
Abstract
A unique electron-accepting analog of vitamin K1 found in photosystem I in several species of oxygenic photosynthetic microorganisms was confirmed to be 5′-hydroxyphylloquinone (1) through stereo-uncontrolled synthesis. Furthermore, the stereochemistry of 1 obtained from Synechococcus sp. PCC 7942 was assigned to be 5′S using proline-catalyzed stereocontrolled reactions.

Polar vitamin K1 (1) was synthesized. Stereochemistry of natural 1 was determined to be (5′S) after a derivation, where the same derivative was also synthesized.
Vitamin K1 (phylloquinone, VK1) functions as the secondary electron acceptor in photosystem I in many photosynthetic organisms.Citation1) A polar analog of VK1 (1) was first isolated from the cyanobacterium Anacystis nidulans (currently known as Synechococcus elongatus or Synechococcus sp. PCC 7942) in 1965,Citation2) and has also been found in Chlorella cells cultured with glucose.Citation3) 1 functions similar to VK1 because the phytyl groups do not have great impact to perform electron transport. The extra 5-hydroxyl group in 1 is thought to allow hydrophobic interactions with peripheral amino-acid residues and to affect its stability in photosystem I. However, its specific role and biochemical synthesis pathway in vivo are still not clear. The extremely small amount of 1 in organisms (one or two molecules per one photosystem I) and its instability upon separation from proteins have made its chemical analysis difficult. Its structure has been proposed to be the 5′-hydroxy analog of VK1 based on 1H NMR analysis,Citation4) and this has been confirmed with MS analysis of its trimethylsilyl derivative, despite this analysis being hindered by many contaminating substances.Citation5)
Although the number of species found to use 1 in photosystem I is still increasing,Citation6) no stereochemical study of this unique VK1 analog has been performed, and thus structural confirmation and assignment of its unknown stereochemistry is still of importance. For example, structural information on 1 may gain an understanding of the molecular evolution of electron transfer in photosystem I.
Herein, we report the synthesis of a stereoisomeric mixture of 1, and asymmetric syntheses performed to aid in the stereochemical assignment of the OH carbon (C′5) in 1 isolated from Synechococcus sp. PCC 7942.
Results and discussion
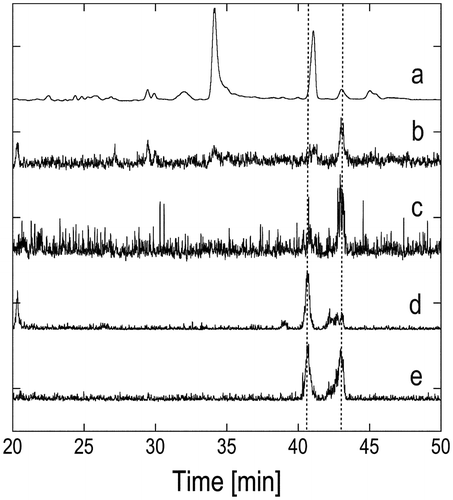
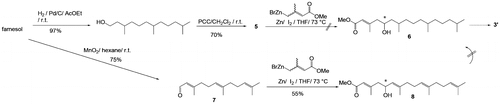
The retrosynthesis of 1 is outlined in Scheme . We planned to introduce the 5′-hydroxy group in 1 via nucleophilic addition to aldehyde 5, where several stereocontrolled asymmetric reactions are available if necessary. Friedel–Crafts condensation of 1-acetoxy-4-hydroxy-2-methyl-naphthalene (2)Citation7) with the tail section, i.e. alcohol 3, followed by a hydrolysis/oxidation process was expected to give target compound 1. Initially, a stereoisomeric mixture of 3 was intended to yield stereochemically uncontrolled 1, and a mixture of 5 was obtained from farnesol in two steps, i.e. hydrogenation and PCC-oxidation (ca. 80%).Citation8) In addition to 5, the unsaturated analog 7 was obtained by oxidation of farnesol (MnO2, quantitative, Scheme ).Citation9,10) Starting either from 5 or 7, a direct five-carbon-unit (four-carbon-chain) extension with a stereoisomeric mixture of methyl 4-bromo-3-methyl-2-butenoate (E/Z = 2:1) was examined.Citation11–14) The reaction of 7 with the zinc derivative of the ester in tetrahydrofuran (THF) resulted in a 55.8% yield of 8, while addition of the same zinc reagent to saturated 5 did not give any adduct, presumably because 5 is less reactive to nucleophiles and is enolizable. Although the starting reagent was an E/Z mixture, the 8 obtained contained only (E)-products, the formation of which can be explained by the conversion of (Z)-8 to a δ-lactone during reaction or extraction followed by removal during purification of methyl ester (E)-8. However, preparation of 6 (or its saturated analog) from (E)-8 via hydrogenation over different catalysts (Pd/C, Pt/Al2O3, etc.) in different solvents failed, and this pathway was abandoned.
Alternatively, a two-step carbon-chain extension procedure consisting of an aldol reaction of the aldehyde 5 with acetone (three-carbon extension to the main-chain) followed by treatment with vinylmagnesium chloride was applied (another two-carbon extension), as shown in Scheme .
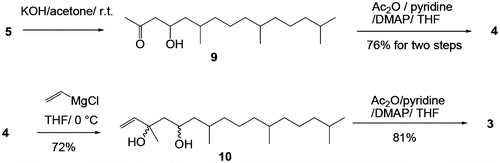
When 5 was treated with KOH in acetone, the expected aldol product 9 was obtained. Adduct 9 was found to be unstable on silica gel columns. Therefore, the extract of the reaction mixture was first treated with acetic anhydride/pyridine/N,N-dimethyl-4-aminopyridine (DMAP), and then purified by column chromatography to give 4 in 76.8% yield for the two steps. The 4 obtained was initially thought to be comprised of a mixture of four diastereomers that could not be distinguished by silica gel thin-layer chromatography (TLC). However, the 1H and 13C NMR spectra of 4 showed two sets of diastereomeric signals (ca. 1:1), which should be due to the relative stereoisomers at positions C4 and C6. This assignment was supported by chiral gas-liquid chromatography (GLC) analysis, in which two peaks (53:47, or approximately 1:1) appeared, and each peak was partially separated (vide infra). Vinylmagnesium chloride addition to 4 in THF resulted in the formation of 10, and subsequent re-acetylation of 10 at the 5-hydroxyl group gave 3 in 67% yield based on 4. Both compounds 10 and 3 were assumed to be mixtures of eight diastereomers, two sets of which were distinguishable on the TLC and in the 1H NMR spectra (ca. < 3:1). The final stage of this synthetic work was connection of the prepared tail section 3 to the naphthoquinone section. For this type of transformation, the Friedel–Crafts reaction of 1,4-dihydroxy-2-methylnaphthalene (the reduced form of vitamin K3) has often been employedCitation15); however, in the present study, the vitamin K4 derivative 1-acetoxy-4-hydroxy-2-methyl-naphthalene (2) was used to isolate the adduct before oxidation at the naphthalene moiety.Citation7) A mixture of 2 and 3 (1:1.1) in a mixture of ethyl acetate and dioxane (1:1) was treated with 6 mol% BF3 at 70 °C for 5 h. 11 was isolated in 58% yield after silica gel column chromatography (Scheme ).
