Trp. These mutant proteins were not expressed in E. coli.
Electrophoretic mobility shift assay (EMSA)
The DNA-binding activity of CbnRHis and its mutants was analyzed by EMSA. Twenty microliters of reaction mixture was prepared on ice. The 20 μL reaction mixture consisted of 20 mM HEPES (pH 7.9), 1 mM EDTA, 1 mM dithiothreitol, 30 mM KCl, 10 mM (NH4)2SO4, 1% Tween 20, 15% glycerol, 1.5 μg of BSA, 0.5 μg of heparin, 195 or 390 nM of purified CbnRHis protein (or its mutant), and 10.2 or 20.3 pM of promoter DNA fragment labeled with DIG. A DNA fragment of the 149 bp cbnR-cbnA intergenic region including the cbnA promoter was amplified by the PCR reaction using pBLcbnR-AB’ as a template. The primers used in the PCR reaction were cbnDigGF1 (5′-GGCGCGCTTTCCGAGTTGGTGATGTGC-3′) and cbnDigGF2 (5′-CCCGGTCTCCTTTGTCGGTTTGCC-3′). The amplified DNA fragment was labeled with digoxigenin (DIG) using a DIG Oligonucleotide 3′-End Labeling Kit, 2nd Generation (Roche). The binding reaction of CbnRHis and DNA was started by the addition of CbnRHis (or its mutant) to the reaction mixture and incubation was conducted for 20 min at room temperature. After 20 min of incubation, the reaction mixtures were loaded on a 5% native polyacrylamide gel prepared with 0.5 × Tris-Borate-EDTA (TBE) buffer and electrophoresis was performed in 0.5 × TBE buffer at 80 V for 90 min. The separated DNA was transferred from the polyacrylamide gel to a Hybond-N + nylon membrane (GE Healthcare) by electroblotting using Mini Trans-Blot Cell (Bio-Rad) at 400 mA for 30 min. The nylon membrane with the transferred DNA was incubated with anti-digoxigenin alkaline phosphatase (Roche) and the transferred DNA fragments were detected by the chemiluminescence method. The results of EMSA were evaluated by visual inspection and are summarized in Table . The symbols “−,” “+,” “++,” and “+++” indicate a band without any shift, a band with less shift than the free promoter DNA band, a band with greater shift than the free promoter DNA band, and a completely shifted band, respectively.
Table 2. Transcriptional activation and DNA-binding activities of CbnR mutants.
Comparison of binding activities of the wild-type CbnR and Gln29 mutants
To compare the binding activities of the wild-type CbnR and Gln29 mutants, a series of EMSA experiments were performed by changing the concentration of wild-type/mutant CbnR from 39.0 to 390 nM (Supplementary Table 2). The reactions contained 20.3 pM of promoter DNA. The chemiluminescence intensities of the EMSA bands were quantified using a CS Analyzer (ATTO). Then, the ratio of intensities of shifted and non-shifted bands was calculated for each experiment, and the averaged ratios of shifted bands/(shifted bands plus non-shifted bands) from triplicate experiments were obtained. Protein concentrations were measured using a Bradford protein assay kit (Bio-Rad) with bovine serum albumin as a standard.
Results
Selection of amino acid residues for mutation
To analyze the functional roles of amino acid residues in DBD and RD, amino acid residues for mutations were selected based on the crystal structures and amino acid sequence alignment with CbnR, CatR, ClcR, CysB, OxyR, OccR, and NahR (Fig. ).Citation6,14) In this study, Glu2, Arg4, Lys7, Asn17, Lys23, His26, Val27, Ser28, Gln29, Pro30, Thr33, Arg34, Gln37, Glu40, Asp42, Arg50, and His52 were selected for mutations of DBD. Since residues Arg4, Gln29, Pro30, Glu40, and Arg50 were highly conserved among the seven related LTTRsCitation6) and exposed to the solvent (Fig. (B and C)), they are likely to interact with promoter DNA. Indeed, all the corresponding residues in BenM except Glu40 have been shown to interact with DNA in the crystal structure of the complex of BenM DBD and DNA (Supplementary Fig. 3).Citation14) Asn17, Ser28, Thr33, Arg34, Gln37, and His52 were also selected because the corresponding residues in BenM interact with DNA (Supplementary Fig. 3).Citation14) In addition, residues located around the above residues, Glu2, Lys7, Lys23, His26, Val27, and Asp42, were also selected since they seemed to affect the interaction between CbnR and DNA.
