
Abstract
Cyclin–cyclin dependent kinase (CDK) complex is negatively regulated by interaction with CDK inhibitors (CKIs). p27 protein is a major CKI in mammals and its down-regulation correlates with malignant transformation. However, some cancer cells express p27 at normal level, suggesting not only quantitative but qualitative control of p27, although little is known about such control. We analyzed the interaction between p27 and cyclin A (CycA)-CDK complex in living human cell lines, using a split yellow fluorescent protein (YFP) system in which the YFP fluorescence solely depends on p27-CycA binding. Introduction of this system into various cancer cell lines revealed that certain cell lines show no detectable YFP fluorescence. Furthermore, these cell lines exhibited reduced p27-CycA interaction as evaluated by immunoprecipitation, while they showed normal co-localization of both proteins. These results suggest that some cancer cells are defective for efficient interaction between p27 and CycA–CDK complex due to qualitative alteration(s).
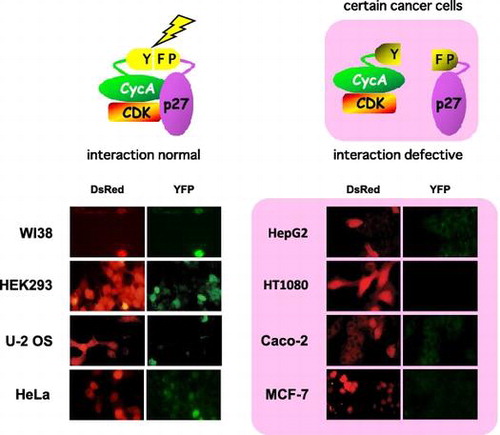
Certain cancer cells are defective for p27-cyclin A (CycA) interaction, evaluated by split YFP assay (detected as green fluorescence). DsRed, a transfection marker.
Proliferation of mammalian cells is strictly controlled by cyclin-CDK complexes which promote cell cycle progression and CDK inhibitors (CKIs) which bind to and repress those CDK complexes. p27Kip1 (hereafter p27) functions as a major CKI in proliferation control responding to extracellular proliferation signals such as serum stimulation, and has been known as a tumor suppressor since its gene disruption leads to a significant increase in body size and carcinogenic frequency, and its low expression level correlates with poor prognosis of cancer.Citation1,2) Thus, proper function of p27 is very important for normal control of cell proliferation and inactivation of p27 could promote carcinogenesis. The negative regulation of proliferation by p27 is mainly ascribed to its cellular abundance, which is controlled largely by proteolytic degradation via ubiquitin-proteasome systemCitation3) and to some extent by transcriptionalCitation4,5) and translational control.Citation6–8) Conversely, a drastic reduction in the level of p27 protein is observed in approximately 50% of human cancer of all types.Citation9)
However, some cancer cells exhibit deregulated proliferation although they express p27 protein as abundantly as normal cells. In many cancer types, p27 protein is localized in cytoplasm where it cannot interact with cyclin-CDKs, while in some cases, it is still localized in nucleus.Citation10) This points to a possibility of qualitative control of p27 function, in addition to the well-known quantitative control. Such qualitative control could include a regulation of p27-cyclin-CDK interaction by post-translational modification(s) and/or other trans-regulatory factor(s). In fact, it has been reported that in budding yeast a certain metabolite of inositol (inositol heptakisphosphate; IP7) is required for a CKI-cyclin-CDK interaction in response to environmental stimuli.Citation11,12) Although such a regulatory factor has not been found in mammalian cells, it is intriguing to speculate that certain change(s) in gene expression and/or metabolic pathway accompanied with carcinogenesis could suppress the interaction of p27 with cyclin-CDK. However, any comprehensive and comparative analysis in various cancer cells to discern whether the p27-cyclin-CDK interaction is suppressed has not been reported to date.
For such a comprehensive analysis, it must be convenient to develop an easy-to-detect assay system in living cells for p27-cyclin-CDK interaction. Bimolecular fluorescence complementation (BiFC) technique has been widely applied to detection of protein–protein interaction.Citation13–19) Regarding CKI–CDK interaction, a BiFC system has been developed for detecting KRP1 (a p27 homolog)-CDKA;1 in plants,Citation17) while no such system has been reported for the mammalian counterparts. Here, we report our construction of a BiFC system using complementing fragments of yellow fluorescent protein (referred to as split YFP hereafter) to detect the interaction of human p27 and cyclin A (CycA)-CDK complex (see Supplementary Fig. S1 online) and analysis of their interaction in various cancer cell lines.