As expected, the deacetylation of the aromatic acetate in 11 was easier than that of the acetate in C5′ position. Although conversion of 11 to 12 under basic conditions didn’t proceed smoothly, 12 was rapidly obtained under the reductive condition using NaBH4 in methanol. However, subsequent deacetylation of 12 was unsuccessful. To perform the double deacetylation while avoiding unfavorable quinone 12 formation, 11 was treated with LiAlH4 in ether. After extraction, 1 was obtained in 25.3% yield as a stereoisomeric mixture originating from 3. The 1H NMR and silica gel TLC analysis results for the as-synthesized 1 are reasonably well matched with previously reported data,Citation3) even though 1 is obtained as a diastereomeric mixture (see Fig. for the NMR spectrum of 1, the minor impurity is the Z-isomer). In addition, the atmospheric-pressure chemical ionization mass spectrum of the trimethylsilyl analog of as-synthesized 1 is identical to that recently reported,Citation6) and is also in agreement with previously reported results.Citation4) As can be seen in Fig. , the as-synthesized 1 shows two sets of isomeric peaks in a ratio of ca. 2:1, which could be attributed to the E/Z isomers at C2′ and C3′ positions as deduced from 2D-NMR analysis (supplementary data). That is, the C1′ methylene protons in the minor product (δ 3.49 and 3.30) exhibits a nuclear Overhauser effect (NOE) with the C4′ methylene, while the major isomer C1′ methylene (δ 3.39) exhibits an NOE with the C3′ methyl. The split in the protons of the minor isomer at the C1′ position could be because of the existence of the chiral center C5′ in the Z-isomer which might locate spatially near to C1′ position than that of E-isomer.
Fig. 1. 600 MHz 1H NMR spectrum of as-synthesized 5′-hydroxyphylloquinone (1) determined in CDCl3 using a JEOL ECA-600.
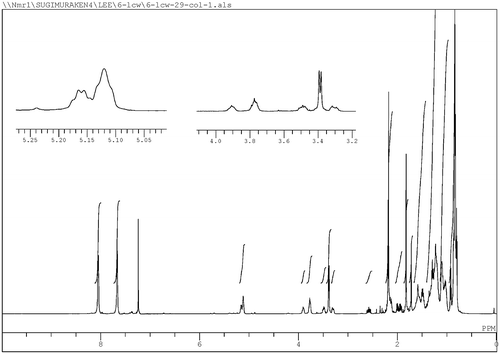
Fig. 2. Chiral GLC charts of stereocontrolled and uncontrolled products 4 derived from industrial VK1. (a) (4S)-enriched 4 derived from l-proline-catalyzed reaction, (b) (4R)-enriched 4 derived from d-proline-catalyzed reaction, (c) (4RS)-4 derived from VK1, and (d) stereoisomeric mixture of 4 prepared in Scheme .
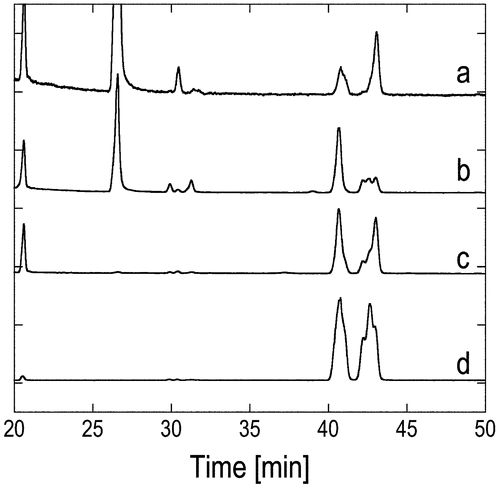
Fig. 3. TIC and mass chromatogram charts of compound 4 derived from natural 1 and stereo-controlled synthetic products 4 derived from industrial VK1. The TIC chart for (a) 4 derived from the natural product. Mass cromatograms at m/z 283 for 4 derived from the (b) natural product, (c) synthetic (4S)-4, (d) synthetic (4R)-4 and (e) synthetic (4RS)-4. The chart of (a) was integrated three times (n = 3) and smoothed using a seven-point weighted moving average.
Scheme 1. Retrosynthesis of 5′-hydroxyphylloquinone (1).
Scheme 2. Preliminary process for the five-carbon-unit extension.
Scheme 3. Preparation of the hydrocarbon chain component.
Scheme 4. Structure of the Friedel–Crafts adduct 11 and its derivative 12.
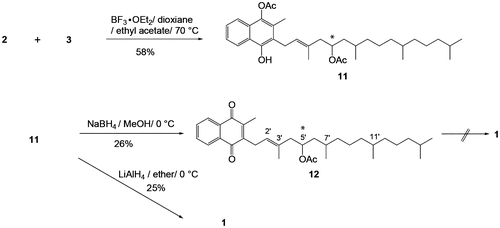
In the second part of the present study, stereochemical assignment at the 5′-position for 1 was performed as follows (Scheme ). Commercially available VK1 (Tokyo Chemical Industry Co., Ltd. Japan) was used as a starting material to yield the optically active aldehyde 5. The assumed absolute configurations of the optically active aldehydes 5 and 4 derived from VK1 are indicated as (3R,7R)-5 and (6R,10R)-4, respectively, in Scheme . Ozonolysis/dimethyl sulfide treatment of VK1 gave the optically active ketone (6R,10R)-13 (97% yield), and a subsequent four-step procedure, i.e. acetalization (82%), dibromination with excess bromine (73%), dehydrobromination with t-BuOK at 70 °C (99%), and ozonolysis gave (+)-5 in 59% yield ([α]20D = 0.3 (c 1.0, CHCl3), lit. [α]25D = 9.0 (synthesized from natural (4′R,8′R)-α-tocopherol, c 1.11, octane),Citation16) [α]20D = 7.6 (synthesized from (R,R)-hexahydrofarnesol [93.45% (3R,7R); 5.81% (3R,7S); 0.75% (3S,7R); < 0.1% (3S,7S)], c 2.265, CHCl3).Citation17) The smaller value of optical rotation in 5 than the literature values would show presence of some structural isomers which are difficult to separate by column chlromatography. Thus, the aldehyde 5 is optically active but not optically pure.