As for RD, Phe98, Thr100, Lys129, Arg147, Arg199, Phe202, Val246, and Pro267, which are located around the putative inducer-binding site, were selected for mutation (Fig. ). While the inducer-binding site of CbnR has not been experimentally determined, the crystal structure of BenM RD in complex with its inducer, cis, cis-muconate,Citation28) and a homology model derived from this crystal structure suggested that the selected residues are located around the bound inducer. Of the eight residues, Phe98, Thr100, Lys129, Arg147, and Phe202 may directly interact with the bound inducer molecule; the corresponding residues in BenM interact with its inducer molecule.Citation28) In addition, Arg199, Val246, and Pro267 were selected for mutation based on the results of the mutational analysis of TfdT, which showed 52.7% amino acid sequence identity with CbnR. Mutations of the corresponding residues in TfdT altered the response to its inducer.Citation29)
Mutational analysis of DBD
Initially, all selected amino acid residues in DBD were substituted with alanine. Transcriptional activity from the cbnA promoter was then analyzed by β-galactosidase assay. While cis, cis-muconate and 2-chloro-cis, cis-muconate were not added to the medium in the β-galactosidase assay, these inducer molecules were generated in the cells from BA and 3-CB contained in the medium.Citation2) The results of the assay showed that the mutants Glu2Ala, Arg4Ala, Lys7Ala, Asn17Ala, His26Ala, Val27Ala, Ser28Ala, Gln29Ala, Pro30Ala, Thr33Ala, and Arg34Ala exhibited no response to the inducer (Fig. (A)); no significant activations of transcriptional activities were observed with the inducer. Mutants Glu40Ala and Arg50Ala also showed no responses to the inducer, but the basal level of transcription was higher than in the former cases. While mutants Gln37Ala, Asp42Ala, and His52Ala demonstrated lower activation levels than the wild type, these mutants showed clear responses to the inducer. Interestingly, Lys23Ala showed an activation level by cis, cis-muconate which was higher than that of the wild type (Fig. (A)).
Fig. 3. β-galactosidase assay for DBD mutants. (A) Results for the seventeen DBD mutants with alanine substitutions. (B) Results for the other mutants of Lys23 and Gln29. White, gray, and black bars show the results from cultures with 10 mM glucose (no inducer in the medium), 10 mM glucose +5 mM BA, and 10 mM glucose +5 mM 3-CB, respectively. The values shown are relative to those for BA of wild-type CbnR. All assays were performed in triplicate cultures. Standard deviations are shown.
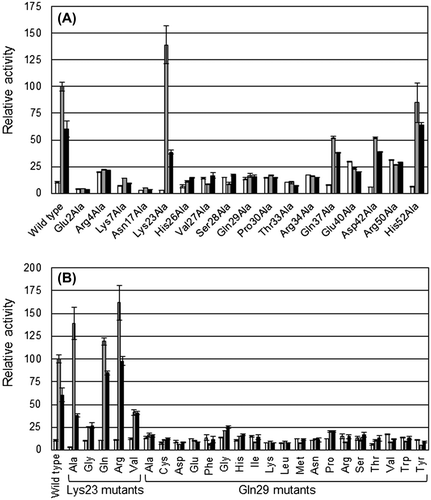
To investigate the mechanistic role of Lys23, additional mutants were prepared for Lys23. In addition, Gln29 was analyzed intensively because this amino acid residue is likely to interact with DNACitation14) and it is one of the most highly conserved residues in LTTRs.Citation6,14) The results of these mutational analyses revealed distinct characteristics of the two amino acid residues (Fig. (B)). Since all mutants of Gln29 lost their transcriptional activation activities, we concluded that Gln29 is essential for the transcriptional activation. On the other hand, the functional role of Lys23 seems to be not only a DNA-binding but also a regulatory one; mutations of Lys23 significantly altered the level of transcriptional activation. While Lys23Gly and Lys23Val could respond to the inducer, the level of transcriptional activation was substantially smaller than that of the wild type. Lys23Ala, Lys23Arg, and Lys23Gln, however, showed comparable or greater transcriptional activity than the wild type (Fig. (B)).
