
Abstract
Lignans are a large class of naturally occurring secondary metabolites which are widely spread within the plant kingdom. Their diverse structures and variety of biological activities have fascinated organic chemists. For synthesizing optically active lignans, we have developed the novel asymmetric dimerization of cinnamic acid derivatives, and applied it to the enantioselective syntheses of furofuran lignans (yangambin, sesamin, eudesmin, caruilignan A) and diarylbutane lignans (sauriols A and B). This review summarizes the methodology of our asymmetric dimerization of cinnamic acid derivatives, and efficient total syntheses of furofuran and diarylbutane lignans reported by our and other groups.
Optically active furofuran and diarylbutane lignans were synthesized in short steps employing the asymmetric dimerization of cinnamic acid derivatives.
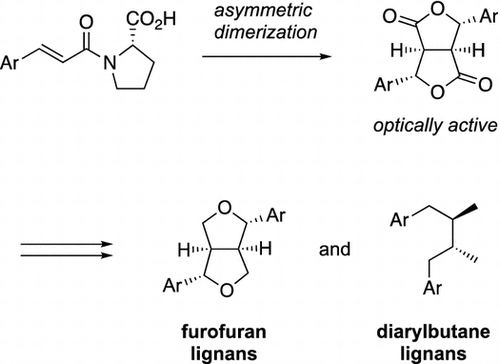
Lignans are a large group of naturally occurring secondary metabolites which are widely distributed in the plant kingdom and derived from the shikimic acid biosynthetic pathway [Citation1−4]. Although the molecular framework of lignans consists only of two phenylpropanoid units linked at C-8/C-8’ positions, lignans exhibit a wide variety of structures and biological activities originating from a different degree of oxidation in the side chains and a different substitution pattern in the aromatic rings. Among the many types of lignans, we focused on furofuran and diarylbutane lignans with unique structural characteristics and stereochemical diversity. Four furofuran lignans and two diarylbutane lignans synthesized by our group are shown in Figure . Yangambin (1), which was first obtained as a dimethylated derivative of lirioresinol-B [Citation5] and then isolated from Chinese medicinal plant, Magnolia fargesii [Citation6], exhibits various pharmacological activities, such as selective inhibition against platelet activating factor [Citation7,8], protective effects against cardiovascular collapse and anaphylactic shock, anti-allergic properties, analgesic activity, depressant effect in the central nervous system [Citation9], and apoptosis induction [Citation10]. Sesamin (2), a major abundant lignan compound present in Sesamum indicum L., is known to possess antihypertensive [Citation11−14], anticancer [Citation15−17], and antioxidant [Citation14] properties. Eudesmin (3) is a component of a Brazilian folk medicine, and shows selective inhibition against platelet activating factor [Citation18], T-cell proliferation [Citation19], antioxidant [Citation20,21], and neuritogenic [Citation22] activities. Caruilignan A (4) was isolated from the extract of traditional Chinese medicine, Artemisia caruifolia, and exhibits a cytotoxicity against Meth-A cells [Citation23]. On the other hand, sauriols A (5) and B (6), which belong to diarylbutane lignans, have been isolated from emergent portions of the southeastern United States freshwater angiosperm Saururus cernuus L. (Saururaceae) and exhibit antifeedant activity against the omnivorous crayfish Procambarus clarkii [Citation24]. This review summarizes our novel methodology for synthesizing lignans in optically active form and short-step syntheses of furofuran and diarylbutane lignans reported by our and other groups.
Figure 1. Furofuran and diarylbutane lignans synthesized by our group.