Scheme 5. Structural determination and asymmetric synthesis of 4.
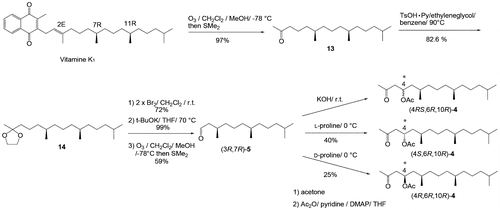
Stereocontrolled construction of a stereogenic center at the 5′ position of 1 was performed by the proline-catalyzed direct intermolecular aldol reaction reported by List et al. with good yields of aromatic aldehydes, α-branched aldehydes, and some amino acid in the presence of excess acetone,Citation18,19) and analyzed by chiral GLC as shown in Fig. . Optically active 5 was treated with acetone in the presence of L-proline, D-proline, or KOH, which should give different ratios of the major isomers (4S)-4 (Fig. (a)), (4R)-4 (Fig. (b)), and (4RS)-4 (Fig. (c)), respectively. The configuration at the 4-position of 4 corresponds to that at the 5′-position of 1. The yield of unbranched aldehydes was low in comparison with that of branched aldehydes, but the enantiomeric excess (ee) was high (>70%) for some substances.Citation12) In this study, 4S- and 4R-enriched products using the proline-catalyzed reaction of optically active 5 were obtained in ~30 and 26% yield, respectively. As seen in Fig. (c) and (d), the two separated peaks are detected corresponding to diastereomers due to the C4 and C6 stereogenic centers, and enantiomers of each diastereomer are partially separated within the broadening of each peak. Judging from the aldol stereoselectivities, the faster peak is assigned to (4R) and the slower peak to (4S). Several small peaks between them (see chart b) are not currently assignable. The broad peaks observed in the chiral GLC analysis of rac-4 (Fig. (d)) derived from farnesol may be because of stereoisomers due to the stereogenic centers at positions C6 and C10. In contrast, relatively sharp signals are observed in the GLC analysis of 4 derived from VK1, as seen in Fig. (c). Several stereoisomers contained in industrial VK1Citation20) are assumed to affect the stereochemical purity of 4.
Stereochemical assignment of natural 1 was performed using gas chromatography-mass spectrometry (GCMS) employing a chiral column (DEX-CB), considering both its amount and purity. Here 1 obtained from Synechococcus sp. PCC 7942 was chemically transformed to 4 to use the same chiral GLC column for GCMS measurements as that used for Fig. . A hexane extract of the culture was purified using a single preparative HPLC step, and was then subjected to acetylation (acetic anhydride/pyridine/DMAP for 120 min) and subsequent ozonolysis/dimethyl sulfide treatment. The 4 obtained from natural 1 was subjected to GCMS analysis, and the total ion mass spectrum (TIC) showed many peaks (Fig. (a)). Although the molecular ion of 4 (M+; m/z 326) was not detected, the signal at m/z 283 was assigned as a reasonable fragment of 4 because the weight-loss of 43 corresponds to an acyl (CH3CO) or isopropyl ((CH3)2CH) group. The mass chromatogram for m/z 283 of 4 obtained from natural 1 was shown in Fig. (b) and compared to these of synthetic 4 were shown in Fig. (b)–(e). The elution pattern of signal from 4 obtained from natural 1 (Fig. (b)) mostly consisted of the main peak synthetic (4S)-4 (Fig. (c)) and the back peak of synthetic (4RS)-4 (Fig. (e)). A small peak was also detected around the front peak (4R); however, this peak position was slightly out of alignment from the main peak of synthetic (4R)-4 (Fig. (d)) and the front peak of synthetic (4RS)-4 (Fig. (e)). As a result, 1 was concluded to have 5′S-configuration. Furthermore, GCMS analysis indicated the possibility that natural 1 has the same stereochemistry at both C7 and C11 as the main component of the industrial VK1, because the main m/z 283 signal of 4 derived from the natural product (Fig. (b)) appears at almost the same retention time as that of the main m/z 283 signal of synthetic (4S)-4 (Fig. (c)).
At the current stage, it is difficult to unambiguously assign the stereochemistry at C7 and C11, but it would be same as the stereochemistries of natural VK1, which was predominantly 2′,3′ -trans-7′R, 11′R-phylloquinone.
Conclusion
The 5′-hydroxy analog of phylloquinone (1) was successfully synthesized as a mixture of the 16 possible stereoisomers, which were only partially distinguishable. 1 was confirmed to have a 5′-OH substituent. For the compound isolated from Synechococcus sp. PCC 7942, the configuration at the 5′-OH position was determined to be 5′S. Furthermore, we have developed here a method to obtain a sufficient amount of the compound for future study of its biological role.
Experimental
General
All new compounds were characterized by NMR spectrometry using a JEOL ECA-600 spectrometer at 600 MHz for the proton spectra and 150 MHz for the carbon spectra, and by IR with a JASCO FT/IR-410 spectrophotometer. High-resolution MS was performed using a JEOL JMS-T100LC with electrospray ionization (ESI). Chiral GLC analysis was performed with a Shimadzu GC-17A equipped with DEX-CB column (i.d. 0.25 μm × 30 m, GL Science). GCMS was performed using a Shimadzu GCMS-QP 2010 equipped with a DEX-CB column. Ozonolysis was performed with a 0-1-2 ozone generator (Nihon Ozone). Vinylmagnesium chloride was obtained as a THF solution (1.46 mol/L) from Kanto Chemicals. All solvents were purified by distillation with proper drying agents. Reactions were carried out under dry nitrogen atmosphere unless noted.