EMSA analysis of DBD mutants
To further investigate the characteristics of the mutations, we performed EMSA on DBD mutants. Intriguingly, there were no absolute correlations between the DNA-binding activity from EMSA and the transcriptional activation activity from the β-galactosidase assay (Table ).
First, we analyzed the mutants that showed no transcriptional activation: Arg4Ala, Lys7Ala, Asn17Ala, Val27Ala, Ser28Ala, Gln29Ala, Pro30Ala, Thr33Ala, Arg34Ala, Glu40Ala, and Arg50Ala. While these non-functional mutants were expected to lose their DNA-binding activity, some of them—namely, Asn17Ala, Gln29Ala, and Pro30Ala—retained their DNA-binding activity (Fig. (A–C), Table ). The mutants showing weak but significant transcriptional activation—Gln37Ala, Asp42Ala, and His52Ala—were expected to retain their DNA-binding activity. Indeed, these mutant proteins possessed DNA-binding activity. While the DNA-binding activity of Gln37Ala is very weak, a faint signal showing the DNA-binding was reproducibly obtained (Fig. (B), Table ).
Fig. 4. Binding assay of CbnR mutants of DBD to the cbnA promoter with EMSA. Panels A, B, and a part of C show the results for the alanine mutants. Panels D, E, F, and a part of C show the results of Lys23 and Gln29 mutants. The reaction contained 390 nM of purified CbnRHis and 10.2 pM (A) or 20.3 pM (B–F) of promoter DNA. N.P. means no CbnRHis protein. Bands indicated by C and F represent the band of complex of promoter DNA and CbnRHis (or mutant) and the band of free promoter DNA, respectively.
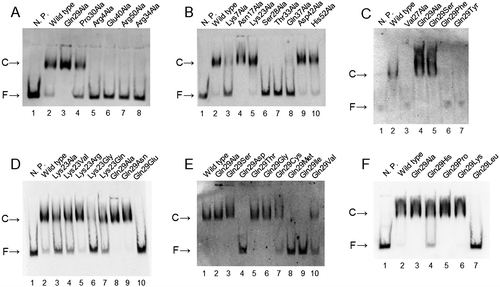
For further analysis of the relationship between DNA-binding activity and the transcriptional activation process by CbnR, we performed EMSA for a series of mutants of Lys23 and Gln29 (Fig. (C–F), Table ). Although none of the Gln29 mutants exhibited any activity to activate transcription in β-galactosidase assay, 10 mutants including Gln29Ala showed DNA-binding activity. Moreover, four mutants, Gln29Ala, Gln29Gly, Gln29Ser, and Gln29Thr, showed stronger affinity to the DNA than the wild-type CbnR (Supplementary Table 2). It is noteworthy that the side chains of the four substituted amino acids are smaller than that of Gln. These results suggest that Gln29 has functional roles beyond merely serving to bind to the promoter DNA. The EMSA for Lys23 mutants also showed that their DNA-binding activity was not simply correlated with their transcriptional activity (Fig. (D), Table ). For example, the Lys23Ala mutant showed strong activity for transcriptional activation, but its DNA-binding activity was relatively weak. Our data thus demonstrated that the DNA-binding activity measured in the present EMSA did not have a strong correlation with the transcriptional activation; the transcriptional activation process of CbnR cannot be simply divided into the two stages of binding to promoter and recruitment of a transcription enzyme.
