
Abstract
Hymenoic acid, isolated from cultures of the fungus, Hymenochaetaceae sp., is a specific inhibitor of DNA polymerase λ. The first synthesis of (S)-(+)-hymenoic acid was achieved by starting from trans-1,4-cyclohexanedimethanol and methyl (R)-(−)-3-hydroxyisobutyrate, and Julia–Kocienski olefination was employed as the key step.
(S)-(+)-Hymenoic acid, an inhibitor of DNA polymerase λ, was synthesized.
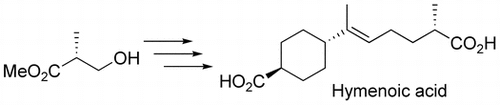
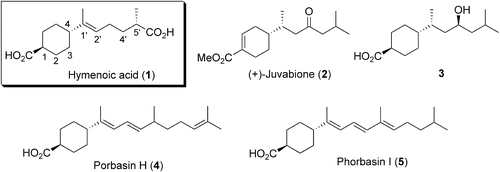
In 2007, Nishida et al. reported the isolation and structure elucidation of hymenoic acid (1) from a fungus, Hymenochaetaceae sp [Citation1]. This fungus had been isolated from a coral collected from Hachijo Island, Japan. Hymenoic acid (1) is a monocyclic sesquiterpene dicarboxylic acid. Its absolute configuration at C-5′ was determined to be S, as shown in Figure . Although the well-known sesquiterpene juvabione (2) and its relatives such as 3 are known to exist [Citation2], only two structurally analogous marine natural products, namely phorbacins H and I (4 and 5), have been reported to date [Citation3]. Nishida et al. also reported that 1 could potently inhibit only the activity of human and plant DNA polymerase (pol) λ [Citation1]. Thus, 1 might be useful not only as a specific inhibitor against pol λ, but also as a lead compound for design of new antitumor agents.
Figure 1. Structures of hymenoic acid (1) and the related natural products.
Inspired by its interesting biological profiles, we initiated studies toward the synthesis of 1 as a continuation of our synthetic studies on pol inhibitors [Citation4,5]. Here, we report the first synthesis of (S)-(+)-1.
Results and discussion
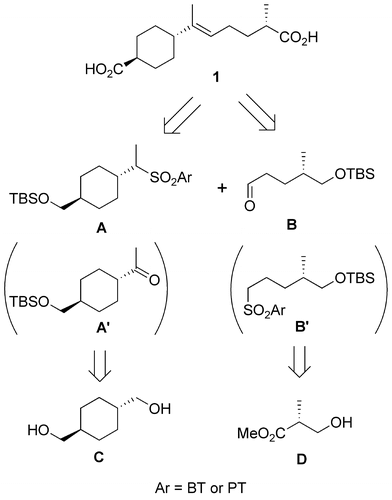
The synthetic plan for 1 is shown in Scheme . The basic skeleton of the target compound 1 would be synthesized using Julia–Kocienski (J–K) olefination [Citation6−9] between a sulfone A and an aldehyde B. Although J–K olefination between A′ and B′ was also possible, this method was not adopted because early model studies on J–K olefination showed that the (E)-selectivity in the coupling between a secondary sulfone and an aldehyde was much better than that between a ketone and a primary sulfone. Intermediates A and B would be preparable from trans-1,4-cyclohexanedimethanol (C) and methyl (R)-(−)-3-hydroxyisobutyrate (D), respectively.
Scheme 1. Synthetic plan for 1.
Scheme illustrates the synthesis of 1. The starting material, trans-1,4-cyclohexanedimethanol 6 (= C), was converted to the known aldehyde 7 (53% in two steps) [Citation10]. Treatment of 7 with MeMgI afforded 8 (89%). The alcohol 8 was converted into the corresponding sulfide, which was immediately oxidized with m-CPBA to give the key 1-phenyl-1H-tetrazol-5-yl (PT) sulfone 9 (= A; 56% in two steps). Despite considerable effort, the corresponding benzothiazole-2-yl (BT) sulfone could not be prepared in an acceptable yield. By starting from methyl (R)-(−)-3-hydroxyisobutyrate 10 (=D), the known aldehyde 12 (= B) was prepared. Although several methods of preparation have been reported, [Citation11−13] we prepared the ester 11 (48% in four steps) according to Chandrasekhar’s procedure [Citation14] and reduced 11 with DIBAL to give 12 (quant.).