Materials and methods
Cells and cell culture
All the cell lines used (except for WI-38) were provided by M. Nakajima. WI-38 cells were provided by RIKEN BioResource Center (Tsukuba, Japan). Cells were grown at 37 °C in Dulbecco’s modified Eagle medium (DMEM; Life Technologies) containing 10% fetal bovine serum (FBS) with 5% CO2.
Construction of the split-YFP vectors
(1) Construction of the cyclin A-YFPN fusion-expressing vector: we amplified the C-terminal region (105 bp) of human cyclin A2 cDNA by PCR using the myc-tagged cyclin A2-carrying vector pCSMTcycACitation20) as a template and the following primers:
Primer 1: 5′-TACCTCAAAGCACCACAG
Primer 2: 5′-CAGATTTAGTGTCTCTGG
And we amplified the N-terminal region (474 bp, corresponding to amino acids 1–158) of enhanced yellow fluorescent protein (EYFP) cDNA fused in-frame to a downstream stop codon (TAA) and an upstream fragment containing a linker sequence coding for 10 amino acids, [Gly-Gly-Gly-Gly-Ser]2, and 18-bp C-terminal region of the cyclin A2 cDNA by PCR, using the EYFP-expression vector pEYFP-C1 (Clontech) as a template and following primers:
Primer 3: 5′-CCAGAGACACTAAATCTGGGCGGCGGCGGCAGCGGCGGCGGCGGCAGCATGGTGAGCAAG
Primer 4: 5′-GGCTCGAGTTAGATGTTGTGGCGGATCTTG
Then we generated a recombinant fragment consisting of [cyclin A2 C-terminal region]- [10-amino acid linker sequence]-[EYFP N-terminal region (YFPN)] by PCR using the above two fragments and the primers 1 and 4. The recombinant fragment was digested with SphI and XhoI and cloned between the SphI and XhoI sites of pCSMTcycA to obtain the vector pCSMTcycA-L10-YN (Fig. (A)).
Fig. 1. Construction and verification of the split YFP system for detecting CycA-p27 interaction in living cells. (A) Construction of the split YFP vectors. PCMV, CMV promoter; MT, 6× myc epitope tag; L10, 10-amino acid linker peptide ([Gly-Gly-Gly-Gly-Ser]2). pCS2YC-L10-hp27 RXL carries five alanine-substitution mutations in the RXL (cyclin-binding) motif of p27. (B) Split-YFP fluorescence is detected specifically upon CycA-p27 interaction. HEK293 cells were transfected with 10 μg each of the indicated split-YFP (MT CycA-YFPN and YFPC-p27) expression vectors, and examined under fluorescent microscope. Nuclei were stained with DAPI. YFP signal was detected as green fluorescence. (C) MT CycA-YFPN and YFPC-p27 proteins form a functional complex. HEK293 cells were transfected with 10 μg each of the split-YFP expression vectors, and the cell extracts were subjected to anti-myc tag immunoprecipitation, followed by Western Blot Analysis using antibodies against the myc tag, p27 and CDK2, and by histone H1 kinase assay (32P-histone H1).