Mutational analysis of RD
The eight amino acid residues selected in RD, Phe98, Thr100, Lys129, Arg147, Arg199, Phe202, Val246, and Pro267, were each substituted with alanine. Then, their transcriptional and cbnA promoter-binding activities were analyzed (Fig. ). Of the eight mutants, Phe98Ala, Lys129Ala, and Phe202Ala showed greatly impaired transcriptional activity by the inducer (Fig. (A)). It is of note that the corresponding residues in BenM directly interact with the inducer molecule,Citation28) suggesting that Phe98, Lys129, and Phe202 in CbnR interact with the inducer. The Thr100Ala mutant was the only case that showed higher transcriptional activation activity with 2-chloro-cis, cis-muconate than with cis, cis-muconate (Fig. (A)). Arg147Ala and Pro267Ala responded to the inducer in the same manner as the wild-type CbnR, suggesting that the two amino acid residues were not involved in inducer recognition. Interestingly, the Arg199Ala and Val246Ala mutants showed significant transcriptional activity even in the absence of the inducer; these two mutants are constitutively active mutants. The transcriptional activities in the absence of the inducer of the two mutants were approximately twofold of those of the wild-type CbnR with the inducer (Fig. (A)). The activities of the two mutants with the inducer were comparable to those of the same mutants without the inducer. In the tetrameric CbnR, there are four Arg199 residues. Two of them are located at the interface of RD and DBD (Fig. (A)), and the other two are located near the local twofold axis of the tetrameric CbnR (Fig. (B)). Mutation of the former Arg199 could change the relationship between RD and DBD, and mutation of the latter Arg199 could change the relative arrangement of the two RDs. Val246 is located near the interface of the two RD domains, which are related by a local twofold axis for RDs (Fig. ). The replacement of Val246 with Ala might change the interaction between two RDs. These facts suggest that the mutations of Arg199Ala and Val246Ala induce a structural change in tetrameric CbnR, leading to the constitutive active state. Tertiary structural analysis would be required to prove this hypothesis.
Fig. 5. β-galactosidase and DNA-binding assays for RD mutants. (A) β-galactosidase activity of cbnR-RD mutants. White, gray, and black bars show the results from cultures with 10 mM glucose (no inducer in the medium), 10 mM glucose + 5 mM BA, and 10 mM glucose + 5 mM 3-CB, respectively. The values shown are relative to the activity for Ben of wild-type CbnR. All assays were performed in triplicate cultures. Standard deviations are shown. (B) Results of EMSA for the Arg199Ala and Val246Ala mutants of CbnR. 195 nM of purified CbnRHis proteins and 20.3 pM of cbnA promoter DNA were used for the assay. N.P. means no proteins. Bands indicated by C and F represent the band of complex of promoter DNA and CbnRHis (or mutant) and the band of free promoter DNA, respectively.
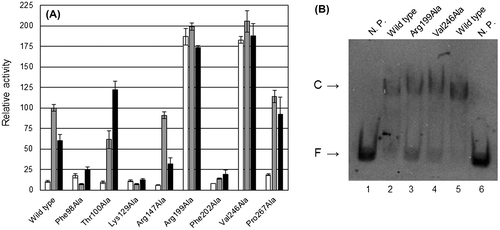
Fig. 6. Summary of the β-galactosidase assay in this study. All results of the β-galactosidase assay are summarized on the crystal structure of tetrameric CbnR. One pair of compact- and extended-form subunits is shown in the cartoon model, and the other pair is shown in surface representations. (A) and (B) are results of the β-galactosidase assay for the RD mutants. Red, yellow, and green residues are those showing less than 25% of the wild-type activity (Phe98Ala, Lys129Ala, and Phe202Ala), those showing 50–100% of the wild-type activity (Thr100Ala, and Arg147Ala), and those showing activity levels comparable to the wild type (Pro267Ala) by mutations, respectively (only activities with cis, cis-muconate are considered). Residues showing a constitutively active character by mutations are shown in blue (Arg199Ala and Val246Ala). (A) and (B) are side and top views of tetrameric CbnR. (C) and (D) are results of the β-galactosidase assay for the DBD mutants. Red and yellow residues are those showing less than 25% of the activity of the wild type and those showing 50–100% of the activity of the wild type by mutation, respectively (only activities with cis, cis-muconate are considered). Lys23, which was more active than the wild type, is shown in cyan. (C) and (D) are side and bottom views of tetrameric CbnR.