Preparation of 8 from 7
A suspension of zinc dust (222 mg, 5 eq.) in THF (0.5 mL) was treated with a trace amount of iodine. After heating this solution to 73 °C, a solution of 7 (156.7 mg) and methyl 4-bromo-3-methyl-2-butenoate (E/Z = 2:1, 142.2 mg, 1.0 equiv.) in THF (5 mL) was added dropwise over 10 min. The mixture was kept at the same temperature for 1 h, and then quenched by a saturated NH4Cl solution and extracted with ether (×3). The extract was dried over Na2SO4, concentrated, and purified on a silica gel column (5 g, elution with 20% ethyl acetate in hexane) to give a 2,3-E/Z mixture of 8 as a colorless oil (126 mg, 55.8% yield). 1H NMR of the major (trans) isomer (CDCl3) δ 5.80 (s, 0.6H), 5.32 (s, 0.4H), 5.31 (d, J = 8.9 Hz, 0.6H), 5.17 (d, J = 8.9 Hz, 0.4H), 5.16–5.07 (m, 2.4H), 4.57 (m, 0.6H), 3.67 (s, 1.2H), 2.39–1.51 (m, 25H).
Aldol reaction of 5 to give 4 through 9
A solution of 5 (2.2 g) in acetone (50 mL) in the presence of 50% KOH (10 mL) was stirred at room temperature for 12 h. The reaction mixture was treated with water, extracted with ethyl acetate (×3), dried over Na2SO4, and concentrated in vacuo to give a mixture of 9 as a colorless oil (5.4 g). The obtained mixture was treated with acetic anhydride (3.3 g) in THF (50 mL) in the presence of pyridine (2.86 g) and DMAP (696 mg), and stirred at room temperature for 4 h. The reaction mixture was treated with water and extracted with ether (×3), dried over Na2SO4, concentrated, and purified by silica gel column chromatography (elution with 6% ethyl acetate in hexane) to give 2.46 g of 4 as a colorless oil (76.8% yield for two steps, diastereomeric mixture); 1H NMR (CDCl3) δ 5.30 (m, 1H), 2.71 (dd, J = 6.87, 6.87 Hz, 0.5H), 2.68 (dd, J = 7.56, 6.87 Hz, 0.5H), 2.59 (dd, J = 5.50, 5.50 Hz, 0.5H), 2.56 (dd, J = 6.87, 6.19 Hz, 0.5H), 2.14 (s, 3H), 2.00 (s, 1.5H), 1.99 (s, 1.5H), 1.61–1.40 (m, 4H), 1.34–1.18 (m, 8H), 1.13–1.03 (m, 5H), 0.88 (d, J = 6.19 Hz, 3H), 0.84 (d, J = 6.19 Hz, 6H), 0.82 (brd, J = 6.87 Hz, 3H); 13C NMR (CDCl3) δ 205.69, 205.64, 170.44, 170.32, 68.96, 68.50, 48.75, 48.23, 41.68, 41.60, 41.44, 39.29, 37.64, 37.29, 37.21, 37.18, 36.75, 32.70, 30.36, 29.48, 29.16, 27.91, 25.70, 24.74, 24.72, 24.20, 24.07, 22.66, 22.56, 21.09, 21.06, 19.87, 19.80, 19.66, 19.60, 19.30, 19.22; IR (neat) 2927, 2870, 1740, 1464, 1365, 1241, 1167, 1021, 972, 939, 833, 736, 650 cm−1; HRMS (ESI) m/z (M+Na+) calcd for C20H38NaO3 349.2719, found 349.2753.
Vinyl Grignard addition to 4 to give 10, and then 3
To a solution of 4 (2.46 g) in THF (50 mL) was added slowly a THF solution of vinylmagnesium chloride (20 mL, 29 mmol) at 0 °C. The resulting mixture was stirred for 1 h at 0 °C, and allowed to warm to room temperature. The reaction mixture was treated with water and then extracted with ether (×3), dried over Na2SO4, concentrated, and purified by silica gel column chromatography (elution with 6% ethyl acetate in hexane) to give 1.69 g of 10 as a colorless oil (72% yield, diastereomer mixture = 1:2 based on NMR); 1H NMR (CDCl3) δ 5.91–5.86 (dd, J = 17.18, 10.31 Hz, 0.33H), 5.85–5.80 (ddm, J = 17.18, 10.31 Hz, 0.67H), 5.28 (d, J = 17.18 Hz, 0.67H), 5.17 (d, J = 17.18 Hz, 0.33H), 5.08 (d, J = 10.31 Hz, 0.67H), 4.96 (d, J = 10.31 Hz, 0.33H), 4.08–4.04 (m, 0.33H), 3.9–3.86 (m, 0.67H), 1.97–1.86 (m, 2H), 1.65–1.42 (m, 6H), 1.32–0.99 (m, 16H), 0.84–0.76 (m, 12H); 13C NMR (CDCl3) δ 146.07, 143.98, 143.94, 112.56, 112.52, 111.20, 77.21, 77.00, 76.79, 74.30, 74.26, 73.65, 68.25, 68.23, 67.83, 46.75, 46.73, 45.75, 45.70, 39.32, 37.37, 37.34, 37.31, 37.28, 37.20, 32.77, 32.75, 32.73, 30.29, 30.25, 29.36, 29.32, 27.95, 24.79, 24.77, 24.25, 22.70, 22.63, 22.61, 22.56, 20.06, 20.00, 19.73, 19.63, 19.32; IR (neat) 3348, 2954, 2927, 2869, 2360, 1714, 1642, 1462, 1377, 1264, 1168, 1029, 994, 922, 853 cm−1; HRMS (ESI) m/z (M+Na+) calcd for C20H40NaO2 335.2950, found 335.2926.
The obtained 10 was treated with acetic anhydride (967 mg) in THF (50 mL) in the presence of pyridine (728 mg) and DMAP (330 mg), and was stirred at room temperature for 5 h. The reaction mixture was treated with water, extracted with ether (×3), dried over Na2SO4, concentrated, and purified by silica gel column chromatography (elution with 10% ethyl acetate in hexane) to give 1.57 g of 3 as a colorless oil (81.7% yield, diastereomer mixture 2:2:1:1 based on NMR); 1H NMR (CDCl3) δ 5.85 (dd, J = 17.18, 11.00 Hz, 0.33H), 5.82 (dd, J = 11.00, 5.50 Hz, 0.33H), 5.79 (dd, J = 11.00, 5.50 Hz, 0.33H), 5.27 (dd, J = 17.18, 1.38 Hz, 0.33H), 5.26 (dd, J = 17.18, 1.38 Hz, 0.33H), 5.19–5.11 (m, 0.33H), 5.15 (d, J = 17.18 Hz, 0.33H), 5.05 (dd, J = 10.31, 1.38 Hz, 0.67H), 5.02–4.95 (m, 0.66H), 4.93 (d, J = 10.31 Hz, 0.33H), 2.86 (s, 0.17H), 2.77 (s, 0.17H), 2.75 (s, 0.33H), 2.68 (s, 0.33H), 2.01 (d, J = 4.12 Hz, 2H), 1.93 (d, J = 2.06 Hz, 1H), 1.88–1.69 (m, 2H), 1.65–1.59 (m, 2H), 1.50–1.02 (m, 19H), 0.87–0.80 (m, 12H); 13C NMR (CDCl3) δ 171.99, 171.44, 171.07, 145.12, 145.08, 144.18, 112.29, 112.25, 110.56, 110.54, 72.38, 72.28, 71.63, 70.79, 70.23, 69.39, 68.94, 47.56, 47.40, 46.96, 46.60, 43.52, 43.44, 43.11, 43.04, 42.85, 42.79, 39.32, 37.63, 37.34, 37.25, 36.97, 36.92, 32.76, 32.73, 29.92, 29.79, 29.51, 29.48, 29.28, 29.14, 28.58, 27.95, 24.78, 24.27, 24.22, 24.10, 22.69, 22.60, 21.47, 21.45, 21.42, 19.82, 19.75, 19.70, 19.63, 19.45, 19.37, 19.27; IR (neat) 3481, 2926, 1737, 1463, 1376, 1246, 1138, 1022, 921 cm−1; HRMS (ESI) m/z (M+Na+) calcd for C22H42NaO3 377.3032, found 377.3017.