Scheme 2. Synthesis of (S)-(+)-hymenoic acid (1).
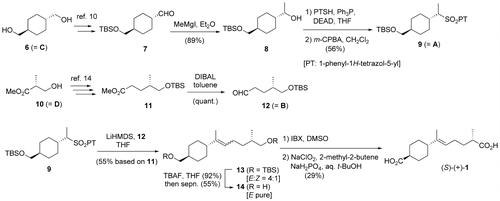
With the two intermediates (9 and 12) in hand, J–K olefination was attempted. As stated in review articles, [Citation8,9] the formation of a trisubstituted olefin via J–K olefination is difficult as there are only a limited number of (E)-selective options available for the coupling between a secondary sulfone and an aldehyde [Citation15−17]. However, because the successful formation of a trisubstituted olefin is usually performed using an alkaline metal hexamethyldisilazide (HMDS) in an ethereal solvent, the coupling between 9 and 12 with three alkaline HMDSs in THF is examined. As a result of this strategy, the desired olefin 13 was obtained (55% yield based on 11; E:Z = ca. 4:1) only when LiHMDS was used as a base. The use of NaHMDS and KHMDS gave no desired adduct. Furthermore, it is important to note that this base dependency is also observed in early model studies. Upon treatment with TBAF, two TBS groups of 13 were removed (92%), and the resulting diol 14 was carefully separated by SiO2 column chromatography to afford (E)-14. Finally, (E)-14 was stepwisely oxidized in the conventional manner to give (S)-(+)-1 (29%). The various spectral data of synthetic (S)-(+)-1 were in good agreement with those of the naturally ocurring 1.
In conclusion, the first synthesis of (S)-(+)-hymenoic acid (1) was accomplished by employing Juila–Kociensky olefination as the key step. This synthetic rout is straightforward and enables the synthesis of a wide range of hymenoic acid analogs. To clarify the structure-activity relationship, the synthesis of hymenoic acid analogs is an ongoing focus of this research.
Experimental
General procedures
1H-NMR spectra were recorded at 300 MHz with a Jeol JNM-AL300 spectrometer. TMS or the residual solvent peak in CDCl3 (at δH = 7.26) or CD3OD (at δH = 3.30), was used as the internal standard. 13C-NMR spectra were recorded at 75 MHz with the Jeol JNM-AL300 spectrometer, the peak for CDCl3 (at δC = 77.0) or CD3OD (at δC = 49.0) being used as the internal standard. Optical rotations were taken with a HORIBA SEPA-300 polarimeter. Mass spectra were measured with a Jeol JMS-SX102A spectrometer. Anhydrous THF (Cat. No. 41001–84) was purchased from Kanto Chemical Co., Inc., and other anhydrous solvents were dried over MS 3A/4A.
1-[trans-4′-(t-Butyldimethylsilyloxymethyl)cyclohexyl]ethanol (8)
To an ice-cooled solution of MeMgI (ca. 1.0 M in Et2O; 35 mL, 35 mmol), a solution of 7 (1.84 g, 7.19 mmol) in dry Et2O (15 mL) was added dropwise. After removal of an ice-bath, the stirring was continued for 1.5 h. The reaction mixture was quenched with sat. NH4Cl aq. and extracted with EtOAc. The organic layer was washed with water and brine, dried (MgSO4), and concentrated under reduced pressure. The residue was chromatographed on SiO2 to give 8 (1.74 g, 89%). NMR δH (CDCl3): 0.03 (6H, s, Si-Me), 0.89 (9H, s, t-Bu), 0.88–1.60 (6H, m), 1.17 (3H, d, J = 6.3 Hz, CH-Me), 1.65–1.94 (4H, m), 3.40 (2H, d, J = 6.3 Hz, O-CH2), 3.56 (1H, m, O-CH). NMR δC (CDCl3): −5.4, 18.4, 20.5, 26.0, 27.7, 28.1, 29.2, 29.3, 40.5, 45.3, 68.7, 72.2. HREIMS m/z [M]+: calcd. for C15H32O2Si, 272.2172; found, 272.2198.
