
Abstract
The secondary metabolite aphidicolin has previously been produced by Aspergillus oryzae after the heterologous expression of four biosynthetic enzymes isolated from Phoma betae. In this study, we examined the subcellular localization of aphidicolin biosynthetic enzymes in A. oryzae. Fusion of green fluorescent protein to each enzyme showed that geranylgeranyl diphosphate synthase and terpene cyclase are localized to the cytoplasm and the two monooxygenases (PbP450-1 and PbP450-2) are localized to the endoplasmic reticulum (ER). Protease protection assays revealed that the catalytic domain of both PbP450s was cytoplasmic. Deletion of transmembrane domains from both PbP450s resulted in the loss of ER localization. Particularly, a PbP450-1 mutant lacking the transmembrane domain was localized to dot-like structures, but did not colocalize with any known organelle markers. Aphidicolin biosynthesis was nearly abrogated by deletion of the transmembrane domain from PbP450-1. These results suggest that ER localization of PbP450-1 is important for aphidicolin biosynthesis.
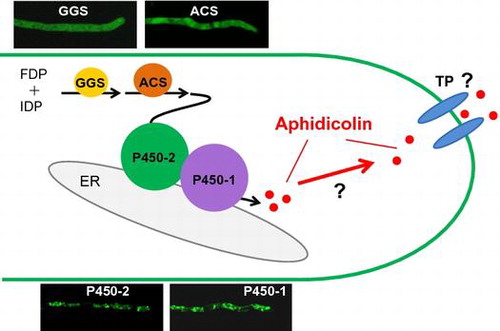
Schematic representation of localization of aphidicolin biosynthetic machinery in A. oryzae.
Aspergillus oryzae is a filamentous fungus that has long been used for the production of traditional Japanese foods such as sake, shoyu (soy sauce), and miso (soybean paste) [Citation1]. A. oryzae has low productivity of its own secondary metabolites and is suitable as a heterologous host for highly pure secondary metabolite production. Therefore, a wide variety of fungal biosynthetic machineries for secondary metabolite production has been expressed in A. oryzae [Citation2–11].
Biosynthesis of some fungal secondary metabolites occurs in specific organelles and biosynthesis of aflatoxin and penicillin has been well characterized [Citation14,15]. In Aspergillus parasiticus, although the early step of aflatoxin synthesis occurs in the peroxisome, three aflatoxin synthetic enzymes (Nor-1, Ver-1, and OmtA) are localized to vesicles and vacuoles [Citation16], and the last two enzymatic steps are completed in vesicles called aflatoxisomes [Citation17]. In Penicillium chrysogenum, the final step of penicillin biosynthesis also occurs in the peroxisome [Citation18]. Interestingly, overexpression of peroxisomal proteins increases penicillin production in P. chrysogenum [Citation19,20]. Therefore, subcellular localization analysis of biosynthetic enzymes is important for improving the production of fungal secondary metabolites.
Aphidicolin is a DNA polymerase inhibitor [Citation12] biosynthesized by geranylgeranyl diphosphate synthase (PbGGS), aphidicolan-16β-ol synthase (PbACS), and two monooxygenases (PbP450-1 and PbP450-2) in Phoma betae [Citation13] (Figure ). Overexpression of these four biosynthetic genes in A. oryzae led to the production of 0.33 mg/L aphidicolin, but it was much lower than P. betae (16.2 mg/L) [Citation3]. Despite overexpression of biosynthetic genes, the low production of aphidicolin by A. oryzae was attributed to mislocalization of the biosynthetic enzymes. Therefore, in this study, we examined the subcellular localization of aphidicolin biosynthetic enzymes expressed in A. oryzae. We also determined membrane topology and examined the effect of transmembrane domain deletion on the subcellular localization of PbP450-1 and PbP450-2 and subsequent effects on aphidicolin production to understand the importance of monooxygenase localization for aphidicolin production in A. oryzae.
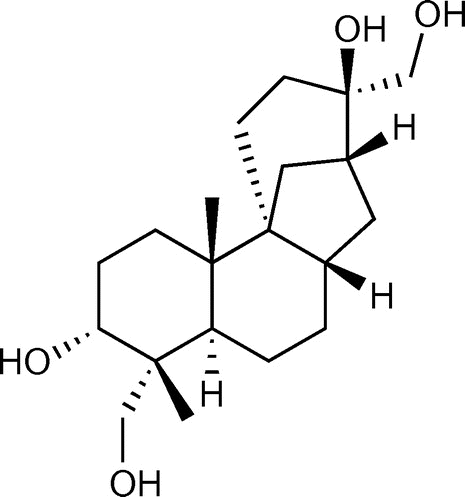
Figure 1. Chemical structure of aphidicolin.
Materials and methods
Strains and media
The A. oryzae niaD300 strain (niaD−) [Citation21], derived from the wild-type strain A. oryzae RIB40 (National Research Institute of Brewing Stock Culture), was used as a recipient strain for expression of GFP-fused aphidicolin biosynthetic enzymes. An A. oryzae strain carrying overexpression cassettes of PbACS, PbGGS, and PbP450-2 [Citation3] was used as a host for overexpression of the transporter gene (PbTP) [Citation13], and the resultant strain (amyBp-PbACS::argB; amyBp-PbGGS::ptrA; amyBp-PbP450-2::sC; amyBp-PbTP::adeA; niaD−) was used for overexpression of PbP450-1. Plasmids were constructed and propagated in Escherichia coli DH5α and HST08 (Takara Bio Inc., Shiga, Japan). The minimal medium (MM) for A. oryzae cultivation was Czapek-Dox medium with 0.6% NaNO3, 0.05% KCl, 0.2% KH2PO4, 0.05% MgSO4, 1% glucose, and trace amounts of FeSO4, ZnSO4, CuSO4, MnSO4, Na2B4O7, and (NH4)6Mo7O24.