Friedel–Crafts reaction of 3 with 2 to give 11
To a solution of 3 (1.3 g, 3.7 mmol) and 2 (0.9 g, 4 mmol, 1.08 eq) in a mixture of dioxane and ethyl acetate (1:1, 60 mL) was added BF3·OEt2 (0.3 mL) at room temperature. The resulting solution was stirred at 70 °C under nitrogen for 6 h. Reaction progress was monitored by TLC on silica gel (elution with 10% ethyl acetate in hexane). The reaction mixture was treated with water and extracted with ethyl acetate (×3). The combined organic layer was washed with water and then brine, dried over MgSO4, filtered, and concentrated under vacuum. Purification on a silica gel column (elution with 6% ethyl acetate in hexane) gave 1.19 g of 11 as a yellowish oil (58% yield); 1H NMR (CDCl3) δ 8.10 (d, J = 8.25 Hz, 1H), 7.61 (d, J = 8.25 Hz, 1H), 7.44–7.38 (m, 2H), 5.88 (d, J = 11.69 Hz, 0.5H), 5.59 (d, J = 11.69 Hz, 0.5H), 5.31–5.29 (m, 1H), 5.17–5.12 (m, 1H), 3.58–3.37 (m, 2H), 2.45 (s, 3H), 2.23 (d, J = 4.81 Hz, 3H), 2.08 (s, 1.5H), 1.89 (s, 1.5H), 1.75 (s, 3H), 1.61 (s, 2H), 1.56–1.47 (m, 3H), 1.39–1.03 (m, 14H), 0.94–0.80 (m, 12H); 13C NMR (CDCl3) δ 170.99, 169.66, 147.46, 138.06 134.42, 126.36, 126.30, 126.05, 125.07, 125.00, 124.80, 124.48, 121.82, 121.74, 120.57, 120.55, 119.99, 70.67, 45.96, 42.25, 39.34, 37.75, 37.73, 37.36, 37.26, 32.77, 32.76, 29.38, 29.29, 27.96, 26.33, 26.10, 24.78, 24.29, 24.25, 24.20, 24.14, 23.89, 22.71, 22.61, 21.23, 20.63, 19.69, 19.63, 19.41, 16.55, 13.50, 13.42; IR (neat) 3456, 3096, 2954, 2926, 2867, 1763, 1737, 1598, 1461, 1364, 1239, 1208, 1182 cm−1; HRMS (ESI) m/z (M+Na+) calcd for C35H52NaO5 575.3712, found 575.3704.
Conversion of 11 into 12
To a solution of 11 (50 mg, 0.09 mmol) in MeOH (3 mL) was added NaBH4 (10 mg, 0.27 mmol, 3 eq.) at 0 °C. The resulting mixture was stirred at room temperature for 24 h. The mixture was treated with water and extracted with ethyl acetate (×3), dried over Na2SO4, and concentrated under vacuum. Purification on a silica gel column (elution with 6% ethyl acetate in hexane) gave 12 mg of 12 as a yellowish oil (26.6% yield); 1H NMR (CDCl3) δ 8.05 (brs, 2H), 7.66 (brs, 2H), 5.21–5.15 (m, 0.5H), 5.12–5.00 (m, 1.5H), 3.41 (dd, J = 14.43, 6.87, 0.5H), 3.33 (d, J = 6.87, 1.5H), 2.60 (m, 0.5H), 2.27 (m, 0.5H), 2.16 (d, J = 13.06 Hz, 3H), 2.02 (d, J = 6.19 Hz, 1.5H), 1.90 (d, J = 4.81 Hz, 1.5H), 1.80 (s, 1H), 1.70 (s, 1H), 1.61 (s, 1H), 1.49–1.47 (m, 2H), 1.35–1.02 (m, 16H), 0.88–0.78 (m, 12H); 13C NMR (CDCl3) δ 185.35, 184.39, 170.55, 145.76, 143.38, 133.75, 133.38, 133.33, 132.07, 126.29, 126.27, 126.21, 122.52, 70.57, 70.11, 45.74, 45.22, 41.54, 39.33, 37.82, 37.77, 37.35, 37.30, 37.27, 37.25, 37.19, 36.79, 36.73, 32.77, 32.74, 32.71, 29.68, 29.53, 29.22, 27.95, 26.12, 25.99, 24.77, 24.33, 24.27, 24.14, 24.10, 23.94, 22.70, 22.61, 21.30, 21.26, 21.12, 21.07, 20.01, 19.71, 19.68, 19.62, 19.30, 16.58, 16.55, 12.76, 12.66; IR (neat) 2959, 2867, 2926, 1737, 1661, 1618, 1596, 1461, 1376, 1295, 1241, 1022 cm−1; HRMS (ESI) m/z (M+Na+) calcd for C33H48NaO4 531.3450, found 531.3483.