![Fig. 1. Construction and verification of the split YFP system for detecting CycA-p27 interaction in living cells. (A) Construction of the split YFP vectors. PCMV, CMV promoter; MT, 6× myc epitope tag; L10, 10-amino acid linker peptide ([Gly-Gly-Gly-Gly-Ser]2). pCS2YC-L10-hp27 RXL carries five alanine-substitution mutations in the RXL (cyclin-binding) motif of p27. (B) Split-YFP fluorescence is detected specifically upon CycA-p27 interaction. HEK293 cells were transfected with 10 μg each of the indicated split-YFP (MT CycA-YFPN and YFPC-p27) expression vectors, and examined under fluorescent microscope. Nuclei were stained with DAPI. YFP signal was detected as green fluorescence. (C) MT CycA-YFPN and YFPC-p27 proteins form a functional complex. HEK293 cells were transfected with 10 μg each of the split-YFP expression vectors, and the cell extracts were subjected to anti-myc tag immunoprecipitation, followed by Western Blot Analysis using antibodies against the myc tag, p27 and CDK2, and by histone H1 kinase assay (32P-histone H1).](/cms/asset/28f813bc-78b0-42a0-aa9d-c64477fe8595/tbbb_a_1391686_f0001_oc.gif)
(2) Construction of the p27-YFPC fusion protein-expressing vector: we amplified the N-terminal region (147 bp) of both human wild-type and RXL mutant (Arg30, Asn31, Leu32, Phe33, and Gly34 substituted with alanines) p27 cDNA fused in-frame to an upstream fragment containing a linker sequence coding for 10 amino acids, [Gly-Gly-Gly-Gly-Ser]2, and the 19-bp C-terminal region of the EYFP cDNA by PCR, using the p27-expression vectors pRevTRE-hp27WT and pRevTRE-hp27RXL (pRevTRE [Clontech] derivatives carrying the full-length wild-type and RXL mutant human p27 cDNA, respectively) as templates, and the following primers:
Primer 5: 5′-CATGGACGAGCTGTACAAGGGCGGCGGCGGCAGCGGCGGCGGCGGCAGCATGTCAAACGTGAGAGTG
Primer 6: 5′-GCAGTGCTTCTCCAAGTC
Also we amplified the fragment consisting of an initiation codon fused in-frame to the C-terminal region (243 bp, corresponding to amino acids 159–239) of EYFP cDNA by PCR using the pEYFP-C1 as a template and the following primers:
Primer 7: 5′-CCGGATCCATGGAGGACGGCAGCGTGCAG
Primer 8: 5′-CTTGTACAGCTCGTCCATG
We then generated recombinant fragments consisting of [EYFP C-terminal region (YFPC)]- [10-amino acid linker sequence]-[wild-type/mutant p27 N-terminal region] by PCR using the above fragments and the primers 6 and 7. The recombinant fragments containing wild-type and mutant p27 alleles were digested with BamHI and SmaI, combined with a p27 cDNA fragment downstream of SmaI (the SmaI-ClaI fragment of pRevTREhp27), and cloned between the BamHI and ClaI sites of pCS2Citation21) to obtain the vector pCS2YC-L10-hp27WT and pCS2YC-L10-hp27RXL, containing wild-type and RXL mutant p27, respectively (Fig. (A)).
Transfection of the split-YFP vectors and detection of the fluorescence
Cells were grown overnight on the cover slips (15 × 15 mm) in 6-well cell culture plates, refed with fresh DMEM containing 10% FBS, and transfected with the above vectors, according to a modified calcium-phosphate method.Citation22) 10 μg each of the split-YFP vectors and 1 μg of a DsRed-expressing vector pDsRed (Clontech) were mixed and used for the transfection per ϕ35 mm well (ca. 105 cells). 16 h after the transfection, cells were refed with fresh DMEM containing 10% FBS, and 24 h after the refeeding, cells on the cover slips were rinsed once with PBS and fixed with 3.7% paraformaldehyde in PBS for 10 min at room temperature. The cover slips were rinsed twice with PBS and stained with 0.2 μg/ml 4′,6-diamidino-2-phenylindole (DAPI) for 5 min. After rinsing with sterile water, the cover slips were air dried and mounted on slide glass with ProLong Gold mounting reagent (Life Technologies).
Western analysis
16 h after the transfection, cells were refed with fresh DMEM containing 10% FBS, and 24 h after the refeeding, cells were harvested using trypsin-EDTA solution (Life Technologies). MG132 (Sigma-Aldrich) was added at final concentration of 10 μM to the medium 8 h before the harvest. Harvested cells were rinsed once with PBS and lysed at 100 °C for 5 min in 1 × SDS sample buffer (62.5 mM Tris-HCl, pH 6.8, 2% SDS, 10% glycerol, 2.5% β-mercaptoethanol, 0.01% bromophenol blue). SDS-PAGE and Western Analysis were performed as describedCitation22) using anti-myc tag monoclonal (PL14; MBL), anti-p27 polyclonal (C-19; Santa Cruz Biotechnology), anti-Cdk2 polyclonal (M2; Santa Cruz Biotechnology), anti-CycA polyclonal,Citation23) and anti-hemagglutinin (HA) tag monoclonal (clone 16B12; Covance) antibodies.