Construction of plasmid DNAs
The nucleotide sequences of all primers used in this study are shown in Table S1. To construct a plasmid for N-terminally GFP-tagged enzymes, a DNA fragment approximately 1 kb upstream of malP [Citation22] was amplified from A. oryzae RIB40 genomic DNA by PCR using PmalPupsen and PmalPantiSalI primers. The amplified fragment was digested with PstI and SalI and replaced with the glaA142 promoter of plasmid pNGA142TglaA, yielding pNPmalPTglaA. The plasmid pNGA142TglaA is a modified pNGA142 [Citation23], with the agdA terminator replaced by the glaA terminator amplified by PCR using the TglaAsenXbaI and TglaAantiSmaI primers. GFP was amplified from the plasmid pAGAR-F [Citation24] by PCR with GFPsenSalI and GFP5GAantiNotI primers. The amplified fragment was digested with SalI and NotI and inserted into pNPmalPTglaA, yielding the plasmid pNMPG(N) with niaD as a fungal selectable marker. For N-terminally GFP-tagged PbGGS, PbGGS was amplified from the plasmid pTAex3-PbGGS [Citation3] by PCR with the primers GGS-fw-NotI and GGS-rv-SpeI. The amplified fragment was digested with NotI and SpeI and inserted into NotI/SpeI-digested pNMPG(N).
For C-terminally GFP-tagged PbACS, PbACS was amplified from the plasmid pTAex3-PbACS [Citation3] by PCR using the ACS-fw-NotI and ACS-rv-SpeI primers and inserted into NotI/SpeI-digested pNMPG(C) [Tanaka et al., unpublished].
For C-terminally GFP-tagged PbP450s, PbP450-1 was amplified from the plasmid pAdeA-PamyB/P450-1/TamyB [Citation3] by PCR using the P450-1-fw-InF and P450-1-rv-InF primers and inserted into SalI/NotI-digested pNMPG(C) by the In-Fusion® HD Cloning Kit (Takara Bio Inc.) according to the manufacturer’s instructions. PbP450-2 was amplified from the plasmid pUSA-PbP450-2 [Citation3] by PCR using the P450-2-fw-SalI and P450-2-rv-NotI primers and inserted into SalI/NotI-digested pNMPG(C).
PbP450 transmembrane domains were predicted by Transmembrane Helix Prediction (http://www.cbs.dtu.dk/services/TMHMM/) [Citation25]. For GFP-PbP450s, PbP450-1 lacking the sequence for an N-terminal transmembrane region (PbP450-1∆TM) was amplified from the plasmid pAdeA-PamyB/P450-1/TamyB by PCR using the P450-1(∆SS)-fw-SpeI and P450-1(∆SS)-rv-SpeI primers and inserted into SpeI-digested pNMPG(C). PbP450-2 lacking the sequence for a transmembrane region was amplified by fusion PCR using the primers P450-2(∆TM)-fw-InF, (∆TM)-N ter.-rv, (∆TM)-C ter.-fw, and P450-2(∆TM)-rv-InF, and the expression plasmid of PbP450-2-GFP (pNMP-P450-2-gfp) as a template. The amplified PCR product was inserted into MscI/NotI-digested pNMP-P450-2-gfp by the In-Fusion® HD Cloning Kit.
For fusion of mCherry at the N-terminus of organelle markers, a DNA fragment of mCherry was amplified from the plasmid pRFP-Ape1 (pTS559) [Citation26] by PCR using the GFPsenSalI and GFP5GAantiNotI primers. The amplified fragment was digested with SalI and NotI and inserted into SalI/NotI-digested pNPmalP [Citation27], yielding pNMPm(N) for fusion of mCherry at the N-terminus. For replacement of niaD with ptrA, the DNA fragment of malP promoter-mCherry-glaA terminator was amplified from pNMPm(N) by PCR and inserted into PstI/SmaI-digested pPTRI (Takara Bio Inc.) by the In-Fusion® HD Cloning Kit, yielding pPMPm(N). The DNA fragments of Use1 (AB279877), Sed5 (AB099696), Nyv1 (AB279871), and Tlg2 (AB279864) were amplified from A. oryzae genomic DNA by PCR using specific primers (Table S1) and inserted into pPMPm(N) by ligation or the In-Fusion® HD Cloning Kit. A DNA fragment of mCherry with nucleotides for a peroxisomal targeting signal (Ser-Arg-Leu) at the 3′-terminus was amplified from pPMPm(N) by PCR using the primers PTS1-fw-EcoRV and PTS1-rv-NotI. The amplified fragment was digested with EcoRV and NotI and inserted into EcoRV/NotI-digested pPMPm(N). For fusion of mCherry at the C-terminus, the DNA fragment of mCherry was amplified from the plasmid pTS559 by PCR using the mCherry5GAsenSpeI and CmCherry-antiXbaI primers and inserted into SpeI/XbaI-digested pNPmalP. The resultant plasmid was digested with SbfI and XmaI to obtain the DNA fragment of malP promoter-mCherry-agdA terminator and inserted into PstI/XmaI-digested pPTRI, yielding pPMPm(C). The DNA fragments of histone H2B (AO090020000006) and PlaA (AO090026000448) were amplified by PCR using A. oryzae genomic or complementary DNA as a template and the primers shown in Table S1. The amplified fragments were inserted into pPMPm(C) by the In-Fusion® HD Cloning Kit. To construct the expression plasmids of mCherry-fused PbP450-1 and PbP450-2, DNA fragments of PbP450-1 and Pb450-2 were amplified by PCR using the primer sets P450-1-fw-InF2 + P450-1-rv-InF2 and P450-2-fw-InF + P450-2-rv-InF, respectively. The amplified fragments were inserted into pPMPm(C) by the In-Fusion® HD Cloning Kit.