Discussion
In this study, we performed a mutational analysis of the amino acid residues in DBD and RD of CbnR. Residues that seemed to interact with DNA or inducer molecules were selected for mutations based on the earlier obtained structural informationCitation6,14) and a homology model. Many of the mutants lost their transcriptional activity, but the molecular mechanism of the functional losses cannot be explained only by the promoter binding activity of CbnR, particularly in the case of the mutations in DBD. In the case of the RD mutants, mutations affecting the CbnR function could be categorized into two types: those affecting inducer binding (Phe98Ala, Lys129Ala, and Phe202Ala) and the others, which would not alter the interaction with the inducer molecule (Arg199Ala and Val246Ala). Interestingly, the mutants of Arg199 and Val246 in TfdT, which correspond to Arg199 and Val246 of CbnR, exhibited altered responses to the inducer.Citation29) While these mutants of TfdT were not constitutively active, the observed change of the response to the inducer suggested that these residues affect the change of the conformation of CbnR and TdfT. In addition, the constitutively active mutants of BenM, CbbR, CysB, and OxyR have also been obtained.Citation9,10,30–33) Notably, most of the constitutively active mutations were located near inter-subunit interfaces. While details of the molecular mechanism cannot be provided in this study due to the lack of structural information, these mutations seem to mimic the conformation of tetrameric LTTR in complex with the inducer molecule.
In contrast to the mutations in RD, the mutational effects of DBD residues should be interpreted based on a multi-step transcription activation model of LTTRs (Fig. ). When analyzing the transcription activity of a mutant, we need to consider not only the binding to RBS but also binding to ABS. While most of the inactive mutants lost their DNA-binding activity, some inactive mutants retained DNA-binding activity. These results cannot be explained by a simple model such that CbnR binding to the promoter DNA is sufficient for the transcriptional activation. Indeed, biochemical analyses have shown that typical LTTRs including CbnR have several DNA-binding states in the process of the transcriptional activation (Fig. ).Citation2,22,34-40) The binding sites of DBD differ depending on whether the inducer is present; a significant shift of the DBD binding site on ABS has been observed upon inducer binding (Fig. ). Therefore, it has been proposed that DBD requires not only the ability to stably bind to RBS, but also an appropriate affinity to ABS to change the interaction with DNA upon inducer binding (Fig. ). It should be noted that the EMSA used in this study can only provide information about the binding affinity to RBS; it cannot provide information regarding a shift of the DBD-interacting site on ABS. Of course, mutants lacking the DNA-binding activity in EMSA are likely to lose the binding activity to RBS, and thereby their ability to activate transcription. However, when a DBD mutant that retains a DNA-binding activity is inactive for transcription, such as Lys7Ala, Asn17Ala, Pro30Ala or several Gln29 mutants including Gln29Ala, the mutated residue seems to be involved in the later steps of transcription activation—i.e. the steps after inducer binding. In the later steps, it has been proposed that quaternary structural change of LTTR upon inducer binding causes a shift of the DBD-binding site on ABS for the transcriptional activation.Citation16,22,38,40) Indeed, there seem to be several molecular steps—namely, inducer binding, a local conformational change around the inducer-binding site, a quaternary structure change of tetrameric LTTR, dissociation of DBDs from site 1 of ABS, and re-interaction of DBD to site 2 of ABS. To understand the molecular mechanism of the later steps of transcriptional activation, further biochemical analyses, such as a foot print assay with and without inducer will be needed. Of course, the crystal structures of the CbnR–DNA complex with and without inducer are required to reveal the mechanism responsible for the shift of the DBD-binding site on ABS (Fig. ), which would provide deep insight into the transcriptional activation process of CbnR as well as other LTTRs.