Immunoprecipitation analysis
For detecting the interaction between endogenous CycA and HA-tagged p27 (HA-p27) proteins, pCSHA-p27, a CS2-based vector carrying HA-tagged human p27 cDNA, was transfected as above into various cell lines. To equalize the HA-p27 expression levels, different amount [1–5 μg, see the legend for Fig. (B)] of pCSHA-p27 was used for each of the cell lines based on their transfection efficiencies and expression capabilities. 16 h after the transfection, cells were refed with fresh DMEM containing 10% FBS, and 24 h after the refeeding, cells were rinsed once with Tris-buffered saline (pH 7.4) and lysed on ice by directly adding RIPA buffer (10 mM Tris-HCl, pH 7.4, 0.15 M NaCl, 0.5% Nonidet P-40, 50 mM NaF, 1 mM Na-vanadate, 1 mM PMSF, 10 μg/ml each of aprotinin, leupeptin, and pepstatin) to the culture dish. Immunoprecipitation and histone H1 kinase assay were performed as describedCitation22) using anti-myc tag monoclonal (PL14) or anti-CycA polyclonalCitation23) antibodies.
Immunofluorescence analysis
Cells grown overnight on the cover slips (15 × 15 mm) in 6-well cell culture plates were transfected as above with pCSMTcycA and pCS2hp27, a CS2-based vector carrying untagged human p27 cDNA. 16 h after the transfection, cells were refed with fresh DMEM containing 10% FBS, and 24 h after the refeeding, cells on the cover slips were rinsed once with phosphate-buffered saline (PBS) and fixed with 4% paraformaldehyde in PBS for 10 min at room temperature. Immunofluorescence detection was performed as describedCitation20) using anti-myc tag monoclonal (PL14) and anti-p27 polyclonal (C-19) antibodies and appropriate secondary antibodies (anti-mouse IgG-FITC and anti-rabbit IgG-Rhodamine; Roche Diagnostics).
Results and discussion
Construction of the split YFP vectors for detection of CycA-p27 interaction in living cells
We chose CycA as a model for p27-interacting cyclins since the crystal structure model of the CycA-Cdk2-p27 tertiary complex has been resolved.Citation24) The model has shown that the C-terminus of CycA protein and the N-terminus of p27 protein are located in close vicinity in the complex (see Supplementary Fig. S1 online). Therefore, we designed the N-terminal YFP domain (YFPN) and the C-terminal YFP domain (YFPC) to be fused to the C-terminus of CycA and the N-terminus of p27, respectively, via a short and flexible linker peptide consisting of [Gly-Gly-Gly-Gly-Ser]2, which has been used successfully in other split YFP systemsCitation15,18) (Fig. (A)). The CycA-YFPN fusion gene was 5′-tagged with 6× myc tag derived from the vector CSMT.Citation21) The resulting myc-tagged CycA-YFPN and YFPC-p27 fusion genes were cloned downstream of the cytomegalovirus immediately early promoter (CMV promoter).Citation21) We also constructed the YFPC-p27 fusion vector carrying missense mutations (Arg30, Asn31, Leu32, Phe33, and Gly34 substituted with alanines; RXL mutant) in the RXL motif of p27, which is essential for binding to CycACitation24) (Fig. (A)). Since the p27 RXL mutant cannot bind to CycA, we used this construct as a negative control.
We transfected human embryonic kidney cell line (transformed with Adenovirus) HEK293 with these split YFP vectors and examined for YFP fluorescence. As shown in Fig. (B), the complemented YFP fluorescence was detected only when both CycA-YFPN and YFPC-p27 (wild type; p27 WT) fusion vectors were co-transfected. Co-transfection of CycA-YFPN and YFPC-p27 RXL mutant (p27 RXL) yielded no YFP signal, indicating that the YFP fluorescence is dependent on CycA-p27 interaction. Next, we asked if these fusion proteins form a functional complex. Cell extracts from HEK293 cells transfected with the split YFP vectors were immunoprecipitated with anti-myc tag antibody and subjected to Western Analysis and histone H1 kinase assay for evaluating CDK activity (Fig. (C)). When CycA-YFPN and YFPC-p27 WT fusion vectors were co-transfected, YFPC-p27 and endogenous CDK2 proteins were co-immunoprecipitated with myc-tagged CycA-YFPN (Fig. (C), lane 8), demonstrating that these fusion proteins form a tertiary complex with CDK2. Moreover, CDK2 was co-immunoprecipitated with CycA-YFPN in the absence of YFPC-p27 WT and the immunoprecipitate exhibited a substantial activity of histone H1 kinase (Fig. (C), lane 6), which was abolished in the complex containing YFPC-p27 WT (lane 8). These indicate that the two fusion proteins form a functionally relevant complex; i.e. CycA-YFPN forms an active complex with CDK2, which in turn is inactivated by binding of YFPC-p27 WT. Therefore, we concluded our split YFP system is useful for easy and specific detection of functional interaction between p27 and CycA–CDK complex in living cells.
