
ABSTRACT
The leaves of Nandina domestica Thunb. exhibited high hydroxynitrile lyase (HNL) activity in (R)-mandelonitrile synthesis. The specific activity of young leaves was significantly higher than that of mature leaves. We isolated two HNLs with molecular mass of 24.9 kDa (NdHNL-S) and 28.0 kDa (NdHNL-L) from the young leaves. Both NdHNLs were composed of two identical subunits, without FAD and carbohydrates. We purified NdHNL-L and revealed its enzymatic properties. The whole deduced amino acid sequence of NdHNL-L was not homologous to any other HNLs, and the specific activity for mandelonitrile synthesis by NdHNL-L was higher than that by other plant HNLs. The enzyme catalyzed enantioselective synthesis of (R)-cyanohydrins, exhibited high activity at pH 4.0, and high stability in the pH range of 3.5–8.0 and below 55°C. Thus, NdHNL-L is a novel HNL with novel amino acid sequence and has a potential for the efficient production of (R)-cyanohydrins.
Graphical Abstract
Leaves of Nandina domestica Thunb. showing high R-hydroxynitrile lyase (HNL) activity.
Hydroxynitrile lyases (HNLs; EC 4.1.2.10, 4.1.2.11, 4.1.2.46, and 4.1.2.47) catalyze degradation of cyanohydrins to form the corresponding aldehydes or ketones, and hydrogen cyanide (cyanogenesis) in the physiological state. They are believed to play a role in the defense systems against insects or microorganisms. For example, cyanohydrins are stored as glycosides in the rubber tree Hevea brasiliensis and are hydrolyzed to cyanohydrins by β-glycosidase in response to physical damage, followed by the formation of hydrogen cyanide and aldehydes or ketones by HNL or via non-enzymatic reactions [Citation1]. The enzymes also catalyze reverse reaction and are industrially utilized for enantioselective production of cyanohydrins because chiral cyanohydrins are valuable building blocks for the synthesis of fine chemicals, pharmaceuticals, and agrochemicals such as diltiazem (cardiac drug) [Citation2] and (-)-aegeline (hypoglycemic agent) [Citation3].
To date, several HNLs have been discovered and characterized to reveal the relationship between structure and function for their industrial use. Recently, Asano et al. performed systematic screening for HNLs of plant origin by assay of chiral mandelonitrile (MAN) formation from benzaldehyde and potassium cyanide using the homogenates of leaves, roots, shoots, spikes, and rhizomes of 163 species of plants, of which 74 were cyanogenic and the remaining were non-cyanogenic plants [Citation4,Citation5]. They detected R-selective HNL (R-HNL) activities in leaves and seeds of Eriobotrya japonica (EjHNL), Prunus mume (PmHNL), Passiflora edulis (PeHNL), Chaenomeles sinensis (CsHNL), and Sorbus aucuparia (SaHNL). The former three enzymes, EjHNL [Citation6], PmHNL [Citation7,Citation8], and PeHNL [Citation9,Citation10], have been well-characterized, including X-ray structure determinations. R-HNLs have also been characterized from Prunus dulcis (P. amygdalus) (PaHNL) [Citation11–Citation14], Granulicella tundricola (GtHNL) [Citation15], Arabidopsis thaliana (AtHNL) [Citation16,Citation17], and Prunus serotina (PsHNL) [Citation18]. Of these, EjHNL, PmHNL, PaHNL and PsHNL contain FAD, whereas PeHNL, GtHNL and AtHNL do not contain FAD. In contrast, S-selective HNLs (S-HNL) have been characterized from tropical rubber tree (Hevea brasiliensis; HbHNL) [Citation19], cassava plant (Manihot esculenta; MeHNL) [Citation5,Citation20,Citation21], Sorghum bicolor (SbHNL) [Citation22], and Baliospermum montanum (BmHNL) [Citation23]. Of these S-HNLs, BmHNL is a non-FAD enzyme. Recently, HNL was discovered and characterized from an invasive millipede, Chamberlinius hualienensis, which showed much higher specific activity in MAN synthesis compared with that by plant HNLs [Citation5,Citation24]. These studies show that HNLs have a wide variety of physicochemical characteristics. HNLs are now classified into six different structure groups: FAD-dependent oxidoreductases [Citation15], α/β-hydrolases [Citation20,Citation25], Zn2+-containing alcohol dehydrogenases [Citation26], dimeric α + β barrel superfamily [Citation10], Bet v1 superfamily [Citation27], and cupin-fold enzymes [Citation15]. This is a typical example of the progress of science with fundamental discovery and highly developed applications stimulated by each other. It also indicates that new types of HNLs are yet to be discovered in the nature.
Semba and our group found that Nandina domestica Thunb produces HNL in the leaves. Therefore, we purified HNL from the leaves and found two HNLs with different molecular mass. In the present paper, we describe the effect of developmental stages on HNL production as well as the enzymatic characteristics, cDNA cloning, and the primary structure of HNL with high molecular mass.