Funding
This work was supported by JSPS KEKENHI (Grant-in-Aid for Scientific Research (C)) [grant number 22580078].
Supplemental data
Supplemental data for this article can be accessed at https://doi.org/10.1080/09168451.2017.1373592.
Disclosure statement
No potential conflict of interest was reported by the authors.
Author contributions
RMor, KT, MT, YY, JS, NK, EA, and MU designed and performed experiments. RMin helped obtain homology modeling data. MF provided advice. RMor drafted the manuscript and TS and NO wrote the final paper. RMor, TS, and NO prepared tables and figures. TS provided the original idea for the research. NO conceived and is responsible for the entire project.
SupTableFinal.pdf
Download PDF (91.9 KB)SupFiguresFinal.pdf
Download PDF (494.4 KB)Notes
Abbreviations: 3-CB, 3-chlorobenzoate; ABS, activation-binding site, Ap: ampicillin; BA, benzoate; Cbpc, carbenicillin; CD, circular dichroism; DBD, DNA-binding domain; DIG, digoxigenin; EMSA, electrophoretic mobility shift assay; Gm, gentamycin; IPTG, isopropyl-β-D(-)-thiogalactopyranoside; LB, Luria–Bertani; LTTR, LysR-type transcriptional regulator; ONPG, o-nitrophenyl-β-D-galactopyranoside; PCR, polymerase chain reaction; RBS, recognition-binding site; RD, regulatory domain; SDS-PAGE, sodium dodecyl sulfate-polyacrylamide gel electrophoresis; TBE, Tris-Borate-EDTA; Vic, viccillin.
Related Research Data
References
- Ogawa N, Miyashita K. Recombination of a 3-chlorobenzoate catabolic plasmid from Alcaligenes eutrophus NH9 mediated by direct repeat elements. Appl Environ Microbiol. 1995;61(11):3788–3795.
- Ogawa N, McFall SM, Klem TJ, et al. Transcriptional activation of the chlorocatechol degradative genes of Ralstonia eutropha NH9. J Bacteriol. 1999;181(21):6697–6705.
- Ogawa N, Miyashita K. The chlorocatechol-catabolic transposon Tn5707 of Alcaligenes eutrophus NH9, carrying a gene cluster highly homologous to that in the 1,2,4-trichlorobenzene-degrading bacterium Pseudomonas sp. strain P51, confers the ability to grow on 3-chlorobenzoate. Appl Environ Microbiol. 1999;65(2):724–731.
- Schell MA. Molecular biology of the LysR family of transcriptional regulators. Annu Rev Microbiol. 1993;47:597–626.10.1146/annurev.mi.47.100193.003121
- Maddocks SE, Oyston PC. Structure and function of the LysR-type transcriptional regulator (LTTR) family proteins. Microbiology. 2008;154(12):3609–3623.10.1099/mic.0.2008/022772-0
- Muraoka S, Okumura R, Ogawa N, et al. Crystal structure of a full-length lysr-type transcriptional regulator, CbnR: unusual combination of two subunit forms and molecular bases for causing and changing DNA bend. J Mol Biol. 2003;328(3):555–566.10.1016/S0022-2836(03)00312-7
- Tropel D, van der Meer JR. Bacterial transcriptional regulators for degradation pathways of aromatic compounds. Microbiol Mol Biol Rev. 2004;68(3):474–500.10.1128/MMBR.68.3.474-500.2004
- Zhou X, Lou Z, Fu S, et al. Crystal structure of ArgP from Mycobacterium tuberculosis confirms two distinct conformations of full-length LysR transcriptional regulators and reveals its function in DNA binding and transcriptional regulation. J Mol Biol. 2010;396(4):1012–1024.10.1016/j.jmb.2009.12.033