Reaction of 11 to give 1
To a suspension of lithium aluminum hydride (34 mg) in dry ether (4 mL) was added an ether solution (2 mL) of 11 (100 mg, 0.18 mmol) at 0 °C. After 3 h, the mixture was treated with water and extracted with ether (×3). The combined organic layer was dried over Na2SO4, and concentrated under vacuum. Purification on a silica gel column (elution with 6% ethyl acetate in hexane) gave 21.3 mg of 1 as a yellowish oil (25.3% yield); 1H NMR (CDCl3) δ 8.06–8.04 (m, 2H), 7.68–7.66 (m, 2H), 5.16–5.12 (m, 1H), 3.91–3.76 (m, 1H), 3.49 (m, 0.3H), 3.39 (d, J = 6.87 Hz, 1.4H), 3.30 (m, 0.3H), 2.19 (d, J = 8.25 Hz, 3H), 2.02–1.92 (m, 1H), 1.83 (s, 2H), 1.72 (s, 1H), 1.59–1.43 (m, 3H), 1.33–1.02 (m, 16H), 0.93–0.80 (m, 12H); 13C NMR (CDCl3) δ 185.32, 184.55, 145.60, 143.52, 134.63, 133.44, 133.42, 133.40, 132.08, 126.33, 126.24, 123.06, 66.73, 66.33, 48.82, 48.31, 44.77, 44.68, 39.34, 38.17, 37.38, 37.33, 37.25, 37.22, 37.05, 32.78, 32.75, 32.75, 29.31, 27.96, 26.36, 26.24, 24.79, 24.36, 24.27, 22.71, 22.62, 19.75, 19.65, 19.28, 16.54, 16.52, 12.77; IR (NaCl, neat) 3500, 2925, 1660, 1596, 1459, 1295, 715 cm−1; HRMS (ESI) m/z (M+Na+) calcd for C31H46NaO3 489.3345, found 489.3391.
Ozonolysis of VK1 to give optically active 13
A three-necked conical flask was equipped with a glass tube to bubble ozone/oxygen gas and charged with a mixture of VK1 (1 g, 2.219 mmol) in a mixture of CH2Cl2 and MeOH (1:1, 200 mL). The solution was cooled to −78 °C (MeOH/dry ice) under O2 and then ozone-containing O2 was bubbled with stirring. After the solution turned pale blue, remaining ozone was removed by purging with O2. At the same temperature, dimethyl sulfide (0.633 mL, 6.657 mmol, 3 eq.) was added and the solution was allowed to warm to room temperature. The resulting solution was concentrated to approximately 50 mL on a rotary evaporator. Water was then added and the mixture was extracted with ether (×3). The combined organic layer was washed with brine, dried over MgSO4, filtered, and concentrated under vacuum. Purification on a silica gel column (elution with 3% ethyl acetate in hexane) gave 0.58 g of 13 as a colorless oil (97.3%); [α]20D=+0.50 (c 0.6, CHCl3) [lit. [α]20D = + 0.59 (c 4.37, ether)Citation21)]; 1H NMR (CDCl3) δ 2.37 (td, J = 2.06, 6.19 Hz, 2H), 2.11 (s, 3H), 1.58–1.48 (m, 4H), 1.39–1.20 (m, 9H), 1.13–1.02 (m, 6H), 0.85–0.81 (m, 12H); 13C NMR (CDCl3) δ 209.42, 44.14, 39.34, 37.39, 37.26, 36.47, 32.77, 32.66, 29.85, 27.97, 24.80, 24.78, 24.42, 22.71, 22.62, 21.42, 19.73, 19.57; IR (neat) 2953, 2926, 2868, 1719, 1462, 1364, 1164 cm−1; EI MS (m/z) calcd for C18H36O 268.28, obsvd 250 (17%), 179 (10%), 165 (21%), 138 (23%), 124 (63%), 109 (81%), 95 (56%), 71 (67%), 58 (100%).
Conversion of 13 to 14
A solution of 13 (550 mg, 2.05 mmol), TsOH·Py (1 g), and ethylene glycol (2.27 mL, 41 mmol, 20 eq.) in benzene (40 mL) was placed in a flask fitted with a Dean-Stark apparatus. The solution was stirred at 90 °˚C under nitrogen for 8 h. The reaction mixture was then treated with aqueous NaHCO3 at 0 °C and extracted with ether (×3). The combined organic layer was washed with brine and dried over MgSO4, filtered, and concentrated under vacuum. Purification on a silica gel column (elution with 3% ethyl acetate in hexane) gave 0.53 g of 14 as a light yellow oil (82.6% yield); [α]20D = + 0.13 (c 0.79, CHCl3); 1H NMR (CDCl3) δ 3.94–3.89 (m, 4H), 1.60–1.48 (m, 3H), 1.38–1.19 (m, 15H), 1.14–1.01 (m, 6H), 0.85–0.80 (m, 12H); 13C NMR (CDCl3) δ 110.21, 64.60, 39.53, 39.36, 37.42, 37.38, 37.34, 37.27, 37.25, 32.79, 27,98, 24.81, 24.79, 24.49, 23.71, 22.72, 22.62, 21.63, 19.74, 19.68; IR (neat) 2926, 1463, 1377, 1253, 1220, 1155, 1069, 946, 869, 736 cm−1; EI MS (m/z) calcd for C20H40O2 312.3, obs 313 (7%), 217 (7%), 165 (11%), 151 (11%), 137 (18%), 124 (38%), 109 (47%), 95 (67%), 71 (93%), 58 (100%).
Dibromination of 14
To a solution of 14 (0.49 g, 1.568 mmol) in CH2Cl2 (10 mL) was added a solution of bromine (0.17 mL, 3.246 mmol, 2.07 eq.) in CH2Cl2 (10 mL). Initially, a small amount of the bromine solution was added in one portion. When the reaction had started, as seen by decolorization, the rest of the bromine solution was added dropwise. The resulting solution was stirred at room temperature under nitrogen for 1 h, and then pyridine (0.26 mL, 3.246 mmol, 2.07 eq.) was added. The reaction mixture was further treated with 20 mL of 10% Na2CO3, washed with water, and then brine. The organic layer was dried over MgSO4, filtered, and concentrated under vacuum. The residue was filtered through a silica gel pad using a glass Büchner funnel. The filtration gave 0.53 g of a pale yellow oil (72.6% yield), which was used for next step without further purification; 1H NMR (CDCl3) δ 4.20–4.10 (m, 6H), 3.85 (d, J = 11.00 Hz, 1H), 3.58 (dd, J = 11.00, 1.38 Hz, 1H), 1.94 (m, 1H), 1.74–1.62 (m, 2H), 1.52–1.45 (m, 1H), 1.40–1.32 (m, 2H), 1.26–1.17 (m, 7H), 1.12–1.05 (m, 5H), 0.89–0.82 (m, 12H); 13C NMR (CDCl3) δ 108.64, 66.98, 66.83, 57.89, 39.34, 37.34, 37.32, 37.25, 36.75, 34.99, 34.80, 34.78, 32.76, 32.12, 29.96, 27.96, 24.77, 22.71, 22.65, 19.73; IR (neat) 2925, 1461, 1379, 1203, 1119, 1038, 951 cm−1.