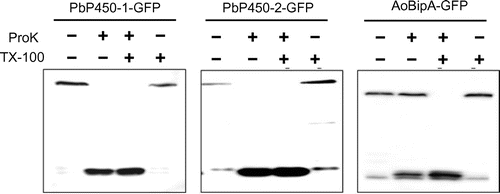
The plasmid for expression of BipA-GFP by malP promoter was constructed by amplifying a DNA fragment of BipA-GFP from plasmid pUNABG [Citation28] with bipA-EGFP-fw-SalI and bipA-EGFP-rv-SpeI primers and inserted into SalI/SpeI-digested pNMPG(N).
For overexpression of PbTP by the amyB promoter, PbTP was amplified from P. betae genomic DNA with TP-fw-NheI and TP-rv-SpeI primers and inserted into NheI/SpeI-digested pAANA1 [Citation29] containing adeA as a fungal selectable marker.
For overexpression of PbP450-1 and PbP450-1∆TM, DNA fragments of PbP450-1 and PbP450-1∆TM were amplified using the primer sets P450-1-fw-InF3 + P450-1-rv-InF3 and P450-1(∆SS)-fw-InF2 + P450-1-rv-InF3, respectively. The amplified fragments were inserted into NotI/SpeI-digested pAPTLN [Citation30] by the In-Fusion® HD Cloning Kit. These plasmids contain niaD as a fungal selectable marker.
Fungal transformation
Plasmid DNAs were introduced into A. oryzae by the PEG-protoplast method [Citation31].
Fluorescence microscopy
Approximately 1 × 105 A. oryzae conidiospores were inoculated onto coverslips (18 × 18 mm) dipped in 500 μL of MM with 1% maltose as the sole carbon source and grown at 30 °C for 20 h. Hyphae were examined by a laser scanning confocal microscope (FV1000-D IX81; Olympus Corp., Tokyo, Japan) at 3000 × magnification.
Protease protection assay
A. oryzae strains expressing GFP fusion proteins were grown for 36 h in liquid MM with 1% casamino acids as the carbon source, followed by the addition of maltose to a final concentration of 1%. After further incubation for 6 h to induce the expression of GFP fusion proteins, the mycelia were harvested by filtration through Miracloth (Merck Millipore, Billerica, MA, USA) and suspended in HSF10 buffer (50 mM HEPES, 200 mM sucrose, 10% Ficoll® PM400 [GE Healthcare UK Ltd., Buckinghamshire, UK], pH 7.3). Mycelia were disrupted by the Multi-beads shocker® (Yasui Kikai, Osaka, Japan) with glass beads, and the debris was removed by filtration through Miracloth. Homogenates were suspended in HK buffer (50 mM HEPES, 100 mM potassium acetate, pH 7.3) containing either 10 μg/mL proteinase K and 1% Triton X-100 or distilled water. After incubation on ice for 15 min, trichloroacetic acid (TCA) was added at a final concentration of 10% to stop the reaction, followed by further incubation on ice for 10 min. After centrifugation at 20,400 ×g for 10 min at 4 °C, the pellet was washed twice with chilled 80% acetone. The pellet was resuspended in Laemmli sample buffer [Citation32], followed by boiling for 5 min. The samples were subjected to western blot analysis with anti-GFP antibody mFX75 (Wako Pure Chemical Industries Ltd., Osaka, Japan).
Estimation of the aphidicolin production
A. oryzae strains were grown in Erlenmeyer flasks with 50 mL MM with 1% tryptone and 3% maltose as the carbon source for 6 days. Then, 20 mL of acetone was added and flasks were incubated, with shaking, for 30 min. Then, 30 mL of ethyl acetate was added to the flask with saturated NaCl, followed by vigorous shaking for 1 h. Debris was removed by filtration through Miracloth and the organic layer was dried over Na2SO4. The amount of extracted aphidicolin was estimated by thin-layer chromatography (CHCl3:methanol = 85:15). TLC plates were sprayed with the anisaldehyde reagent (15 g p-methoxybenzaldehyde, 250 mL ethanol, 2.5 mL conc. H2SO4) and heated at 100 °C for 15 min.
Extraction of GFP-fused PbP450s and mutants
A. oryzae strains expressing GFP-fused PbP450 mutants that lacked the transmembrane region were grown for 36 h in liquid MM containing 1% casamino acids as the carbon source, followed by the addition of maltose to a final concentration of 1%. After further incubation for 6 h, mycelia were harvested and ground to a fine powder in liquid nitrogen using a mortar and pestle. Powdered mycelia were suspended in protein extraction buffer (50 mM Tris–HCl [pH 8.0], 1% Triton X-100, 100 mM NaCl, 200 μM PMSF, 200 μM pepstatin A, complete EDTA-free protease inhibitor [Sigma-Aldrich Co. LLC, St. Louis, MO, USA], 5 mM EDTA, and 0.5% SDS) and incubated at room temperature for 15 min. After centrifugation at 3000 ×g for 1 min, the supernatant was collected and concentrated by TCA precipitation. The pellet was suspended in Laemmli sample buffer, followed by boiling for 5 min.