Asymmetric dimerization of cinnamic acid derivatives
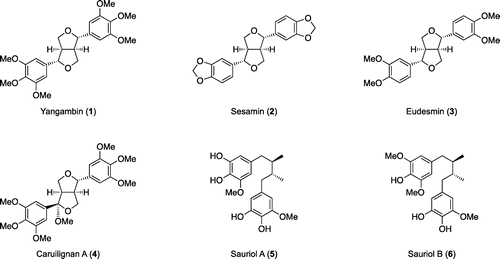
In order to achieve the efficient synthesis of optically active lignans, we focused on the dimerization reaction of cinnamic acids. Although oxidizing reagents such as FeCl3 [Citation25], Tl(OTFA)3 [Citation26], PbO2 [Citation27], NaIO4 [Citation28] are known to oxidize cinnamic acids 7 to racemic dilactones 8 (Scheme (a)), asymmetric versions under these conditions have not been developed yet. We, therefore, designed to equip the carboxyl group with L-proline as a chiral auxiliary to control the newly introduced four asymmetric centers (Scheme (b)) [Citation29]. At first, 3,4,5-trimethoxycinnamic acid derivative 9a was chosen as a substrate to examine the reaction conditions (Scheme (c)). Detailed experiments showed that the dimerization reaction only proceeds under the conditions using PbO2 reported by Yuzikhin et al. [Citation27]. When examining the reaction temperature (–20 or –78 °C), it was found that desired dilactone 10a could be obtained with higher yield and higher enantiomeric purity at lower temperature (–78 °C). Because of the low solubility of dilactone 10a, enantiomeric purity was determined by chiral HPLC analysis of 11, which was obtained from 10a by hydrogenolysis and methyl ester formation. Furthermore, the absolute stereochemistry was determined by comparing the sign of specific rotation with known antipodal compound of (–)-12 [Citation30].
Scheme 1. Asymmetric dimerization of cinnamic acid derivatives.
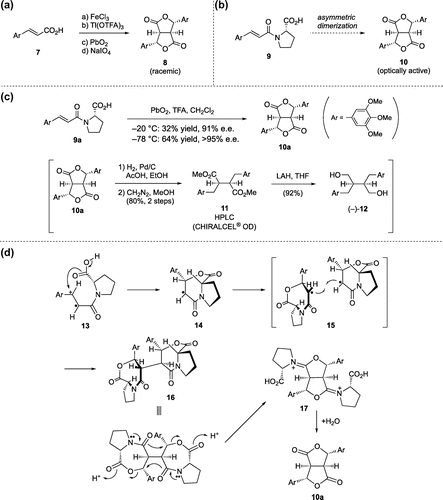
The presumed mechanism of the asymmetric dimerization reaction is illustrated in Scheme (d). Initial one-electron oxidation of 9a generates a radical cation (13), which was trapped intramolecularly by the carboxyl group from α-face to form a radical intermediate of seven-membered lactone (14). Radical–radical coupling of 14 takes place on the opposite side of the OC=O group to afford 16. Finally, the reconstruction of the ring system (16 to 17) occurred under strong acidic conditions with TFA, and 10a was obtained by hydrolytic loss of proline during aqueous workup. We reason that the higher yield of 10a at –78 °C compared with –20 °C (Scheme (c)) is due to the increased facial selectivity of radical–radical coupling of 14 at lower temperature.
Intrigued by these successful results, we next examined the application of this method to other substrates. However, when compounds 9b-d were subjected to Yuzikhin’s conditions (PbO2, TFA, CH2Cl2), only a trace amount of the desired dilactones (10b-d) was obtained. Therefore, we decided to investigate electrochemical oxidation conditions that can adjust the oxidation potential suitable for each substrate [Citation31]. After screening suitable reaction conditions, oxidative dimerization of each substrate was found to proceed under Ronlan’s electrochemical conditions [Citation32] (Pt anode, n-Bu4N•BF4, CH2Cl2/TFA). Chemical yield and enantiomeric excess of dilactones (10a-d) obtained under Ronlan’s conditions were summarized in Table . It should be noted that enantiomeric excess was determined by chiral HPLC analysis of 18a-d (18a = 1: yangambin, 18b = 2: sesamin, 18c = 3: eudesmin), which were prepared from 10a-d by reduction with Ca(BH4)2 and subsequent ether formation. Initially, the reaction of 3,4,5-trimethoxycinnamic acid derivative 9a was performed at various temperatures (r.t., 0, –20, –40 °C), and the best results (52% yield, 91% e.e.) were obtained at –40 °C. Since electric current was sometimes hard to be kept the same flow at –40 °C, reactions of other substrates were carried out at 0 or –20 °C. The reaction of 3,4-methylenedioxycinnamic acid derivative 9b at –20 °C gave dilactone 10b in 24% yield and with 83% e.e. On the other hand, dilactones 10c,d were obtained in low chemical yield (8–10%), while enantiomeric excess of dilactone 10c was tolerable (85–87% e.e.). The low chemical yields of 10c,d were due to decompositions of the starting materials 9c,d under the oxidation conditions. Interestingly, in our experiments, chemical yields tended to be better when the substrates with more oxygenated benzene ring were used.
Table 1. Electrochemical oxidation of cinnamic acid derivatives.