5-{1-[trans-4′-(t-Butyldimethylsilyloxymethyl)cyclohexyl]ethanesulfonyl}-1-phenyl-1H-tetrazole (9)
To an ice-cooled solution of 8 (270 mg, 0.993 mmol) in dry THF (10 mL), 5-mercapto-1-phenyl-1H-tetrazole (0.53 g, 3.0 mmol), Ph3P (0.79 g, 3.0 mmol) and DEAD (2.2 M in toluene; 1.4 ml, 3.1 mmol) were added successively under Ar. After stirring overnight at room temperature, the reaction mixture was diluted with water and extracted with EtOAc. The organic layer was washed with brine, dried (MgSO4), and concentrated under reduced pressure to give the residue (0.36 g). To a solution of this residue in CH2Cl2 (10 mL), m-CPBA (75%, 0.58 g, 2.5 mmol) was added at 0 °C. After stirring overnight at room temperature, the reaction mixture was diluted with sat. NaHCO3 aq. and extracted with CH2Cl2. The organic layer was washed with 10% Na2S2O3 aq., sat. NaHCO3 aq., and brine, dried (MgSO4), and concentrated under reduced pressure. The residue was chromatographed on SiO2 to give 9 (257 mg, 56% in two steps). NMR δH (CDCl3): 0.03 (6H, s, Si-Me), 0.89 (9H, s, t-Bu), 0.88–1.60 (6H, m), 1.45 (3H, d, J = 7.2 Hz, CH-Me), 1.65–1.98 (4H, m), 2.27 (1H, m), 3.38 (2H, d, J = 6.3 Hz, O-CH2), 3.98 (1H, dq, J = 2.4, 7.2 Hz, SO2-CH), 7.53–7.69 (5H, m, Ph). NMR δC (CDCl3): −5.4, 9.5, 18.4, 25.9, 26.9, 28.8, 29.5, 30.8, 36.4, 39.9, 65.2, 68.3, 125.4, 129.6, 131.4, 133.3, 153.3. HREIMS m/z [M]+: calcd. for C22H36N4O3SSi, 464.2277; found, 464.2273.
(S)-5-(t-Butyldimethylsilyloxy)-4-methylpentanal (12)
To a solution of 11 (78 mg, 30 mmol) in toluene (2 mL), DIBAL (1.0 M in toluene; 0.33 mL, 33 mmol) was slowly added at −78 °C under Ar. After stirring for 2 h at −78 °C, the reaction mixture was quenched with MeOH and then diluted with hexane. The resulting mixture was filtered through Celite, and the filtrate was concentrated under reduced pressure to give 12 (88 mg, quant.). This was used for the next step immediately.
(5′S)-trans-1-(t-Butyldimethylsilyloxymethyl)-4-(6′-t-butyldimethylsilyloxy-1′,5′-dimethyl-1′-hexenyl)cyclohexane (13)
To a solution of 9 (140 mg, 0.301 mmol) in dry THF (2 mL), LiHMDS (1.09 M in THF; 0.45 mL, 0.49 mmol) was added dropwise at −78 °C under Ar. After stirring for 30 min, a solution of 12 (88 mg) in THF (1 mL) was added, and the mixture was stirred overnight with gradual warming to room temperature. It was then quenched with sat. NH4Cl aq. and extracted with Et2O. The organic layer was washed with water and brine, dried (MgSO4), and concentrated under reduced pressure. The residue was chromatographed on SiO2 to give a mixture of (E)- and (Z)-13 (77 mg, 55% based on 11 in two steps, E:Z = ca. 4:1). [α]D21 = −0.58 (c 1.04, CHCl3). NMR δH (CDCl3): 0.03, 0.04 (12H, each s, Si-Me), 0.84–0.90 (12H, m), 0.90–2.40 (15H, m), 1.56 (3H, br s, 1′-Me), 3.32–3.48 (4H, m, O-CH2), 5.05 (1/5H, t, J = 6.9 Hz, Z-2′-H), 5.11 (4/5H, t, J = 6.9 Hz, E-2′-H). NMR δC (CDCl3): −5.34, −5.32, 14.3, 16.71, 16.73, 18.36, 18.40, 19.6, 24.7, 25.3, 25.97, 26.00, 29.7, 29.8, 30.4, 31.3, 33.4, 33.8, 35.4, 35.5, 39.8, 40.3, 40.4, 47.4, 68.3, 68.4, 68.87, 68.90, 122.9, 124.7, 139.7, 139.9. HREIMS m/z [M]+: calcd. for C27H56O2Si2, 468.3819; found, 468.3818.