Results
Subcellular localization of aphidicolin biosynthetic enzymes in A. oryzae
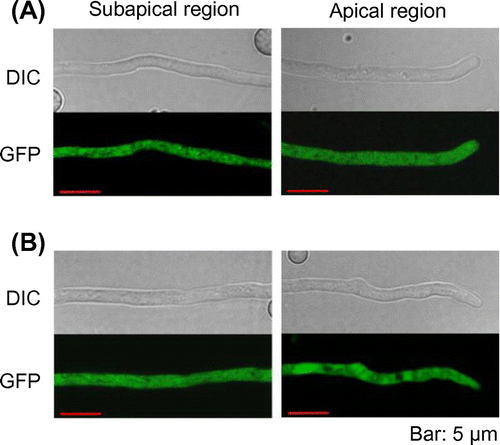
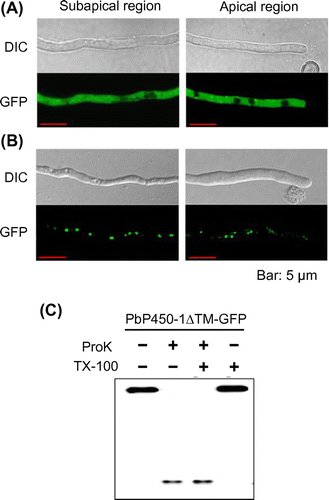
To determine subcellular localization of PbGGS and PbACS, GFP was fused to the C-terminus of both enzymes. In this study, the promoter of the maltose permease gene (malP) [Citation22], which allows gene expression at moderate levels, was used for the expression of GFP-fused proteins to avoid mislocalization of the enzymes due to overexpression. After incubation in maltose-containing medium for 20 h, GFP fluorescence in hyphae was examined by laser scanning confocal microscopy. Fluorescence in the PbACS-GFP expressing strain was seen clearly in the cytoplasm (Figure (A)). In contrast, fluorescence of PbGGS-GFP could not be observed (data not shown). Thus, we constructed an additional A. oryzae strain expressing PbGGS with GFP at the N-terminus (GFP-PbGGS). In this strain, GFP fluorescence was clearly observed in the cytoplasm (Figure (B)). These results indicated that both PbGGS and PbACS were localized in the cytoplasm in A. oryzae.
Figure 2. Subcellular localization of PbGGS and PbACS in A. oryzae. Hyphae of strains expressing GFP-PbGGS (A) and PbACS-GFP (B) were grown for 20 h in minimal medium (MM) containing 1% maltose as the sole carbon source. GFP fluorescence at subapical and apical regions was observed by confocal microscopy.
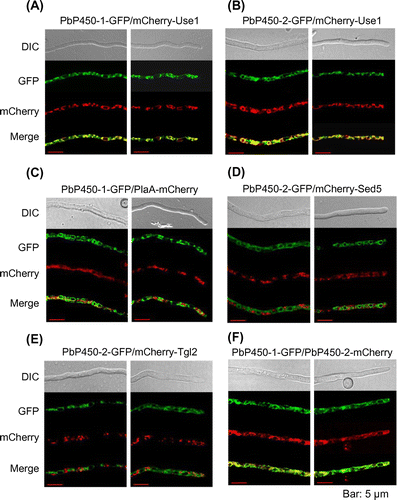
When the amino acid sequences of aphidicolin biosynthetic enzymes were analyzed by TMHMM [Citation25], one transmembrane domain was predicted at the N-terminal region of both PbP450s (Figure S1), which prompted us to examine the subcellular localization of these enzymes. GFP fluorescence of both C-terminally GFP-tagged PbP450-1 (PbP450-1-GFP) and PbP450-2 (PbP450-2-GFP) was observed at tubular organelles (Figure ). To identify these organelles, SNARE proteins (Use1, Sed5, and Tlg2) [Citation33] and phospholipase A2 (PlaA) [Citation34] were expressed as a fusion protein with mCherry in the PbP450-GFP expressing strains. These proteins were used as organelle markers for the endoplasmic reticulum (ER; Use1), Golgi apparatus (Sed5), endosome (Tlg2), and mitochondrion (PlaA). Fluorescence of both PbP450-1-GFP and PbP450-2-GFP was completely merged with fluorescence of mCherry-Use1 (Figure (A) and (B)), whereas mCherry fluorescence of PlaA, Sed5, and Tlg2 fusion proteins was not observed in the regions with GFP fluorescence (Figure (C)–(E)). In addition, fluorescence of GFP and mCherry was completely merged when these fusion proteins were co-expressed with PbP450-1 and PbP450-2 (Figure (F)). These results indicated that PbP450-1 colocalized with PbP450-2 in the ER.
Figure 3. Subcellular localization of PbP450-1 and PbP450-2 in A. oryzae. Hyphae of strains expressing PbP450-1-GFP (A and C) and PbP450-2-GFP (B, D, and E) with mCherry-fused organelle markers were examined by confocal microscopy. (F) Colocalization analysis of GFP-fused PbP450-1 and mCherry-fused PbP450-2. Fluorescence images of apical and subapical regions are shown in the right and left panels, respectively.
Membrane topology analysis of PbP450-1 and PbP450-2
To investigate the membrane topology of PbP450-1 and PbP450-2, we performed the protease protection assay using C-terminally GFP-fused proteins (PbP450-1-GFP and PbP450-2-GFP). In this assay, when the C-terminal region of PbP450 is exposed to the cytoplasm, most would be degraded by proteinase K and only free GFP that is highly resistant to proteinase K would remain. In contrast, most of the GFP fusion protein is protected from proteinase K digestion when the C-terminal region of PbP450 is located in the ER lumen. This protection is lost by solubilization of the microsomes with Triton X-100. To confirm whether the intact microsomes were isolated by homogenization of the fungal cells, a GFP fusion protein of the ER-localized chaperone BipA [Citation28] that is resistant to protease digestion without the treatment of detergents was used as a control. Detection of intact BipA-GFP from the homogenate after treatment with proteinase K suggested that the intact microsomes were successfully prepared by our homogenization method (Figure ). In contrast to BipA-GFP, both PbP450-1-GFP and PbP450-2-GFP were sensitive to proteinase K without Triton X-100 treatment (Figure ). This result suggested that both PbP450s are located in the ER and their C-terminal regions, including the catalytic domain, are exposed to the cytoplasmic side.
Figure 4. Protease protection assay of GFP-fused Pb450s. Mycelia grown for 36 h in MM containing 1% casamino acids as the sole carbon source were transferred to fresh MM containing 1% maltose and incubated for 6 h. The homogenates were prepared from harvested mycelia and treated with proteinase K with or without Triton X-100. Protected proteins from proteinase K digestion were detected by western blot analysis using an anti-GFP antibody.