Synthetic approaches to furofuran lignans
Selected examples of concise enantioselective syntheses of furofuran lignans including our synthesis of yangambin (1) and caruilignan A (4) are presented in this section.
| (1) | Synthesis of (–)-sesamin and (–)-eudesmin by Kise et al. | ||||
In 2003, Kise et al. reported the synthesis of (–)-sesamin [(–)-2] and (–)-eudesmin [(–)-3] using the stereoselective oxidative coupling of diaroylacetate derivatives with NaH-Br2 as a key step (Scheme (a)) [Citation33]. They selected 19, which was prepared from (+)-camphor in three steps, as a chiral auxiliary. Diaroylacetates 20a,b, precursors for the intramolecular oxidative coupling, were synthesized by condensation of diacetate 19 with aromatic aldehydes and Jones oxidation of the resulting hydroxy group (61% yield for 20a, 67% yield for 20b). The key oxidative coupling of 20a proceeded by the successive treatment with NaH (2.5 eq.) and Br2 (1.0 eq.) in DMF/THF (1:1) to give the coupled product 21a in 83% yield with good stereoselectivity [(R,R):(R,S):(S,S) = 96:3:1]. Diaroylacetate 20b was also transformed to 21b in 78% yield with [(R,R):(R,S):(S,S) = 97:2:1]. Their detailed experiments and DFT calculations suggest that (R,R)-isomer is most thermodynamically stable and accumulates preferentially after equilibration of the three isomers. Finally, (–)-2 and (–)-3 were synthesized from 21a and 21b via LAH reduction and subsequent treatment with PPTS in benzene (43% yield for (–)-2, 38% yield for (–)-3).
| (2) | Synthesis of (–)-sesamin by Roy et al. | ||||
Scheme 2. Synthetic approaches to furofuran lignans.
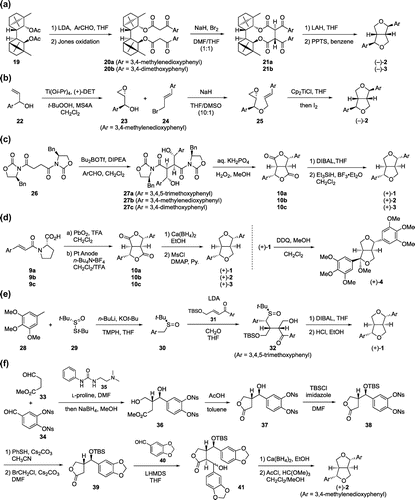
Three-step synthesis of (–)-sesamin [(–)-2] was accomplished by Roy et al. in 2005 (Scheme (b)) [Citation34]. Their key step involved stereoselective radical cyclization of chiral epoxy olefinic ether using bis(cyclopentadienyl)titanium(III) chloride (Cp2TiCl) as a radical initiator. Chiral epoxy alcohol 23 (98% e.e.), which was prepared from allyl alcohol 22 by the Sharpless kinetic resolution method [Citation35], was condensed with cinnamyl bromide 24 to give the key intermediate 25 in 82% yield. Finally, treatment of 25 with Cp2TiCl in THF at 60 °C and further stirring with an excess of iodine for one hour afforded (–)-2 in 91% yield.
| (3) | Synthesis of (+)-yangambin, (+)-sesamin and (+)-eudesmin by Park et al. | ||||
In 2005 and 2006, Park et al. reported the efficient synthesis of (+)-yangambin [(+)-1], (+)-sesamin [(+)-2] and (+)-eudesmin [(+)-3] based on the dianion aldol condensation strategy (Scheme (c)) [Citation36,37]. Their synthesis started from N-acyloxazolidinone 26, which was prepared by condensation of succinyl chloride and (S)-(–)-4-benzyl-2-oxazolidinone in 89% yield. Stereoselective aldol condensation of 26 and aromatic aldehydes proceeded by treatment with n-Bu2BOTf [Citation38] and DIPEA in CH2Cl2 at –78 °C to give unstable products 27a-c. Subsequent intramolecular cyclization of 27a-c using aqueous KH2PO4 and H2O2 in methanol gave 10a-c (81% yield and 20:1 diastereomeric selectivity for 10a, 91% yield and 19:1 diastereomeric selectivity for 10b, 84% yield and 18:1 diastereomeric selectivity for 10c). Finally, reduction of dilactones 10a-c with DIBAL gave the corresponding dilactols, which were treated with Et3SiH and BF3•OEt2 to afford (+)-1, (+)-2 and (+)-3 in 26, 27 and 23% yields over two steps, respectively.