YFPC-p27 RXL mutant protein did not bind to CycA-YFPN (Fig. (C), lane 9), which verifies the mutant phenotype and is consistent with the result shown in Fig. (B). We also observed a relatively weak association of CDK2 with CycA-YFPN in the absence of YFPC-p27 WT, compared to the case in the presence of YFPC-p27 WT (Fig. (C), lanes 6 and 8). This might derive from a steric hindrance caused by the YFPN domain fused to CycA, and binding of YFPC-p27 WT might circumvent it by bridging CycA and CDK2 via p27 domainCitation24) and also via stable association of the split YFP domains.
Evaluation of CycA-p27 interaction by split YFP system in various human cancer cell lines
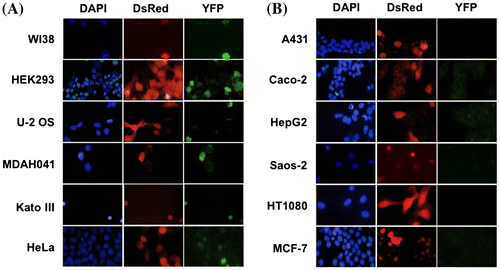
We then introduced our split YFP system (CycA-YFPN and YFPC-p27 WT expression vectors) into various cancer cell lines to assess the efficacy of interaction between p27 and CycA among the cell lines. U-2 OS (human osteosarcoma), MDAH041 (human Li-Fraumeni syndrome fibroblasts), KATO III (human gastric carcinoma), HeLa (human uterine cervix carcinoma), A431 (human epidermoid carcinoma), Caco-2 (human colorectal adenocarcinoma), HepG2 (human hepatocellular carcinoma), Saos-2 (human osteosarcoma), HT1080 (human fibrosarcoma), MCF7 (human breast adenocarcinoma) cells, along with WI-38 (human normal diploid fibroblasts) and HEK293 cells, were co-transfected with the split YFP vectors and a DsRed (a marker for transfected cells)-expressing vector. As shown in Fig. (A), cancer cell lines U-2 OS, MDAH041, Kato III, and Hela, as well as human normal cell line WI-38 and HEK293 cells, exhibited YFP fluorescence in the DsRed-positive cells. By contrast, other cancer cell lines A431, Caco-2, HepG2, Saos-2, HT1080, and MCF7 showed no detectable YFP signal in the DsRed-positive cells (Fig. (B)), suggesting the interaction of p27 and CycA-CDK is reduced in these cells.
Fig. 2. Evaluation of CycA-p27 interaction by split-YFP fluorescence in various cancer cell lines. WI-38, HEK293, U-2 OS, MDAH041, Kato Ⅲ, HeLa (A) and A431, Caco-2, HepG2, Saos-2, HT1080, MCF-7 (B) cells were co-transfected with 10 μg each of the split-YFP expression vectors and 1 μg of DsRed-expressing vector, and examined under fluorescent microscope. Nuclei were stained with DAPI. Note that the cells in (B) showed no detectable YFP signal.