Effect of transmembrane domain deletion on PbP450 localization
To verify whether putative transmembrane domains of PbP450-1 and PbP450-2 contributed to ER localization, we examined the subcellular localization of PbP450-1 and PbP450-2 mutants lacking the putative transmembrane domain. As shown in Figure (A), fluorescence of PbP450-2∆TM-GFP, lacking amino acid residues 27-47, was diffused in the cytoplasm (Figure (B)). Fluorescence of PbP450-1∆TM-GFP, lacking 21 N-terminal amino acids, was observed as dot-like structures. Fluorescence of PbP450-1∆TM-GFP was not merged with mCherry-Use1 (Figure S2). This result indicated that PbP450-1∆TM-GFP was not localized to the ER. In addition, PbP450-1∆TM-GFP was not colocalized with other mCherry-fused organelle markers, such as Histone H2B (nucleus) [Citation35], Nyv1 (vacuole) [Citation33]), peroxisomal targeting signal 1 (PTS1) (peroxisome), Sed5 (Golgi apparatus), and Tlg2 (endosome) (Figure S2). To examine whether the GFP-fused mutant proteins were degraded due to deletion of the transmembrane regions, the PbP450 mutants were subjected to western blot analysis. Full-length mutant proteins that showed the expected molecular mass were detected and no degraded protein bands were observed (Figure S3). In protease protection assays, PbP450-1∆TM-GFP was degraded by proteinase K without treatment with Triton X-100 (Figure (C)), suggesting that this truncated protein was not located inside any organelles. These results suggested that transmembrane domains of PbP450-1 and PbP450-2 were required for ER-targeting of these enzymes, whereas other regions of PbP450-1 may be involved in the targeting of another organelle.
Figure 5. Subcellular localization of PbP450-1 and PbP450-2 mutants lacking the transmembrane domain. Hyphae of PbP450-2∆TM-GFP (A) and PbP450-1∆TM-GFP (B) expressing strains were examined by confocal microscopy. (C) Protease protection assay of Pb450-1∆TM-GFP.
Effect of transmembrane domain deletion from PbP450-1 on aphidicolin production
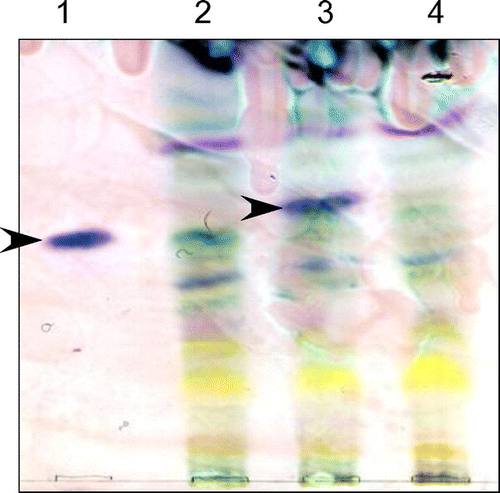
To examine the effect of PbP450-1 ER localization on aphidicolin production, plasmids for overexpression of PbP450-1 and PbP450-1∆TM using the α-amylase promoter were introduced into an A. oryzae strain overexpressing PbACS, PbGGS, and PbP450-2. Although a transporter in the aphidicolin biosynthetic cluster [Citation13] was also overexpressed in this strain, this had no apparent effect on aphidicolin production (data not shown). After cultivation of strains overexpressing PbP450-1 and PbP450-1∆TM in maltose-containing medium for 6 days, aphidicolin was extracted from the culture supernatant and mycelia with ethyl acetate. Then, aphidicolin was detected by thin-layer chromatography. The signal corresponding to aphidicolin was clearly observed in the strain that expressed intact PbP450-1 (Figure ). However, this signal was undetectable in the strain that expressed PbP450-1∆TM (Figure ). This result suggested that localization of PbP450-1 to the ER membrane was required for aphidicolin biosynthesis.
Figure 6. Detection of aphidicolin production by thin-layer chromatography. A. oryzae strains were cultured in MM with 1% tryptone and 3% maltose for 6 days. Aphidicolin was extracted from the culture supernatant and mycelium with ethyl acetate. The reagent product of aphidicolin (lane 1) was used as a control. An A. oryzae strain expressing PbGGS, PbACS, Pb450-2, and PbTP (lane 2) was used as a host for the expression of intact PbP450-1 (lane 3) and PbP450-1∆TM (lane 4). Arrowheads indicate the signal corresponding to aphidicolin.
Discussion
A. oryzae is often used to express biosynthetic genes involved in secondary metabolite synthesis to identify the function of biosynthetic enzymes and to produce a sufficient amount of secondary metabolites of interest [Citation36]. However, the subcellular localization of biosynthetic enzymes heterologously expressed in filamentous fungi, including A. oryzae, has not been documented. In this study, we examined the subcellular localization of aphidicolin biosynthetic enzymes and the effect of transmembrane domain deletion of monooxygenases on subcellular localization and aphidicolin production.
PbGGS and PbACS synthesize aphidicolan-16β-ol from isopentenyl diphosphate (IDP) and farnesyl diphosphate (FDP) in A. oryzae [Citation3]. In this study, subcellular localization analysis revealed that both PbGGS and PbACS diffused in the cytoplasm when expressed alone in A. oryzae. In contrast, two monooxygenases that catalyze the hydroxylation of aphidicolan-16β-ol were localized to the ER membrane. Since IDP and FDP are provided from the ER-localized mevalonate pathway [Citation37], cytosolic localization of PbGGS and PbACS would be inefficient for aphidicolin biosynthesis. This may account for the lower production of aphidicolin observed in A. oryzae compared to the original host, P. betae. In this context, we are interested in the subcellular localization of aphidicolin biosynthetic enzymes, including a putative transporter, PbTP, in P. betae.