| (4) | Synthesis of (+)-yangambin, (+)-sesamin, (+)-eudesmin and (+)-caruilignan A by our group. | ||||
As mentioned above, we synthesized dilactones 10a-c in optically active form employing our asymmetric dimerization reaction of cinnamic acid derivatives 9a-c (Scheme (d)) [Citation29,31]. That is, dilactone 10a was obtained in 64% yield, with > 95% e.e. under Yuzikhin’s conditions [Citation27] (PbO2, TFA, CH2Cl2), whereas dilactones 10b,c were obtained in 24% and 8% yields, with 83% and 87% e.e., respectively, under Ronlan’s electrochemical conditions [Citation32] (Pt anode, n-Bu4N•BF4, CH2Cl2/TFA). Dilactones 10a-c were converted to (+)-yangambin [(+)-1], (+)-sesamin [(+)-2] and (+)-eudesmin [(+)-3] by reduction with Ca(BH4)2 and subsequent ether formation (71% yield for (+)-1, 43% yield for (+)-2, 43% yield for (+)-3). Furthermore, oxidation of (+)-1 with DDQ in the presence of methanol afforded (+)-caruilignan A [(+)-4] in quantitative yield based on the recovery of (+)-1.
| (5) | Synthesis of (+)-yangambin by Syed et al. | ||||
In 2014, Syed et al. succeeded in the enantioselective total synthesis of (+)-yangambin [(+)-1] (Scheme (e)) [Citation39]. The key transformation was a one-pot reaction that involved conjugate addition of enantioenriched sulfoxide to enone and subsequent aldol reaction with formaldehyde. Sulfoxide 30 (90% e.e.), needed for the key one-pot reaction, was prepared by the addition of (3,4,5-trimethoxybenzyl)lithium to (R)-29 [Citation40] (>90% e.e.) under the superbasic conditions [Citation41] using n-BuLi, KOt-Bu and 2,2,6,6-tetramethylpiperidine (TMPH) in 47% yield. Deprotonation of sulfoxide 30 with LDA and addition of enone 31 in THF at –78 °C, and trapping of the resultant enolate with formaldehyde afforded 32 (90% e.e.) in 72% yield with excellent stereocontrol. DIBAL reduction of 32 gave the corresponding diol as a 3:2 mixture of diastereomers. Finally, heating the crude diol in ethanol with a catalytic amount of HCl resulted in removal of the TBS group and formation of the tetrahydrofuran rings to afford (+)-1 (83% e.e.) in 54% yield over two steps.
| (6) | Synthesis of (+)-sesamin by Kan et al. | ||||
In 2015, Kan et al. reported the synthesis of (+)-sesamin [(+)-2] employing the L-proline-catalyzed asymmetric cross-aldol reaction as a key step (Scheme (f)) [Citation42]. The key L-proline-catalyzed cross-aldol reaction of aliphatic aldehyde 33 with aromatic aldehyde 34 smoothly proceeded in the presence of bifunctional urea 35 [Citation43]. Reduction of the unstable aldol product with NaBH4 and treatment with AcOH provided 37 in 67% yield over two steps with good stereoselectivity (dr = 10:1, 97% e.e.). It should be noted that addition of urea 35 in the cross-aldol reaction contributes to shortening the reaction time and increasing the enantiomeric excess. After protection of the secondary alcohol of 37 as a TBS ether and subsequent removal of Ns groups, compound 39 was obtained by treatment with bromochloromethane and Cs2CO3 in 52% three steps yield. Aldol reaction of 39 with 40 proceeded from the less-hindered β-face of the lactone ring to give 41 in 62% yield with 1.3:1 diastereomeric selectivity at newly generated benzylic alcohol. After reduction of lactone 41 (a mixture of diastereomers) with Ca(BH4)2, tetrahydrofuran rings were constructed by treatment with anhydrous HCl and trimethyl orthoformate in methanol to afford (+)-2 in 69% yield over two steps.