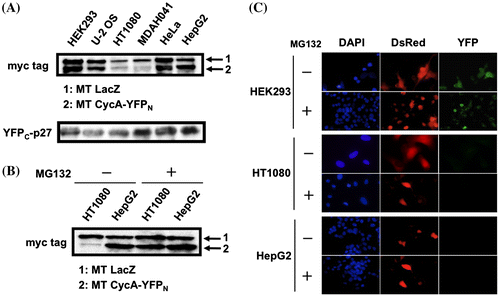
Since efficiency of the transfection using a conventional Ca-phosphate method varies among different cell lines, we selected six cell lines (HEK293, U-2 OS, HT-1080, MDAH041, HeLa, and HepG2), which exhibit relatively high efficiencies in Ca-phosphate transfection, for the further analyses. First, we checked the expression level of both YFP fusion proteins in these cell lines when co-transfected with myc-tagged LacZ-expressing vector as a control. As shown in Fig. (A), CycA-YFPN protein was expressed at different levels, relatively at a higher level in HepG2 and a lower level in HT1080 and MDAH041. YFPC-p27 WT protein was expressed almost equivalently, except for relatively high-level expression in MDAH041 and HepG2 cells. The YFP fluorescence in these cells, however, did not correlate to the different expression levels of both fusion proteins since MDAH041 cells exhibited the YFP signal in spite of a low CycA-YFPN expression while HepG2 cells did not even with a high-level expression of both fusion proteins (Figs. and ). We further analyzed the relationship between the expression level of the fusion proteins and the YFP fluorescence in HT1080 cells, which showed a reduced CycA-YFPN expression as low as in MDAH041. Fig. (B) shows that the reduced CycA-YFPN expression in HT1080 was restored by the addition of a potent proteasome inhibitor MG132, revealing that the reduced expression is due to an increased proteasomal degradation of CycA-YFPN protein. Nonetheless, as shown in Fig. (C), the addition of MG132 did not restore the YFP fluorescence in HT1080, as well as in HepG2. Therefore, the defect for split YFP complementation in these cells is apparently not ascribed to the low expression level of the YFP fusion proteins.
Fig. 3. Absence of YFP signal in HT1080 and HepG2 cells is independent of the expression level of split-YFP fusion proteins. (A) HEK293, U-2 OS, HT1080, MDAH041, HeLa, HepG2 cells were co-transfected with 10 μg each of the split-YFP and myc-tagged (MT) LacZ expression vectors. The cell extracts were immunoblotted using antibodies against myc tag and p27. (B) HT1080 and HepG2 cells were co-transfected with 10 μg each of the split-YFP and MT LacZ expression vectors in the absence and presence of 10 μM proteasome inhibitor MG132 and immunoblotted using antibodies against myc tag. (C) HEK293, HT1080 and HepG2 cells were co-transfected with 10 μg each of the split-YFP and 1 μg of DsRed-expressing vectors in the absence and presence of 10 μM MG132, and examined under fluorescent microscope. Nuclei were stained with DAPI.
CycA-p27 interaction is reduced in certain cancer cell lines
We then analyzed the interaction of the YFP fusion proteins in those six cell lines by immunoprecipitation. Cells were co-transfected with both split YFP vectors (in the presence of MG132 for HT1080 and MDAH041 cells to compensate for the CycA-YFPN expression level), and the cell extracts were immunoprecipitated with anti-myc tag antibody. As shown in Fig. (A), we observed relatively small amounts of YFPC-p27 protein were co-immunoprecipitated with myc-tagged CycA-YFPN in HT1080 and HepG2 cells, suggesting that the defect for split YFP complementation in these cells is due to a reduced interaction between the YFP fusion proteins.
Fig. 4. Co-immunoprecipitation of CycA and p27 proteins in various cancer cell lines. (A) HEK293, U-2 OS, HT1080, MDAH041, HeLa, and HepG2 cells were co-transfected with 10 μg each of the split-YFP expression vectors (10 μM MG132 was added for HT1080 and MDAH041), and the cell extracts were subjected to anti-myc tag immunoprecipitation, followed by Western Analysis using antibodies against myc tag and p27. (B) The above six cell lines were transfected with 1 μg (HEK293), 2 μg (HT1080), 3 μg (U-2 OS, HeLa) or 5 μg (MDAH041, HepG2) of HA-p27 expression vector to equalize the expression level, and the cell extracts were subjected to anti-CycA immunoprecipitation, followed by Western Analysis using antibodies against CycA, HA tag, and Cdk2. See Supplementary Fig. S4 for quantified data.