Previously, the subcellular localization of fungal geranylgeranyl diphosphate synthases involved in terpene biosynthesis was examined in Penicillium paxilli and Fusarium fujikuroi. In these strains, geranylgeranyl diphosphate synthases are localized to punctate organelles other than the peroxisome [Citation38,39]. Plant geranylgeranyl diphosphate synthases are localized to plastids, mitochondria, and the ER [Citation40,41]. Therefore, there is a possibility that PbGGS is mislocalized to the cytosol in A. oryzae, and subcellular localization analysis of PbGGS in P. betae is important to understand the localization of geranylgeranyl diphosphate synthases in filamentous fungi.
Membrane topology analysis of PbP450-1 and PbP450-2 revealed that both PbP450s were anchored at the ER membrane via their N-terminal transmembrane domains and their catalytic domains were located in the cytoplasm. These data are supported by P450 membrane topology evidence [Citation42]. Protease protection assays are a major method to determine the membrane topology in many organisms. GFP is a suitable tag for protease protection assays since resistance of GFP to protease allows detection of free GFP as a degradation product of fusion proteins. GFP-based protease protection assays to detect degradation are widely used to monitor autophagosome biogenesis [Citation43]. This study indicates that protease protection assays using GFP is a useful method to easily determine the topology of membrane proteins in filamentous fungi.
Deletion of a putative transmembrane domain from PbP450-2 led to diffusion in the cytoplasm. In contrast, fluorescence of PbP450-1∆TM-GFP was observed as dot-like structures, and this fluorescence was not merged with mCherry fluorescence of any organelle markers. Aggregated GFP accumulates in cytosolic inclusions and appear as dot-like fluorescence [Citation44]. However, as shown in Figure S2, detection of intact PbP450-1∆TM as a sharp band by western blot analysis indicated that PbP450-1∆TM was not degraded, suggesting that the dot-like fluorescence observed was not aggregated GFP. Protease protection assays suggested that PbP450-1∆TM was not located in organelles. P450 lacking a transmembrane domain is retained in the membrane fraction of E. coli and yeast after heterologous expression [Citation45–47]. In addition, the hydrophobic surface of P450 is also involved in interactions with the membrane surface [Citation48–51]. Identification of PbP450-1∆TM localization sites will provide valuable information to understand the role of the P450 hydrophobic surface in subcellular localization.
PbP450-1 hydroxylates two distant positions in the final step of aphidicolin biosynthesis [Citation13]. Aphidicolin biosynthesis was almost completely lost by deletion of the putative transmembrane domain from PbP450-1. Although possible structural changes associated with transmembrane domain deletion cannot be ruled out, deletion of the transmembrane domain from P450 had no apparent effect on the catalytic activity [Citation45,47,52,53]. Therefore, we concluded that localization of PbP450-1 to the ER is important for aphidicolin biosynthesis, whereas the relationship between subcellular localization and catalytic activity of P450 is not fully understood. In this study, we did not examine the functionality of the PbP450-2 mutant in aphidicolin biosynthesis. However, based on the results of the truncated PbP450-1 mutant, localization of PbP450-2 to the ER seems to be responsible for aphidicolin biosynthesis. To the best of our knowledge, this is the first report that examined the membrane topology and the effect of the presence of putative transmembrane domains on the function of P450s in filamentous fungi.
In conclusion, we revealed the subcellular localization of aphidicolin biosynthetic enzymes and the importance of the transmembrane domain of PbP450s for ER localization and aphidicolin biosynthesis. A comparison of the subcellular localization of aphidicolin biosynthetic enzymes between P. betae and A. oryzae will provide important information to improve the production of aphidicolin and other secondary metabolites in the heterologous host A. oryzae.
Author contributions
KG and HO conceived and designed the experiments with assistance from RF, AM, and TS. AB performed the experiments with assistance from MT. MT, TS, and KG analyzed the data. MT and KG wrote the paper.
Disclosure statement
The authors declare that they have no conflict of interest.
Funding
This work was supported by a Grant-in-Aid for Scientific Research on Innovative Areas [JSPS KAKENHI grant number 22108007], from the Ministry of Education, Culture, Sports, Science and Technology of Japan.
Supplemental data
Supplemental data for this article can be accessed at https://doi.org/10.1080/09168451.2017.1399789
SM_localization_BBB_Suppl_Fig..pdf
Download PDF (423.4 KB)SM_machinery_localization_BBB_TableS1.pdf
Download PDF (148.3 KB)Acknowledgments
We thank Jun-ichi Maruyama for providing the plasmid pUNABG.