- Craven SH, Ezezika OC, Haddad S, et al. Inducer responses of BenM, a LysR-type transcriptional regulator from Acinetobacter baylyi ADP1. Mol Microbiol. 2009;72(4):881–894.10.1111/mmi.2009.72.issue-4
- Ruangprasert A, Craven SH, Neidle EL, et al. Full-length structures of benm and two variants reveal different oligomerization schemes for LysR-Type transcriptional regulators. J Mol Biol. 2010;404(4):568–586.10.1016/j.jmb.2010.09.053
- Monferrer D, Tralau T, Kertesz MA, et al. Structural studies on the full-length LysR-type regulator TsaR from Comamonas testosteroni T-2 reveal a novel open conformation of the tetrameric LTTR fold. Mol Microbiol. 2010;75(5):1199–1214.10.1111/mmi.2010.75.issue-5
- Tsai CS, Chen CS, Winans SC. Most mutant OccR proteins that are defective in positive control hold operator DNA in a locked high-angle bend. J Bacteriol. 2011;193(19):5442–5449.10.1128/JB.05352-11
- Taylor JL, De Silva RS, Kovacikova G, et al. The crystal structure of AphB, a virulence gene activator from Vibrio cholerae, reveals residues that influence its response to oxygen and pH. Mol Microbiol. 2012;83(3):457–470.10.1111/mmi.2012.83.issue-3
- Alanazi AM, Neidle EL, Momany C. The DNA-binding domain of BenM reveals the structural basis for the recognition of a T-N11-A sequence motif by LysR-type transcriptional regulators. Acta Crystallogr D Biol Crystallogr. 2013;69(10):1995–2007.10.1107/S0907444913017320
- Jo I, Chung IY, Bae HW, et al. Structural details of the OxyR peroxide-sensing mechanism. Proc Natl Acad Sci USA. 2015;112(20):6443–6448.10.1073/pnas.1424495112
- Lerche M, Dian C, Round A, et al. The solution configurations of inactive and activated DntR have implications for the sliding dimer mechanism of LysR transcription factors. Sci Rep. 2016;6:6602.10.1038/srep19988
- Tyrrell R, Verschueren KH, Dodson EJ, et al. The structure of the cofactor-binding fragment of the LysR family member, CysB: a familiar fold with a surprising subunit arrangement. Structure. 1997;5(8):1017–1032.10.1016/S0969-2126(97)00254-2
- Muraoka S, Okumura R, Uragami Y, et al. Purification and crystallization of a Lysr-Type transcriptional regulator cbnr from ralstonia eutropha Nh9. Protein Pept Lett. 2003;10(3):325–329.10.2174/0929866033478942
- Sambrook J, Fritsch EF, Maniatis T. Molecular cloning: a laboratory manual. 2nd ed. Cold Spring Harbor (NY): Cold Spring Harbor Laboratory Press; 1989.
- Parales RE, Harwood CS. Regulation of the pcaIJ genes for aromatic acid degradation in Pseudomonas putida. J Bacteriol. 1993;175(18):5829–5838.10.1128/jb.175.18.5829-5838.1993
- Aldrich TL, Frantz B, Gill JF, et al. Cloning and complete nucleotide sequence determination of the catB gene encoding cis, cis-muconate lactonizing enzyme. Gene. 1987;52(2–3):185–195.10.1016/0378-1119(87)90045-X
- McFall SM, Parsek MR, Chakrabarty AM. 2-chloromuconate and ClcR-mediated activation of the clcABD operon: in vitro transcriptional and DNase I footprint analyses. J Bacteriol. 1997;179(11):3655–3663.10.1128/jb.179.11.3655-3663.1997
- Hanahan D. Studies on transformation of Escherichia coli with plasmids. J Mol Biol. 1983;166(4):557–580.10.1016/S0022-2836(83)80284-8
- Horton RM. PCR-mediated recombination and mutagenesis. SOEing together tailor-made genes. Mol Biotechnol. 1995;3(2):93–99.