Synthetic approaches to diarylbutane lignans
Chemical synthesis and biological evaluation of diarylbutane lignans were studied in detail by Yamauchi et al. Yamauchi’s synthesis of (–)- and meso-dihydroguaiaretic acid (49) as well as our synthesis of sauriols A (5) and B (6) are presented in this section.
| (1) | Synthesis of (–)-sauriols A and B by our group | ||||
In 2006, we achieved the first enantioselective synthesis of (–)-sauriols A [(–)-5] and B [(–)-6] starting from dilactone 10a in six steps (Scheme (a)) [Citation44]. Dilactone 10a (>95% e.e.) was transformed into diol 12 by successive hydrogenolysis of the lactone ring, methyl esterification and LAH reduction. Recrystallization of diol 12 enhanced the enantiomeric purity to give 12 with 100% e.e. Mesylation of the diol and reduction with LAH afforded the key intermediate 42 in 86% two steps yield. Cleavage of two methyl ethers of each benzene ring in 42 was achieved by treatment with BBr3 for five minutes at 0 °C to give (–)-5 in 78% yield. On the other hand, treatment of 42 with BBr3 overnight at –78 °C afforded (–)-6 in 27% yield along with the major 4,4’-didemethylated product (51%).
| (2) | Synthesis of (–)-dihydroguaiaretic acid by Yamauchi et al. | ||||
Scheme 3. Synthetic approaches to diarylbutane lignans.
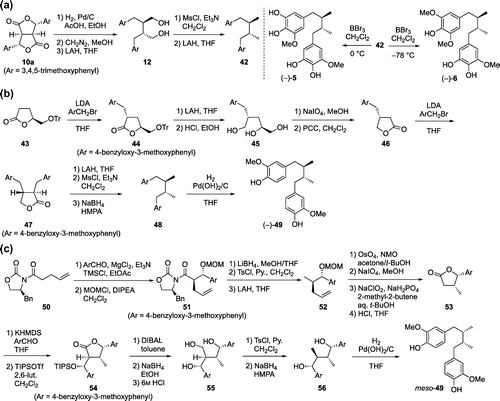
Yamauchi’s synthesis of (–)-dihydroguaiaretic acid [(–)-49], which exhibits antioxidant and antimicrobial activities, in 2008 is shown in Scheme (b) [Citation45]. Enantiomerically pure lactone 43 [Citation46] was selected as a starting material. Stereoselective alkylation of lactone 43 with 4-benzyloxy-3-methoxybenzyl bromide gave 44 in 41% yield, which was converted to triol 45 by LAH reduction and removal of the trityl group in 77% yield over two steps [Citation47]. Oxidative cleavage of the glycol followed by PCC oxidation afforded lactone 46 in 90% yield over two steps [Citation47]. Second alkylation of 46 with 4-benzyloxy-3-methoxybenzyl bromide by using LDA proceeded with excellent stereoselectivity, and 47 was obtained in 64% yield. Lactone 47 was converted to 48 in 50% yield through a three-step sequence including LAH reduction, mesylation of the diol and deoxygenation with NaBH4. Finally, debenzylation of 48 using H2 and Pd(OH)2 gave (–)-49 in quantitative yield with 99% e.e.
| (3) | Synthesis of meso-dihydroguaiaretic acid by Yamauchi et al. | ||||
Following the synthesis of (–)-dihydroguaiaretic acid [(–)-49], Yamauchi et al. accomplished the total synthesis of meso-dihydroguaiaretic acid (meso-49) in 2009 (Scheme (c)) [Citation48]. Evans’ anti-aldol reaction [Citation49] of 50 with 4-benzyloxy-3-methoxybenzaldehyde afforded the anti-aldol adduct in 99% yield [Citation47], whose hydroxyl group was protected as a methoxymethyl ether to give 51 in 96% yield. A chiral auxiliary of 51 was removed by reduction with LiBH4 in 60% yield, and the resulting alcohol was converted to 52 by tosylation and subsequent LAH reduction in 88% two steps yield. Oxidative cleavage of the double bond in 52, followed by transformation into carboxylic acid and cleavage of the methoxymethyl ether afforded lactone 53 in 97% yield over four steps. Next, aldol condensation between 53 and 4-benzyloxy-3-methoxybenzaldehyde gave a mixture of erythro/threo isomers (1:3) in 97% yield, which were separated after conversiton to triisopropyl silyl ether to give the threo product 54 in 40% yield. DIBAL reduction of 54 followed by NaBH4 reduction and treatment with 6 M HCl afforded triol 55 in 77% yield over three steps. After deoxygenation of 55 through a two-step sequence (83% yield), debenzylation of 56 using H2 and Pd(OH)2 gave meso-49 in 46% yield.