Related Research Data
References
- Machida M, Yamada O, Gomi K. Genomics of Aspergillus oryzae: learning from the history of koji mold and exploration of its future. DNA Res. 2008;15:173–183.10.1093/dnares/dsn020
- Heneghan MN, Yakasai AA, Halo LM, et al. First heterologous reconstruction of a complete functional fungal biosynthetic multigene cluster. ChemBioChem. 2010;11:1508–1512.10.1002/cbic.v11:11
- Fujii R, Minami A, Tsukagoshi T, et al. Total biosynthesis of diterpene aphidicolin, a specific inhibitor of DNA polymerase α: heterologous expression of four biosynthetic genes in Aspergillus oryzae. Biosci Biotechnol Biochem. 2011;75:1813–1817.10.1271/bbb.110366
- Tagami K, Liu C, Minami A, et al. Reconstitution of biosynthetic machinery for indole-diterpene paxilline in Aspergillus oryzae. J Am Chem Soc. 2013;135:1260–1263.10.1021/ja3116636
- Tagami K, Minami A, Fujii R, et al. Rapid reconstitution of biosynthetic machinery for fungal metabolites in Aspergillus oryzae: total biosynthesis of aflatrem. ChemBioChem. 2014;15:2076–2080.10.1002/cbic.201402195
- Matsuda Y, Wakimoto T, Mori T, et al. Complete biosynthetic pathway of anditomin: nature’s sophisticated synthetic route to a complex fungal meroterpenoid. J Am Chem Soc. 2014;136:15326–15336.10.1021/ja508127q
- Ugai T, Minami A, Fujii R, et al. Heterologous expression of highly reducing polyketide synthase involved in betaenone biosynthesis. Chem Commun. 2015;51:1878–1881.10.1039/C4CC09512J
- Matsuda Y, Iwabuchi T, Wakimoto T, et al. Uncovering the unusual D-ring construction in terretonin biosynthesis by collaboration of a multifunctional cytochrome P450 and a unique isomerase. J Am Chem Soc. 2015;137:3393–3401.10.1021/jacs.5b00570
- Liu C, Tagami K, Minami A, et al. Reconstitution of biosynthetic machinery for highly elaborated indole diterpene penitrem. Angew Chem Int. 2015;54:5748–5752.10.1002/anie.201501072
- Ye Y, Minami A, Mándi A, et al. Genome mining for sesterterpenes using bifunctional terpene syntheses reveals a unified intermediate of di/sesterterpenes. J Am Chem Soc. 2015;137:11846–11853.10.1021/jacs.5b08319
- Bailey AM, Alberti F, Kilaru S, et al. Identification and manipulation of the pleuromutilin gene cluster from Clitopilus passeckerianus for increased rapid antibiotic production. Sci Rep. 2016;6:25202.10.1038/srep25202
- Ikegami S, Taguchi T, Ohashi M, et al. Aphidicolin prevents mitotic cell division by interfering with the activity of DNA polymerase-alpha. Nature. 1978;275:458–460.10.1038/275458a0
- Toyomasu T, Nakaminami K, Toshima H, et al. Cloning of a gene cluster responsible for the biosynthesis of diterpene aphidicolin, a specific inhibitor of DNA polymerase alpha. Biosci Biotechnol Biochem. 2004;68:146–152.10.1271/bbb.68.146
- Lim FY, Keller NP. Spatial and temporal control of fungal natural products synthesis. Nat Prod Rep. 2014;31:1277–1286.10.1039/C4NP00083H
- Kistler HC, Broz K. Cellular compartmentalization of secondary metabolism. Front Microbiol. 2015;6:68.
- Hong SY, Linz JE. Functional expression and sub-cellular localization of the early aflatoxin pathway enzyme Nor-1 in Aspergillus parasiticus. Mycol Res. 2009;113:591–601.10.1016/j.mycres.2009.01.013
- Chanda A, Roze LV, Kang S, et al. A key role for vesicles in fungal secondary metabolism. Proc Nat Acad Sci USA. 2009;106:19533–19538.10.1073/pnas.0907416106
- Bartoszewska M, Opaliński L, Veenhuis M, et al. The significance of peroxisomes in secondary metabolite biosynthesis in filamentous fungi. Biotechnol Lett. 2011;33:1921–1931.10.1007/s10529-011-0664-y
- Kiel JA, van der Klei IJ, van den Berg MA, et al. Overproduction of a single protein, Pc-Pex11p, results in 2-fold enhanced penicillin production by Penicillium chrysogenum. Fungal Genet Biol. 2005;42:154–164.10.1016/j.fgb.2004.10.010
- Opalinski L, Kiel JA, Homan TG, et al. Penicillium chrysogenum Pex14/17p—a novel component of the peroxisomal membrane that is important for penicillin production. FEBS J. 2014;277:3203–3218.
- Minetoki T, Nunokawa Y, Gomi K, et al. Deletion analysis of promoter elements of the Aspergillus oryzae agdA gene encoding α-glucosidase. Curr Genet. 1996;30(5):432–438.10.1007/s002940050153
- Hasegawa S, Takizawa M, Suyama H, et al. Characterization and expression analysis of a maltose-utilizing (MAL) cluster in Aspergillus oryzae. Fungal Genet Biol. 2010;47:1–9.10.1016/j.fgb.2009.10.005
- Minetoki T, Tsuboi H, Koda A, et al. Development of high expression system with the improved promoter using the cis-acting element in Aspergillus species. J Biol Macromol. 2003;3:89–96.
- Makita T, Katsuyama Y, Tani S, et al. Inducer-dependent nuclear localization of a Zn(II)2Cys6 transcriptional activator, AmyR, in Aspergillus nidulans. Biosci Biotechnol Biochem. 2009;73:391–399.10.1271/bbb.80654
- Krogh A, Larsson B, von Heijne G, et al. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol. 2001;305:567–580.10.1006/jmbi.2000.4315
- Shintani T, Reggiori F. Fluorescence microscopy-based assays for monitoring yeast Atg protein trafficking. Methods Enzymol. 2008;451:43–56.10.1016/S0076-6879(08)03204-7
- Hiramoto T, Tanaka M, Ichikawa T, et al. Endocytosis of a maltose permease is induced when amylolytic enzyme production is repressed in Aspergillus oryzae. Fungal Genet Biol. 2015;82:136–144.10.1016/j.fgb.2015.05.015
- Maruyama J, Kikuchi S, Kitamoto K. Differential distribution of the endoplasmic reticulum network as visualized by the BipA-EGFP fusion protein in hyphal compartments across the septum of the filamentous fungus, Aspergillus oryzae. Fungal Genet Biol. 2006;43:642–654.10.1016/j.fgb.2005.11.007
- Zhang S, Ban A, Ebara N, et al. Self-excising Cre/mutant lox marker recycling system for multiple gene integrations and consecutive gene deletions in Aspergillus oryzae. J Biosci Bioeng. 2017;123(4):403–411.10.1016/j.jbiosc.2016.11.001
- O Hatamoto, G Umitsuki, M Machida, et al., inventor; Noda Institute for Scientific Research, Kikkoman Corporation, assignee. Recombinant vector capable of increasing secretion of Koji mold protease. United States patent US 7842799 B2. 2010 Nov 30.