(2S,5E)-6-[trans-4-(Hydroxymethyl)cyclohexyl]-2-methylhept-5-en-1-ol (14)
To a solution of 13 (93 mg, 0.20 mmol) in THF (3 mL), TBAF (1.0 M in THF; 0.6 mL, 0.6 mmol) was added. After stirring for 2 h at room temperature, the reaction mixture was diluted with sat. NH4Cl aq. and extracted with EtOAc. The organic layer was washed with brine, dried (MgSO4), and concentrated under reduced pressure. The residue was chromatographed on SiO2 to give a mixture of (E)- and (Z)-14 (44 mg, 92%). Further chromatographic purification of this mixture was carefully performed to give the pure (E)-14 (24 mg, 55%). [α]D23=−5.8 (c 0.33, CHCl3). NMR δH (CDCl3): 0.93 (3H, d, J = 6.6 Hz, 2-Me), 0.96–1.06 (2H, m), 1.10–1.30 (2H, m), 1.39–1.58 (2H, m), 1.60 (3H, s, 6-Me), 1.75–2.15 (8H, m), 3.43 (1H, dd, J = 10.2, 6.3 Hz), 3.46 (2H, d, J = 6.0 Hz), 3.51 (1H, dd, J = 10.2, 5.8 Hz), 5.12 (1H, t, J = 6.9 Hz, 5-H). NMR δC (CDCl3): 14.4, 16.5, 25.1, 29.6, 31.1, 33.2, 35.4, 40.3, 47.2, 68.3, 68.7, 122.6, 140.0. HREIMS m/z [M]+: calcd. for C15H28O2, 240.2089; found, 240. 2097.
(S)-(+)-Hymenoic acid (1)
To a solution of 14 (12.3 mg, 51.2 μmol) in DMSO (0.2 mL), IBX (57.8 mg, 0.206 mmol) in DMSO (0.7 mL) was added. After stirring at room temperature for 4 h, the reaction mixture was diluted with water and extracted with EtOAc. The organic layer was washed with brine, dried (MgSO4), and concentrated under reduced pressure to give the crude products, which was immediately dissolved into t-BuOH (0.2 ml) and water (0.2 mL). To this solution were added 2-methyl-2-butene (0.55 mL, 5.1 mmol), NaH2PO4 (30.5 mg, 0.196 mmol), NaOCl2 (26.4 mg, 0.292 mmol). After stirring for 3.5 h at room temperature, the reaction mixture was diluted with sat. NH4Cl aq. and extracted with CH2Cl2. The organic layer was washed brine, dried (MgSO4), and concentrated under reduced pressure. The residue was chromatographed on SiO2 to give (S)-(+)-1 (4.0 mg, 29% in two steps). [α]D25 = + 7.9 (c 0.080, CH3OH). It should be noted that the absolute value of specific rotation of the synthesized 1 has fluctuated considerably due to unknown reason. NMR δH (CD3OD): 1.13 (3H, d, J = 6.9 Hz), 1.21–1.34 (2H, m), 1.37–1.49 (3H, m), 1.58 (3H, s), 1.62–1.77 (3H, m), 1.84 (1H, tt, J = 11.7, 3.0 Hz), 1.99–2.06 (4H, m), 2.21 (1H, tt, J = 12.0, 3.6 Hz), 2.33–2.42 (1H, m), 5.15 (1H, t, J = 6.9 Hz). NMR δC (CD3OD): 14.4, 17.7, 26.5, 30.5, 32.1, 35.0, 40.3, 44.4, 47.9, 123.3, 141.5, 180.1, 180.8. HREIMS m/z [M]+: calcd. for C15H24O4, 268.1675; found, 268.1682.
Author contribution
H.T. designed the synthetic route and wrote the manuscript. K.T. and M.M. conducted the synthetic experiments with the aid of M.K. M.K. specially contributed to analytical works.
Disclosure statement
No potential conflict of interest was reported by the authors.