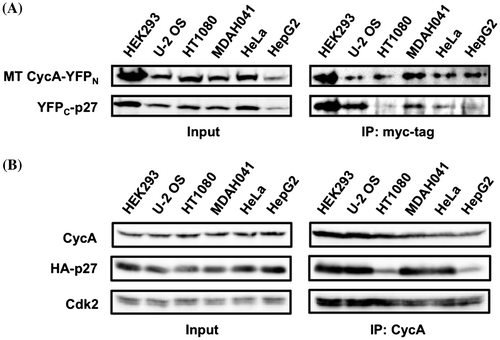
These results point to a possibility that HT1080 and HepG2 cells are intrinsically defective for efficient interaction between p27 and CycA-CDK, which prompted us to evaluate the interaction of native CycA and p27 proteins, which are not fused to YFP domains. We checked the endogenous CycA and p27 protein levels in those six cell lines and observed no obvious difference in CycA level but a reduced level of p27 protein per cell in HEK293, MDAH041, and HeLa (see Supplementary Fig. S2 online). Therefore, we transfected a various amount of HA-tagged p27 (HA-p27) expression vector into those cell lines to equalize the HA-p27 level. Fig. (B) shows that the overall expression levels of endogenous CycA and exogenous HA-p27 proteins (“Input”) in the six cell lines are equivalent. These cell extracts were immunoprecipitated with anti-CycA antibody. As shown in Fig. (B) (“IP: CycA”), HA-p27 protein was co-immunoprecipitated with endogenous CycA and Cdk2 at comparable levels in HEK293, U-2 OS, MDAH041, and HeLa cells, but at significantly reduced levels in HT1080 and HepG2 (see Supplementary Fig. S4 online). These results suggest that the interaction between p27 and CycA proteins is impaired in these cell lines.
In this report, we developed a simple split YFP system for detecting p27 and CycA interaction in living cells as YFP fluorescence signal, and observed that some cancer cell lines do not exhibit the fluorescence signal. Among these cell lines, HT1080 and HepG2 was found defective for the YFP signal independently of the expression level of the split YFP fusion proteins, and moreover, they showed a reduced interaction between p27 and CycA proteins in co-immunoprecipitation assay. One possible cause of this reduced interaction is mislocalization of the exogenous p27 or CycA protein in these cells. p27 must be localized in nucleus to function as a CKI and its cytoplasmic localization has been frequently observed in cancer cells.Citation10) However, immunofluorescence analysis revealed that subcellular localizations of both the exogenous p27 and CycA proteins were almost exclusively nuclear in various cell lines including HT1080 and HepG2 (see Supplementary Fig. S3 online). Therefore, the reduced p27-CycA interaction in these cells is most likely due to qualitative alteration(s) of either p27 or CycA protein via post-translational modification(s) and/or other trans-regulatory factor(s). Although little is known about such qualitative alteration(s) and the underlying mechanism, it would be important to evaluate the frequency of this defect in p27-CycA interaction among more varieties of cancer cell lines. Our split YFP system would be useful for that purpose since this assay system can readily detect the interaction in living cells and can be applied to high-throughput analyses.
Author contributions
Taku Chibazakura initiated the project and the conception. Yuichi Asano and Taku Chibazakura performed all the experiments and analyzed the data. Taku Chibazakura wrote the article. All authors read and approved the manuscript.
Disclosure statement
No potential conflict of interest was reported by the authors.
Funding
This work was supported by Nodai Advanced Research Project (Tokyo University of Agriculture).
Supplementary material
The supplementary material for this paper is available online at https://doi.org/10.1080/09168451.2017.1391686.
Chibazakura___Asano_Suppl_BBB__final_.pdf
Download PDF (5.1 MB)Acknowledgments
We thank Motowo Nakjima and RIKEN BioResource Center for materials, Ayaka Takei for her technical assistance.