- Gomi K, Iimura Y, Hara S. Integrative transformation of Aspergillus oryzae with a plasmid containing the Aspergillus nidulans argB gene. Agric Biol Chem. 1987;51:2549–2555.
- Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685.10.1038/227680a0
- Kuratsu M, Taura A, Shoji JY, et al. Systematic analysis of SNARE localization in the filamentous fungus Aspergillus oryzae. Fungal Genet Biol. 2007;44:1310–1323.10.1016/j.fgb.2007.04.012
- Takaya K, Higuchi Y, Kitamoto K, et al. A cytosolic phospholipase A2-like protein in the filamentous fungus Aspergillus oryzae localizes to the intramembrane space of the mitochondria. FEMS Microbiol Lett. 2009;301:201–209.10.1111/fml.2009.301.issue-2
- Maruyama J, Nakajima H, Kitamoto K. Visualization of nuclei in Aspergillus oryzae with EGFP and analysis of the number of nuclei in each conidium by FACS. Biosci Biotechnol Biochem. 2001;65:1504–1510.10.1271/bbb.65.1504
- Alberti F, Foster GD, Bailey AM. Natural products from filamentous fungi and production by heterologous expression. Appl Microbiol Biotechnol. 2017;101:493–500.10.1007/s00253-016-8034-2
- Koning AJ, Roberts CJ, Wright RL. Different subcellular localization of Saccharomyces cerevisiae HMG-CoA reductase isozymes at elevated levels corresponds to distinct endoplasmic reticulum membrane proliferations. Mol Biol Cell. 1996;7:769–789.10.1091/mbc.7.5.769
- Saikia S, Scott B. Functional analysis and subcellular localization of two geranylgeranyl diphosphate synthases from Penicillium paxilli. Mol Genet Genomics. 2009;282:257–271.10.1007/s00438-009-0463-5
- Albermann S, Linnemannstöns P, Tudzynski B. Strategies for strain improvement in Fusarium fujikuroi: overexpression and localization of key enzymes of the isoprenoid pathway and their impact on gibberellin biosynthesis. Appl Microbiol Biotechnol. 2013;97:2979–2995.10.1007/s00253-012-4377-5
- Okada K, Saito T, Nakagawa T, et al. Five geranylgeranyl diphosphate synthases expressed in different organs are localized into three subcellular compartments in Arabidopsis. Plant Physiol. 2000;122:1045–1056.10.1104/pp.122.4.1045
- Beck G, Coman D, Herren E, et al. Characterization of the GGPP synthase gene family in Arabidopsis thaliana. Plant Mol Biol. 2013;82:393416.
- Black SD. Membrane topology of the mammalian P450 cytochromes. FASEB J. 1992;6:680–685.
- Nair U, Thumm M, Klionsky DJ, et al. GFP-Atg8 protease protection as a tool to monitor autophagosome biogenesis. Autophagy. 2011;7:1546–1550.10.4161/auto.7.12.18424
- Choe YJ, Park SH, Hassemer T, et al. Failure of RQC machinery causes protein aggregation and proteotoxic stress. Nature. 2016;531:191–195.10.1038/nature16973
- Cullin C. Two distinct sequences control the targeting and anchoring of the mouse P450 1A1 into the yeast endoplasmic reticulum membrane. Biochem Biophys Res Commun. 1992;184:1490–1495.10.1016/S0006-291X(05)80051-8
- Pernecky SJ, Larson JR, Philpot RM, et al. Expression of truncated forms of liver microsomal P450 cytochromes 2B4 and 2E1 in Escherichia coli: influence of NH2-terminal region on localization in cytosol and membranes. Proc Nat Acad Sci USA. 1993;90:2651–2655.10.1073/pnas.90.7.2651
- Sagara Y, Barnes HJ, Waterman MR. Expression in Escherichia coli of functional cytochrome P450c17 lacking its hydrophobic amino-terminal signal anchor. Arch Biochem Biophys. 1993;304:272–278.10.1006/abbi.1993.1349
- Cosme J, Johnson EF. Engineering microsomal cytochrome P450 2C5 to be a soluble, monomeric enzyme. Mutations that alter aggregation, phospholipid dependence of catalysis, and membrane binding. J Biol Chem. 2000;275:2545–2553.10.1074/jbc.275.4.2545
- Williams PA, Cosme J, Sridhar V, et al. The crystallographic structure of a mammalian microsomal cytochrome P450 monooxygenase: structural adaptations for membrane binding and functional diversity. Mol Cell. 2000;5:121–131.10.1016/S1097-2765(00)80408-6
- Ozalp C, Szczesna-Skorupa E, Kemper B. Identification of membrane-contacting loops of the catalytic domain of cytochrome P450 2C2 by tryptophan fluorescence scanning. Biochemistry. 2006;45:4629–4637.10.1021/bi051372t
- Mast N, Liao WL, Pikuleva IA, et al. Combined use of mass spectrometry and heterologous expression for identification of membrane-interacting peptides in cytochrome P450 46A1 and NADPH-cytochrome P450 oxidoreductase. Arch Biochem Biophys. 2009;483:81–89.10.1016/j.abb.2009.01.002
- Kempf AC, Zanger UM, Meyer UA. Truncated human P450 2D6: expression in Escherichia coli, Ni(2+)-chelate affinity purification, and characterization of solubility and aggregation. Arch Biochem Biophys. 1995;321:277–288.10.1006/abbi.1995.1396
- Li YC, Chiang JY. The expression of a catalytically active cholesterol 7α-hydroxylase cytochrome P450 in Escherichia coli. J Biol Chem. 1991;266:19186–19191.