Materials and methods
Chemicals and cultivation of N. domestica
Benzaldehyde and racemic-MAN were purchased from Sigma-Aldrich (St. Louis, MO, USA). (R)-MAN was purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA), and (S)-MAN was synthesized using MeHNL according to the method of Semba et al [Citation21]. N. domestica was purchased from a flower shop in Toyama and grown outdoors. For purification of HNL from N. domestica (NdHNL), the harvested young leaves were stored at −20°C until purification.
Screening of HNL from N. domestica cultivars
Shredding leaves (0.5 g) of N. domestica cultivars were suspended in 0.5 ml of 10 mM KPB, pH 6.0, and disrupted by a Multi-beads shocker (Yasui Kikai, Osaka) for 5 min (1 min x 5). The supernatant solution obtained by centrifugation at 16,100 x g for 20 min was used for assay of HNL activity under the following standard assay conditions for MAN synthesis.
Assay of NdHNL activity
Enzyme activity was assayed by measuring the reaction velocity of optically active MAN synthesis from benzaldehyde and cyanide and that for MAN degradation.
Synthesis activity
HNL activity in MAN synthesis was assayed at 25°C using 50 mM benzaldehyde, 100 mM KCN, and 0.3 M sodium citrate buffer, pH 4.0, according to the method of Asano et al [Citation4]. In brief, the reaction mixture containing benzaldehyde and enzyme was incubated at 25°C for 3 min, and the reaction was initiated by addition of KCN and incubated for 5 min. The reaction was stopped by adding 100 μL of the reaction mixture into 900 μL of the solvent, containing n-hexane:2-propanol [85:15 (v/v)]. Each enzymatic reaction was subtracted from the non-enzymatic reaction. The amount of benzaldehyde and (R)- and (S)-MAN were determined at 254 nm using a HPLC system equipped with a Chiralcel OJ-H column (particle size = 5 μm; i.d. = 4.6 mm × 250 mm; Daicel Corporation, Osaka, Japan) and a Prominence UV-Vis Detector SPD-20A (Shimadzu, Kyoto, Japan). For HPLC, the solvent, n-hexane:2-propanol, 85:15 (v/v) was used as the mobile phase at a flow rate of 1 mL min−1. One unit of enzyme activity was defined as the amount of enzyme catalyzing the formation of one μmol of (R)-MAN per min. All experiments were performed in triplicates.
Degradation activity
HNL activity for MAN degradation to benzaldehyde was assayed by monitoring the absorbance at 280 nm using an UV-Vis spectrophotometer [Citation19]. The reaction mixture containing 2 mM racemic MAN and 50 mM sodium citrate buffer, pH 5.5, was incubated at 30°C for 3 min. The reaction was started by addition of the enzyme solution, and the absorbance change at 280 nm was immediately measured at 30°C for 2 min. Because racemic MAN slowly decomposes under this condition, the rate of MAN cleavage without the enzyme was simultaneously determined for each assay and subtracted from the total rate. One unit of enzyme activity in MAN degradation was defined as the amount of enzyme catalyzing the production of one μmol of benzaldehyde per min.
Purification of enzyme
All procedures were performed at 4–10°C, and the buffer used was potassium phosphate buffer, pH 6.0, containing 5 mM 2-mercaptoethanol, unless otherwise stated.
Preparation of crude enzyme solution
Young leaves of N. domestica (50 g) were suspended in 200 mL of 10 mM buffer and crushed for 2–3 min using a juicer mixer. Then, the debris was removed by filtration with a gauze and centrifugation (16,000 × g for 30 min). This step was performed 10 times, and the supernatants obtained were used as the crude enzyme solution (total of 2 L). The crude enzyme solution was dialyzed against 10 mM buffer until the color of the enzyme solution changed to yellow.
Ammonium sulfate fractionation
Solid ammonium sulfate (418 g) was added to the dialyzed enzyme solution to 35% saturation, and the resulting precipitate was removed by centrifugation at 16,000 × g for 30 min. Then, ammonium sulfate concentration of the supernatant was increased up to 60% saturation. The resulting precipitate was collected by centrifugation at 16,000 × g for 30 min and dissolved in 10 mM buffer. Then, the enzyme solution was dialyzed against 10 mM buffer.
DEAE-Toyopearl column chromatography
The dialyzed enzyme solution was applied to a DEAE-Toyopearl column (9 × 2.8 cm diameter) equilibrated with 10 mM buffer. After the column was washed with 10 mM buffer, the adsorbed enzyme was eluted by a linear gradient of 10 mM buffer and 10 mM buffer containing 0.3 M NaCl (250 mL each), and active fractions were collected.