Conclusion
In this review, I describe the overview of recent synthetic studies on optically active furofuran and diarylbutane lignans. Lignans have attracted considerable attention of chemists because of their wide range of biological activities, and therefore development of the novel method for synthesizing optically active lignans in short steps is highly required. In order to respond to these demands, we have developed the asymmetric dimerization of cinnamic acid derivatives by using PbO2 or electrochemical oxidation. This method enabled us to achieve the enantioselective syntheses of furofuran lignans (yangambin, sesamin, eudesmin, caruilignan A) and diarylbutane lignans (sauriols A and B). In addition, several elegant syntheses of these lignans are also discussed. Finally, I hope that our research on lignans will contribute to the progress of chemistry and biology.
Disclosure statement
No potential conflict of interest was reported by the author.
Acknowledgments
I am deeply grateful to Professor Hidenori Watanabe (The University of Tokyo), Emeritus Professor Kenji Mori (The University of Tokyo), Emeritus Professor Takeshi Kitahara (The University of Tokyo), Professor Ken Ishigami (Tokyo University of Agriculture) for their continuous encouragement and valuable discussion. I also thank Dr. Akiko Furuta for the electrochemical oxidation of cinnamic acid derivatives.
Notes
* This review was written in response to the author’s receipt of the JSBBA Award for Young Scientists in 2017.
References
- Ward RS. Lignans, neolignans, and related compounds. Nat Prod Rep. 1993;10:1–28.10.1039/np9931000001
- Ward RS. Lignans, neolignans and related compounds. Nat Prod Rep. 1999;16:75–96.10.1039/a705992b
- Pan J-Y, Chen S-L, Yang M-H, et al. An update on lignans: natural products and synthesis. Nat Prod Rep. 2009;26:1251–1292.10.1039/b910940d
- Teponno RB, Kusari S, Spiteller M. Recent advances in research on lignans and neolignans. Nat Prod Rep. 2016;33:1044–1092.10.1039/C6NP00021E
- Dickey EE. Liriodendrin, a new lignan diglucoside from the inner bark of yellow poplar (Liriodendron tulipifera L.). J Org Chem. 1958;23:179–184.10.1021/jo01096a007
- Kakisawa H, Chen YP, Hsü HY. Lignans in flower buds of Magnolia fargesii. Phytochemistry. 1972;11:2289–2293.10.1016/S0031-9422(00)88392-2
- Castro-Faria-Neto HC, Bozza PT, Cruz HN, et al. Yangambin: a new naturally-occurring platelet-activating factor receptor antagonist: binding and in vitro functional studies. Planta Med. 1995;61:101–105.10.1055/s-2006-958025
- Castro-Faria-Neto HC, Bozza PT, Cruz HN, et al. Yangambin: a new naturally-occurring platelet-activating factor receptor antagonist: in vivo pharmacological studies. Planta Med. 1995;61:106–112.10.1055/s-2006-958026
- Marques RCP, de Medeiros SRB, Dias CS, et al. Evaluation of the mutagenic potential of yangambin and of the hydroalcoholic extract of Ocotea duckei by the Ames test. Mutat Res. 2003;536:117–120.10.1016/S1383-5718(03)00040-8
- Hausott B, Greger H, Marian B. Naturally occurring lignans efficiently induce apoptosis in colorectal tumor cells. J Cancer Res Clin Oncol. 2003;129:569–576.10.1007/s00432-003-0461-7
- Matsumura Y, Kita S, Morimoto S, et al. Antihypertensive effect of sesamin. I. Protection against deoxycorticosterone acetate-salt-induced hypertension and cardiovascular hypertrophy. Biol Pharm Bull. 1995;18:1016–1019.10.1248/bpb.18.1016
- Kita S, Matsumura Y, Morimoto S, et al. Antihypertensive effect of sesamin. II. Protection against two-kidney, one-clip renal hypertension and cardiovascular hypertrophy. Biol Pharm Bull. 1995;18:1283–1285.10.1248/bpb.18.1283
- Matsumura Y, Kita S, Tanida Y, et al. Antihypertensive effect of sesamin. III. Protection against development and maintenance of hypertension in stroke-prone spontaneously hypertensive rats. Biol Pharm Bull. 1998;21:469–473.10.1248/bpb.21.469
- Nakano D, Itoh C, Takaoka M, et al. Antihypertensive effect of sesamin. IV. Inhibition of vascular superoxide production by sesamin. Biol Pharm Bull. 2002;25:1247–1249.10.1248/bpb.25.1247
- Hirose N, Doi F, Ueki T, et al. Suppressive effect of sesamin against 7,12-dimethylbenz[a]anthracene induced rat mammary carcinogenesis. Anticancer Res. 1992;12:1259-1265.