Dehydrobromination
To a solution of the dibromide (0.53 g, 1.127 mmol) in THF (20 mL) was added t-BuOK (379 mg, 3.38 mmol, 3 eq.) at room temperature. The solution was stirred at 70 °C under nitrogen for 3 h. Reaction was monitored by TLC (SiO2, elution with 10% ethyl acetate in hexane). The reaction mixture was filtered through a silica plug using a Büchner funnel twice, dried over MgSO4, and concentrated under vacuum to afford 0.507 g of a pale yellowish oil (99% yield), which was used for the next step without further purification; 1H NMR (CDCl3) δ 5.90 (td, J = 15.12, 7.56 Hz, 1H), 5.42 (d, J = 15.12 Hz, 1H), 4.05 (shm, 2H), 3.93 (shm, 2H), 3.46 (s, 2H), 2.05 (m, 1H), 1.88 (m, 1H), 1.51–1.46 (m, 2H), 1.35–1.15 (m, 8H), 1.12–1.00 (m, 5H), 0.86–0.81 (m, 12H); 13C NMR (CDCl3) δ 133.32, 127.74, 106.05, 65.57, 39.34, 39.24, 37.30, 37.26, 36.87, 36.81, 32.79, 32.75, 27.97, 24.80, 24.78, 24.43, 22.71, 22.62, 19.70, 19.56; IR (neat) 2953, 2925, 1666, 1462, 1378, 1208, 1093, 1040, 974 cm−1.
Ozonolysis to give 5
A three-necked conical flask was equipped with a glass tube to bubble ozone/oxygen gas and charged with a solution of the dehydrobromination product (0.5 g, 1.284 mmol) in a mixture of CH2Cl2 and MeOH (1:1, 30 mL). The solution was cooled to −78 °C (MeOH/dry ice) under O2 and then ozone-containing O2 was bubbled with stirring. After the solution turned pale blue, the remaining ozone was removed by purging with O2. At the same temperature, dimethyl sulfide (0.37 mL, 6.657 mmol, 3 eq.) was added, and the solution was allowed to warm to room temperature. The resulting solution was concentrated to approximately 5 mL by rotary evaporation. Water was added, and the mixture was extracted with ether (×3). The combined organic layer was washed with brine, dried over MgSO4, filtered, and concentrated under vacuum. Purification on a silica gel column (elution with 3% ethyl acetate in hexane) gave 0.17 g of (3R,7R)-5 as a colorless oil (59.3% yield); [α]20D = + 0.3 (c 1.0, CHCl3) [lit. [α]25D = 9.0 (octane)Citation16)]; 1H NMR (CDCl3) δ 9.74 (t, J = 2.06 Hz, 1H), 2.37 (ddt, J = 15.8, 5.5, 1.4 Hz, 1H), 2.21 (ddt, J = 15.8, 7.6, 5.5 Hz, 1H), 2.03 (m, 1H), 1.49 (m, 1H), 1.36–1.02 (m, 13H), 0.94 (d, J = 7.56 Hz, 2H), 0.84 (d, J = 6.19, 7H), 0.82 (dd, J = 6.19, 2.06, 3H); 13C NMR (CDCl3) δ 203.31, 51.05, 39.23, 37.31, 37.25, 37.22, 32.73, 27.96, 24.77, 24.69, 24.38, 24.32, 22.70, 22.61, 19.71; IR (neat) 2927, 2870, 1710, 1464, 1412, 1381, 1366, 1296, 1232, 1170, 939 cm−1.
Preparation of (4S)-4 and (4R)-4 major isomer mixture
A solution of l-proline (8 mg, 0.07 mmol) in acetone (3 mL) was stirred at 0 °C for 10 min. To this, 5 (50 mg, 0.22 mmol) prepared from VK1 was added, and the reaction mixture was warmed to room temperature and stirred for 3 d. Reaction was monitored by TLC (SiO2, elution with 10% ethyl acetate in hexane). Then, the reaction mixture was treated with aqueous NH4Cl and extracted with ethyl acetate (×3), dried over Na2SO4, and concentration under vacuum gave a mixture of 9 as a colorless oil (56.6 mg). The crude product was treated with acetic anhydride (35 mg) in THF (3 mL) in the presence of pyridine (30 mg) and DMAP (7 mg), and was stirred at room temperature for 2 h. The reaction mixture was treated with water, extracted with ether (×3), dried over Na2SO4, concentrated, and purified by silica gel column chromatography (elution with 6% ethyl acetate in hexane) to give 29 mg of (4S)-4 as a colorless oil (40.4% yield for two steps). This compound contained many impurities due to the lower yield and lower selectivity.
A solution of d-proline (8 mg, 0.07 mmol) in acetone (3 mL) was stirred at 0 °C for 10 min. 5 (50 mg, 0.22 mmol) was added and the reaction mixture was warmed to room temperature and stirred for 3 d. Reaction progress was monitored by silica gel TLC (elution with 10% ethyl acetate in hexane). The reaction mixture was treated with NH4Cl aq. and extracted with ethyl acetate (×3), dried over Na2SO4, and concentration under vacuum gave a mixture of 9 as colorless oil (56.8 mg). The obtained mixture was treated with acetic anhydride (35 mg) in THF (3 mL) in the presence of pyridine (27 mg) and DMAP (7.3 mg), and was stirred at room temperature for 2 h. The reaction mixture was treated with water and extracted with ether (×3), dried over Na2SO4, concentrated, and purified by silica gel column chromatography (elution with 6% ethyl acetate in hexane) to give 18.3 mg of (4R)-4 as a colorless oil (25.5% yield for two steps). This compound contained many impurities due to the lower yield and lower selectivity.
Preparation of 4 from natural product
Synechococcus sp. PCC 7942 cells were grown in a BG11 medium (7.5 L × 2) containing 5 mM glucose under light for 6 d and harvested by centrifugation. The cells were suspended in pure water (60 mL) and divided into two tubes. After lyophilization, hexane was added, and the mixture was sonicated and stirred (×3). The mixture was then centrifuged, filtered, and concentrated under vacuum. A small amount of methanol was added and the mixture was roughly purified by HPLC (column: ODS, gradient elution with a two solutions; solution A: 0.02 M ammonium acetate and 80% methanol, solution B: ethyl acetate/methanol (3:7)). The combined mixture was concentrated under vacuum and treated with acetic anhydride (excess), pyridine (excess), and DMAP in THF (10 mL). After 2 h, the reaction mixture was treated with water and extracted with ether (×3), dried over Na2SO4, and concentrated. The concentrated mixture was treated with ozone (the reaction process was the same as that previously performed for ozonolysis). The ether extract of the crude mixture was used for GCMS analysis without further purification.