- Amann E, Ochs B, Abel kJ. Tightly regulated tac promoter vectors useful for the expression of unfused and fused proteins in Escherichia coli. Gene. 1988;69(2):301–315.10.1016/0378-1119(88)90440-4
- Farinha MA, Kropinski AM. Construction of broad-host-range plasmid vectors for easy visible selection and analysis of promoters. J Bacteriol. 1990;172(6):3496–3499.10.1128/jb.172.6.3496-3499.1990
- Miller JH. Experiments in molecular genetics. Cold Spring Harbor (NY): Cold Spring Harbor Laboratory Press; 1972.
- Ezezika OC, Haddad S, Clark TJ, et al. Distinct effector-binding sites enable synergistic transcriptional activation by BenM, a LysR-type regulator. J Mol Biol. 2007;367(3):616–629.10.1016/j.jmb.2006.09.090
- Lang GH, Ogawa N. Mutational analysis of the inducer recognition sites of the LysR-type transcriptional regulator TfdT of Burkholderia sp. NK8. Appl Microbiol Biotechnol. 2009;83(6):1085–1094.10.1007/s00253-009-1960-5
- Colyer TE, Kredich NM. In vitro characterization of constitutive CysB proteins from Salmonella typhimurium. Mol Microbiol. 1996;21(2):247–256.10.1046/j.1365-2958.1996.6301347.x
- Lochowska A, Iwanicka-Nowicka R, Plochocka D, et al. Functional dissection of the LysR-type CysB transcriptional regulator. Regions important for DNA binding, inducer response, oligomerization, and positive control. J Biol Chem. 2001;276(3):2098–2107.10.1074/jbc.M007192200
- Choi H, Kim S, Mukhopadhyay P, et al. Structural basis of the redox switch in the OxyR transcription factor. Cell. 2001;105(1):103–113.10.1016/S0092-8674(01)00300-2
- Dangel AW, Gibson JL, Janssen AP, et al. Residues that influence in vivo and in vitro CbbR function in Rhodobacter sphaeroides and identification of a specific region critical for co-inducer recognition. Mol Microbiol. 2005;57(5):1397–1414.10.1111/j.1365-2958.2005.04783.x
- Parsek MR, Kivisaar M, Chakrabarty AM. Differential DNA bending introduced by the Pseudomonas putida LysR-type regulator, CatR, at the plasmid-borne pheBA and chromosomal catBC promoters. Mol Microbiol. 1995;15(5):819–829.10.1111/j.1365-2958.1995.tb02352.x
- Wang L, Winans SC. High angle and ligand-induced low angle DNA bends incited by OccR Lie in the same plane with OccR bound to the interior angle. J Mol Biol. 1995;253(1):32–38.10.1006/jmbi.1995.0533
- Vichivanives P, Bird TH, Bauer CE, et al. Multiple regulators and their interactions in vivo and in vitro with the cbb regulons of Rhodobacter capsulatus. J Mol Biol. 2000;300(5):1079–1099.10.1006/jmbi.2000.3914
- Kovacikova G, Skorupski K. Overlapping binding sites for the virulence gene regulators AphA, AphB and cAMP-CRP at the Vibrio cholerae tcpPH promoter. Mol Microbiol. 2001;41(2):393–407.10.1046/j.1365-2958.2001.02518.x
- Bundy BM, Collier LS, Hoover TR, et al. Synergistic transcriptional activation by one regulatory protein in response to two metabolites. Proc Natl Acad Sci USA. 2002;99(11):7693–7698.10.1073/pnas.102605799
- Wallecha A, Correnti J, Munster V, et al. Phase variation of Ag43 is independent of the oxidation state of OxyR. J Bacteriol. 2003;185(7):2203–2209.10.1128/JB.185.7.2203-2209.2003
- Lochowska A, Iwanicka-Nowicka R, Zaim J, et al. Identification of activating region (AR) of Escherichia coli LysR-type transcription factor CysB and CysB contact site on RNA polymerase alpha subunit at the cysP promoter. Mol Microbiol. 2004;53(3):791–806.10.1111/j.1365-2958.2004.04161.x