Related Research Data
References
- Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev. 1999;13:1501–1512.10.1101/gad.13.12.1501
- Besson A, Dowdy SF, Roberts JM. CDK inhibitors: cell cycle regulators and beyond. Dev Cell. 2008;14:159–169.10.1016/j.devcel.2008.01.013
- Lu Z, Hunter T. Ubiquitylation and proteasomal degradation of the p21(Cip1), p27(Kip1) and p57(Kip2) CDK inhibitors. Cell Cycle. 2010;9:2342–2352.10.4161/cc.9.12.11988
- Servant MJ, Coulombe P, Turgeon B, et al. Differential regulation of p27(Kip1) expression by mitogenic and hypertrophic factors: involvement of transcriptional and posttranscriptional mechanisms. J Cell Biol. 2000;148:543–556.10.1083/jcb.148.3.543
- Garrett-Engele CM, Tasch MA, Hwang HC, et al. A mechanism misregulating p27 in tumors discovered in a functional genomic screen. PLoS Genet. 2007;3:e219.10.1371/journal.pgen.0030219
- Hengst L, Reed SI. Translational control of p27Kip1 accumulation during the cell cycle. Science. 1996;271:1861–1864.10.1126/science.271.5257.1861
- Millard SS, Yan JS, Nguyen H, et al. Enhanced ribosomal association of p27(Kip1) mRNA is a mechanism contributing to accumulation during growth arrest. J Biol Chem. 1997;272:7093–7098.10.1074/jbc.272.11.7093
- Le Sage C, Nagel R, Agami R. Diverse ways to control p27Kip1 function: miRNAs come into play. Cell Cycle. 2007;6:2742–2749.10.4161/cc.6.22.4900
- Viglietto G, Motti ML, Fusco A. Understanding p27(kip1) deregulation in cancer: down-regulation or mislocalization. Cell Cycle. 2002;1:394–400.10.4161/cc.1.6.263
- Slingerland J, Pagano M. Regulation of the Cdk inhibitor p27 and its deregulation in cancer. J Cell Physiol. 2000;183:10–17.10.1002/(ISSN)1097-4652
- Lee YS, Mulugu S, York JD, et al. Regulation of a cyclin-CDK-CDK inhibitor complex by inositol pyrophosphates. Science. 2007;316:109–112.10.1126/science.1139080
- Lee YS, Huang K, Quiocho FA, et al. Molecular basis of cyclin-CDK-CKI regulation by reversible binding of an inositol pyrophosphate. Nat Chem Biol. 2008;4:25–32.10.1038/nchembio.2007.52
- Hu CD, Chinenov Y, Kerppola TK. Visualization of interactions among bZIP and Rel family proteins in living cells using bimolecular fluorescence complementation. Mol Cell. 2002;9:789–798.10.1016/S1097-2765(02)00496-3
- Grinberg AV, Hu CD, Kerppola TK. Visualization of Myc/Max/Mad family dimers and the competition for dimerization in living cells. Mol Cell Biol. 2004;24:4294–4308.10.1128/MCB.24.10.4294-4308.2004
- Remy I, Montmarquette A, Michnick SW. PKB/Akt modulates TGF-beta signalling through a direct interaction with Smad3. Nat Cell Biol. 2004;6:358–365.10.1038/ncb1113
- Arabi A, Wu S, Ridderstrale K, et al. C-Myc associates with ribosomal DNA and activates RNA polymerase I transcription. Nat Cell Biol. 2005;7:303–310.10.1038/ncb1225
- Jakoby MJ, Weinl C, Pusch S, et al. Analysis of the subcellular localization, function, and proteolytic control of the arabidopsis cyclin-dependent kinase inhibitor ICK1/KRP1. Plant Physiol. 2006;141:1293–1305.10.1104/pp.106.081406
- Qi Q, Sahu N, August A. Tec kinase itk forms membrane clusters specifically in the vicinity of recruiting receptors. J Biol Chem. 2006;281:38529–38534.10.1074/jbc.M609180200
- Wang KZ, Wara-Aswapati N, Boch JA, et al. TRAF6 activation of PI 3-kinase-dependent cytoskeletal changes is cooperative with Ras and is mediated by an interaction with cytoplasmic Src. J Cell Sci. 2006;119:1579–1591.10.1242/jcs.02889
- Chibazakura T, McGrew SG, Cooper JA, et al. Regulation of cyclin-dependent kinase activity during mitotic exit and maintenance of genome stability by p21, p27, and p107. Proc Natl Acad Sci USA. 2004;101:4465–4470.10.1073/pnas.0400655101
- Turner DL, Weintraub H. Expression of achaete-scute homolog 3 in Xenopus embryos converts ectodermal cells to a neural fate. Genes Dev. 1994;8:1434–1447.10.1101/gad.8.12.1434
- Clurman BE, Sheaff RJ, Thress K, et al. Turnover of cyclin E by the ubiquitin-proteasome pathway is regulated by cdk2 binding and cyclin phosphorylation. Genes Dev. 1996;10:1979–1990.10.1101/gad.10.16.1979
- Marraccino RL, Firpo EJ, Roberts JM. Activation of the p34 CDC2 protein kinase at the start of S phase in the human cell cycle. Mol Biol Cell. 1992;3:389–401.10.1091/mbc.3.4.389
- Russo AA, Jeffrey PD, Patten AK, et al. Crystal structure of the p27Kip1 cyclin-dependent-kinase inhibitor bound to the cyclin A-Cdk2 complex. Nature. 1996;382:325–331.10.1038/382325a0