Author contribution
Makiko Kosugi and Changwoo Lee carried out the organic synthesis and chemical analyses. Yasuhiro Kashino performed separation and analyses of the polar analog of VK1 from Synechococcus sp. PCC 7942. Tomonori Misaki and Morifumi Fujita contributed to the data interpretation. Takashi Sugimura designed the analytical strategy and contributed to the data interpretation. Makiko Kosugi, Tomonori Misaki, Yasuhiro Kashino, Morifumi Fujita, and Takashi Sugimura wrote the manuscript.
Funding
This work was supported by Japan Science and Technology Agency [GCOE Program].
Supplemental data
Copies of 2D NMR Spectra (COSY and NOESY) for as-synthesized 5′-hydroxyphylloquinone (1). Available at: https://doi.org/10.1080/09168451.2017.1385381.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supporting_information.pdf
Download PDF (160.4 KB)Acknowledgments
We thank Prof Tatsuo Omata in Nagoya University for providing the strain of Synechocystis sp. PCC 7942. This work was supported by GCOE Program from MEXT (to TS).
Related Research Data
References
- Jordan P, Fromme P, Witt H, et al. Three-dimensional structure of cyanobacterial photosystem I at 2.5 Å resolution. Nature. 2001;411:909–917.10.1038/35082000
- Henninger MD, Bhagavan HN, Crane FL. Comparative studies on plastoquinones: I. Evidence for three quinones in the blue-green alga, Anacystis nidulans. Arch Biochem Biophys. 1965;110:69–74.10.1016/0003-9861(65)90155-4
- Henninger MD. Hydrogenase in chlorella: qualitative differences in quinone composition. Biochem Biophys Res Commun. 1965;19:233–236.10.1016/0006-291X(65)90510-3
- Allen CF, Franke H, Hirayama O. Identification of a plastoquinone and two naphthoquinones in Anacystis nidulans by NMR and mass spectroscopy. Biochem Biophys Res Commun. 1967;26:562–568.10.1016/0006-291X(67)90102-7
- Law A, Thomas G, Threlfall DR. 5′-Monohydroxyphylloquinone from Anacystis and Euglena. Phytochemistry. 1973;12:1999–2004.10.1016/S0031-9422(00)91523-1
- Ozawa S, Kosugi M, Kashino Y, et al. 5′-monohydroxyphylloquinone is the dominant naphthoquinone of PSI in the green alga Chlamydomonas reinhardtii. Plant cell Physiol. 2012;53:237–243.10.1093/pcp/pcr168
- Knierzinger A, Walther W, Weber B, et al. Eine neue Methode zur stereochemischen Analyse offenkettiger terpenoider Carbonyl-Verbindungen. Helv Chim Acta. 1990;73:1087–1107.10.1002/hlca.v73:4
- Cane DE, Iyengar R, Shiao MS. Cyclonerodiol biosynthesis and the enzymic conversion of farnesyl to nerolidyl pyrophosphate. J Am Chem Soc. 1981;103:914–931.10.1021/ja00394a032
- Lei B, Fallis AG. Regioselective control of allyl anions with cadmium chloride: α vs. γ condensation with aldehydes. Tetrahedron Lett. 1986;27:5193–5196.
- Lei B, Fallis AG. Cadmium chloride mediated regiocontrol of dienolates and ketene thioacetals: γ condensation with aldehydes. Can J Chem. 1991;69:1450–1456.10.1139/v91-214
- Suhara Y, Wada A, Tachibana Y, et al. Structure-activity relationships in the conversion of vitamin K analogues into menaquinone-4. Substrates essential to the synthesis of menaquinone-4 in cultured human cell lines. Bioorg Med Chem. 2010;18:3116–3124.10.1016/j.bmc.2010.03.035
- Hirschmann R, Miller R, Wendler NL. The synthesis of vitamin K1. J Am Chem Soc. 1954;76:4592–4594.10.1021/ja01647a026
- Isler O, Rüegg R, Chopard-dit-Jean LH, et al. Synthese und Isolierung von vitamin K2 und isoprenologen Verbindungen. Helv Chim Acta. 1958;41:786–807.10.1002/hlca.v41:3
- Suhara Y, Hirota Y, Nakagawa K, et al. Design and synthesis of biologically active analogues of vitamin K2: evaluation of their biological activities with cultured human cell lines. Bioorg Med Chem. 2008;16:3108–3117.10.1016/j.bmc.2007.12.025
- Burnett AR, Thomson RH. Naturally occurring quinones. Part XII. Extractives from Tabebuia chrysantha nichols and other bignoniaceae. J Chem Soc C. 1968;50:850–853.10.1039/j39680000850
- Mayer H, Schudel P, Rüegg R, et al. Uber die chemie des vitamins. Die total synthese von (2R,4′R,8R)- und (2S,4′R,8′R)-α-tocophenol. Helv Chim Acta. 1963;46:650–671.10.1002/hlca.19630460225
- Termath AO, Velder J, Stemmler RT, et al. Total synthesis of (2 RS)-α-tocopherol through Ni-catalyzed 1,4-addition to a chromenone intermediate. Eur J Org Chem. 2014;2014:3337–3340.10.1002/ejoc.v2014.16
- List B, Lerner RA, Barbas CF III. Proline-catalyzed direct asymmetric aldol reactions. J Am Chem Soc. 2000;122:2395–2396.10.1021/ja994280y
- List B, Pojarliev P, Biller WT, et al. The proline-catalyzed direct asymmetric three-component mannich reaction: scope, optimization, and application to the highly enantioselective synthesis of 1,2-amino alcohols. J Am Chem Soc. 2002;124:827–833.10.1021/ja0174231
- Berger TA, Berger BK. Chromatographic resolution of 7 of 8 stereoisomers of vitamin K1 on an amylose stationary phase using supercritical fluid chromatography. Chromatographia. 2013;76:549–552.10.1007/s10337-013-2428-4
- Suga T, Ohta S, Nakai A, et al. Glycinoprenols: novel polyprenols possessing a phytyl residue from the leaves of soybean. J Org Chem. 1989;54(14):3390–3393.10.1021/jo00275a026