- Hibasami H, Fujikawa T, Takeda H, et al. Induction of apoptosis by Acanthopanax senticosus HARMS and its component, sesamin in human stomach cancer KATO III cells. Oncol Rep. 2000;7:1213–1219.
- Miyahara Y, Komiya T, Katsuzaki H, et al. Sesamin and episesamin induce apoptosis in human lymphoid leukemia Molt 4B cells. Int J Mol Med. 2000;6:43–49.
- Pan JX, Hensens OD, Zink DH, et al. Lignans with platelet activating factor antagonist activity from magnolia biondii. Phytochemistry. 1987;26:1377–1379.10.1016/S0031-9422(00)81816-6
- Cho JY, Yoo ES, Baik KU, et al. Eudesmin inhibits tumor necrosis factor-α production and T cell proliferation. Arch Pharm Res. 1999;22:348–353.10.1007/BF02979056
- Lee J, Lee D, Jang DS, et al. Two new stereoisomers of tetrahydrofuranoid lignans from the flower buds of Magnolia fargesii. Chem Pharm Bull. 2007;55:137–139.10.1248/cpb.55.137
- Mbaze LM, Lado JA, Wansi JD, et al. Oxidative burst inhibitory and cytotoxic amides and lignans from the stem bark of Fagara heitzii (Rutaceae). Phytochemistry. 2009;70:1442–1447.10.1016/j.phytochem.2009.08.007
- Yang YJ, Park JI, Lee H, et al. Effects of (+)-eudesmin from the stem bark of Magnolia kobus DC. var. borealis Sarg. on neurite outgrowth in PC12 cells. Arch Pharm Res. 2006;29:1114–1118.10.1007/BF02969301
- Ma C-M, Nakamura N, Min BS, et al. Triterpenes and lignans from artemisia caruifolia and their cytotoxic effects on Meth-A and LLC tumor cell lines. Chem Pharm Bull. 2001;49:183–187.10.1248/cpb.49.183
- Kubanek J, Fenical W, Hay ME, et al. Two antifeedant lignans from the freshwater macrophyte Saururus cernuus. Phytochemistry. 2000;54:281–287.10.1016/S0031-9422(00)00076-5
- Cartwright NJ, Haworth RD. The constituents of natural phenolic resins. Part XIX. The oxidation of ferulic acid. J Chem Soc. 1944;535–537.10.1039/jr9440000535
- Taylor EC, Andrade JG, Rall GJH, et al. Thallium in organic synthesis. 60. 2,6-Diaryl-3,7-dioxabicyclo[3.3.0]octane-4,8-dione lignans by oxidative dimerization of 4-alkoxycinnamic acids with thallium(III) trifluoroacetate or cobalt(III) trifluoride. J Org Chem. 1981;46:3078–3081.10.1021/jo00328a018
- Yuzikhin OS. Vasil’ev AV, Rudenko AP. Oxidation of aromatic compounds: VIII. Oxidative dehydrodimerization of cinnamic acid derivatives in the system CF3COOH-CH2Cl2-PbO2. Russ. J Org Chem. 2000;36:1743–1754.
- Tazaki H, Taguchi D, Hayashida T, et al. Stable isotope-labeling studies on the oxidative coupling of caffeic acid via o-quinone. Biosci Biotechnol Biochem. 2001;65:2613–2621.10.1271/bbb.65.2613
- Mori N, Watanabe H, Kitahara T. Simple and efficient asymmetric synthesis of furofuran lignans yangambin and caruilignan A. Synthesis. 2006;2006(3):400–404.
- Shibuya H, Takeda Y, Zhang RS, et al. Indonesian nedicinal plants. IV. On the constituents of the bark of Fagara rhetza (Rutaceae). (2). Lignan glycosides and two apioglucosides. Chem Pharm Bull. 1992;40:2639–2646.10.1248/cpb.40.2639
- Mori N, Furuta A, Watanabe H. Electochemical asymmetric dimerization of cinnamic acid derivatives and application to the enantioselective syntheses of furofuran lignans. Tetrahedron. 2017;72:8393–8399.