Acknowledgments
We thank Prof. Koji Kuramochi (Tokyo University of Science) for his helpful discussion. We also thank Mr. Kazuto Imaizumi (Kobe University) for his contribution in the early phase of this work.
References
- Nishida M, Ida N, Horio M, et al. Hymenoic acid, a novel specific inhibitor of human DNA polymerase λ from a fungus of Hymenochaetaceae sp. Bioorg Med Chem. 2008;16:5115–5122.10.1016/j.bmc.2008.03.021
- Numata A, Hokimoto K, Takemura T, et al. Plant constituents biologically active to insects. II. Juvabione analogs from Abies sachalinensis Mast. (1). Chem Pharm Bull. 1983;31:436–442.10.1248/cpb.31.436
- Lee HS, Park SY, Sim CJ, et al. Phorbasins G–I: three new diterpenoids from the sponge Phorbas gukulensis. Chem Pharm Bull. 2008;56:1198–1200.10.1248/cpb.56.1198
- Takikawa H, Nozawa D, Kayo A, et al. Synthesis of sulfobacin A, B and flavocristamide A, new sulfonolipids isolated from Chryseobacterium sp. J Chem Soc Perkin Trans. 1999;1:2467–2477.10.1039/a904258j
- Takikawa H, Kamatani N, Nakanishi K, et al. Synthetic studies on kohamaic acids: synthesis of structurally simplified analogs of kohamaic acid A. Biosci Biotechnol Biochem. 2008;72:3071–3074.10.1271/bbb.80629
- Baudin JB, Hareau G, Julia SA, et al. A direct synthesis of olefins by reaction of carbonyl compounds with lithio derivatives of 2-[alkyl- or (2′-alkenyl)- or benzyl-sulfonyl]-benzothiazoles. Tetrahedron Lett. 1991;32:1175–1178.10.1016/S0040-4039(00)92037-9
- Blakemore PR, Cole WJ, Kocienski PJ, et al. A stereoselective synthesis of trans-1,2-disubstituted alkenes based on the condensation of aldehydes with metallated 1-phenyl-1H-tetrazol-5-yl sulfones. Synlett. 1998;26–28.10.1055/s-1998-1570
- Blakemore PR. The modified Julia olefination: alkene synthesis via the condensation of metallated heteroarylalkylsulfones with carbonyl compounds. J Chem Soc Perkin Trans. 2002;1:2563–2585.10.1039/b208078h
- Aïssa C. Mechanistic manifold and new developments of the Julia–Kocienski reaction. Eur J Org Chem. 2009;12:1831–1844.
- Fotsop DF, Roussi F, Leverrier A, et al. Biomimetic total synthesis of meiogynin A, an inhibitor of Bcl-xL and Bak interaction. J Org Chem. 2010;75:7412–7415.10.1021/jo101088 h
- Marshall JA, Yanik MM. Synthesis of a C1–C21 subunit of the protein phosphatase inhibitor tautomycin: a formal total synthesis. J Org Chem. 2001;66:1373–1379.10.1021/jo0056951
- Molli SD, Qi J, Yajima A, et al. Structure-activity relationship of α hormones, the mating factors of phytopathogen Phytophthora. Bioorg Med Chem. 2012;20:681–686.10.1016/j.bmc.2011.12.015
- Williams BD, Smith AB III. Total synthesis of (±)-18-epi-latrunculol A. Org Lett. 2013;15:4584–4587.10.1021/ol4021892
- Chandrasekhar S, Reddy CR. Towards a synthesis of epothilone A: asymmetric synthesis of C(1)-C(6) and C(7)-C(15) fragments. Tetrahedron: Asymmetry. 2002;13:261–268.10.1016/S0957-4166(02)00095-2
- Huang H, Panek JS. Total synthesis of callipeltoside A. Org Lett. 2004;6:4383–4385.10.1021/ol0480325
- Werneburg M, Hertweck C. Chemoenzymatic total synthesis of the antiproliferative polyketide (+)-(R)-aureothin. ChemBioChem. 2008;9:2064–2066.10.1002/cbic.v9:13
- Tan D-X, Xu Z-J, Chen H-J, et al. Synthesis and configurations of (−)-furospongin-1 and (+)-dihydrofurospongin-2. Eur J Org Chem. 2016;5:946–957.