- Ronlan A, Bechgaard K, Parker VD. Electrochemistry in media of intermediate acidity. VI. Coupling reactions of simple aryl ethers. Acta Chem Scand. 1973;27:2375–2382.10.3891/acta.chem.scand.27-2375
- Kise N, Fujimoto A, Moriyama N, et al. Stereoselective intramolecular coupling of diaroylacetates of (1R,1′R)-exo, exo′-3,3′-biisoborneol by oxidation with Br 2. Tetrahedron Asymmetry. 2003;14:2495–2497.10.1016/S0957-4166(03)00572-X
- Banerjee B, Roy SC. Concise enantioselective synthesis of furan lignans (–)-dihydrosesamin and (–)-acuminatin and furofuran lignans (–)-sesamin and (–)-methyl piperitol by radical cyclization of epoxides. Synthesis. 2005;2005(17):2913–2919.
- Gao Y, Hanson RM, Klunder JM, et al. Catalytic asymmetric epoxidation and kinetic resolution: modified procedures including in situ derivatization. J Am Chem Soc. 1987;109:5765–5780.10.1021/ja00253a032
- Kim J-C, Kim K-H, Jung J-C, et al. An efficient asymmetric synthesis of furofuran lignans: (+)-sesamin and (–)-sesamin. Tetrahedron Asymmetry. 2006;17:3–6.10.1016/j.tetasy.2005.11.007
- Jung J-C, Kim J-C, Moon H-I, et al. Stereoselective total synthesis of furofuran lignans through dianion aldol condensation. Tetrahedron Lett. 2006;47:6433–6437.10.1016/j.tetlet.2006.06.127
- Van Horn DE, Masamune S. Stereoselective synthesis of β-hydroxy-α-methylketones via Z- and E-vinyloxyboranes generated directly from cyclohexyl ethyl ketone. Tetrahedron Lett. 1979;20:2229–2232.10.1016/S0040-4039(01)93683-4
- Syed MK, Murray C, Casey M. Stereoselective synthesis of lignans of three structural types from a common intermediate, enantioselective synthesis of (+)-yangambin. Eur J Org Chem. 2014;5549-5556.10.1002/ejoc.v2014.25
- Weix DJ, Ellman JA. Improved synthesis of tert -butanesulfinamide suitable for large-scale production. Org Lett. 2003;5:1317–1320.10.1021/ol034254b
- Syed MK, Casey M. Enantioselective synthesis of benzyl tert-butyl sulfoxides. Eur J Org Chem. 2011;7207-7214.10.1002/ejoc.v2011.35
- Inai M, Ishikawa R, Yoshida N, et al. Stereocontrolled total synthesis of optically active furofuran lignans. Synthesis. 2015;47:3513–3521.
- Poe SL, Bogdan AR, Mason BP, et al. Use of bifunctional ureas to increase the rate of proline-catalyzed α-aminoxylations. J Org Chem. 2009;74:1574–1580.10.1021/jo802461w
- Mori N, Watanabe H, Kitahara T. Simple synthesis of enantiomerically pure sauriols A and B. Biosci Biotechnol Biochem. 2006;70:1750–1753.10.1271/bbb.60094
- Yamauchi S, Masuda T, Sugahara T, et al. Antioxidant activity of butane type lignans, secoisolariciresinol, dihydroguaiaretic acid, and 7,7′-oxodihydroguaiaretic acid. Biosci Biotechnol Biochem. 2008;72:2981–2986.10.1271/bbb.80461
- Tomioka K, Mizuguchi H, Koga K. Stereoselective reactions. V. Design of the asymmetric synthesis of lignan lctones. Synthesis of optically active podorhizon and deoxypodorhizon by 1,3-asymmetric induction. Chem Pharm Bull. 1982;30:4304–4313.10.1248/cpb.30.4304
- Yamauchi S, Hayashi Y, Nakashima Y, et al. Effect of benzylic oxygen on the antioxidant activity of phenolic lignans. J Nat Prod. 2005;68:1459–1470.10.1021/np050089s
- Kawaguchi Y, Yamauchi S, Masuda K, et al. Antimicrobial activity of stereoisomers of butane-type lignans. Biosci Biotechnol Biochem. 2009;73:1806–1810.10.1271/bbb.90167
- Evans DA, Tedrow JS, Shaw JT, et al. Diastereoselective magnesium halide-catalyzed anti -aldol reactions of chiral N -acyloxazolidinones. J Am Chem Soc. 2002;124:392–393.10.1021/ja0119548