Butyl-Toyopearl column chromatography
To the eluate obtained from DEAE-Toyopearl column chromatography, ammonium sulfate was added to 30% saturation, and the enzyme solution was applied to a Butyl-Toyopearl column (4.5 × 2.8 cm diameter) equilibrated with 10 mM buffer containing 1.33 M ammonium sulfate. After the column was washed with 10 mM buffer containing 1.33 M ammonium sulfate, the adsorbed enzyme was eluted by a linear gradient of 10 mM buffer containing 1.33 M ammonium sulfate and 10 mM buffer containing 0.42 M ammonium sulfate (100 mL each). The active fractions were combined and dialyzed against 10 mM buffer.
GigaCapQ-Toyopearl column chromatography
The dialyzed enzyme solution was applied to a GigaCapQ-Toyopearl column (9 × 2 cm diameter) equilibrated with 10 mM buffer. After the column was washed with 10 mM buffer, the adsorbed enzyme was eluted by a linear gradient of 10 mM buffer containing 80 mM NaCl and 0.3 M NaCl (75 mL each). The active fractions were combined and concentrated by ultrafiltration (Amicon Ultra-15, Merck Millipore).
Substrate specificity and kinetic analysis
Substrate specificity was analyzed under standard assay conditions of cyanohydrin synthesis using 50 mM aldehyde and 100 mM KCN. The kinetic parameters (Km and Vmax) were calculated by a non-linear least-squares curve fitting against plots of the initial velocity vs [S] using the Michaelis–Menten equation.
The reaction products were identified by HPLC according to the method established previously [Citation19], and conversion yield (%) was calculated by the following equation:
{nitrile compounds (mM)/aldehydes (mM)} × 100.
Enantiomeric excess (e.e.; %) was calculated by following equation: {(R)-nitrile compound (mM) − (S)-nitrile compound (mM)}/{(R)-nitrile compound (mM) + (S)-nitrile compound (mM)} × 100.
Effects of temperature and pH on enzyme activity and stability
Effect of temperature on enzyme activity was evaluated by measuring the MAN synthetic activity at pH 4.0 and a temperature range of 5–50°C. Thermal stability was analyzed under standard assay conditions of MAN synthesis after enzyme solution was incubated at pH 6.0 for 30 min at 15–80°C without the substrate. Effect of pH on the synthetic activity was evaluated under standard assay conditions using different buffers and pH values. The pH stability was determined under standard assay conditions for MAN synthesis after the enzyme solution was incubated at 30°C for 30 min at a pH range of 3.0–10.0 without the substrate.
Amino acid sequence analysis of the enzyme
The N-terminal amino acid sequence was determined using a gas-phase protein sequencer equipped with an on-line reverse-phase chromatography system (Procise492HT Protein Sequencer, Applied Biosystems, CA, USA) for identification of PTH-amino acids.
The internal amino acid sequences of peptides digested by trypsin or Lys-C were analyzed as follows: a) Trypsin-digested peptide: After the purified enzyme was separated on 12% SDS-PAGE, the protein was excised and incubated with trypsin (sequence grade-modified trypsin). The trypsin-digested peptides were separated by a nanoUPLC equipped with a trap column (nanoACQUITY UPLC Symmetry C18 Trap Column, Waters, Milford, MA, USA) and a reverse phase capillary column (ACQUITY UPLC Peptide CSH C18 nanoACQUITY Column 10K psi, Waters) using acetonitrile-water containing 0.1% formic acid. The molecular mass of each peptide was determined by an electrospray ionization/quadrupole time-of-flight mass spectrometer (nanoACQUITY UPLC-SYNAPT G2-Si, Waters). The peptide sequence was analyzed using the Biolynx software suite (Waters). b) Lys-C digested peptide: The purified enzyme (1.0 mg) was dissolved in 280 µL of 25 mM Tris-HCl buffer (containing 6 M guanidine HCl and 2 mM EDTA; pH 8.5); 20 µL of 1.4 M DTT was added to the mixture and incubated with 10 µU of Lys-C (Roche Diagnostics GmbH, Mannheim, Germany) at 30°C for 24 h. The peptides generated by Lys-C digestion were separated by a C4 column (4.6 x 150 mm; Vydac, CA, USA) using 0.1% trifluoroacetic acid (TFA) in water and 0.1% TFA in acetonitrile. The amino acid sequence was analyzed by the gas-phase protein sequencer method.
cDNA cloning
Total RNA was isolated from fresh young leaves of N. domestica (60 mg) using Plant RNA Reagent (Invitrogen, Carlsbad, CA, USA) and re-purified using an RNeasy Mini Kit (Qiagen, Hilden, Germany) according to the manufacture’s protocol. After quality verification using the Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA, USA), purified total RNA was sequenced with no biological replicates at the Beijing Genomics Institute using Hiseq 2000 (Illumina, Hayward, CA, USA) equipped with a paired-end flow cell. The RNA-seq data of N. domestica were assembled using Trinity version 2.20 (https://github.com/trinityrnaseq/trinityrnaseq/wiki) [Citation28] with default parameters. The assembled sequences were deposited in our local BLAST server. A partial cDNA sequence encoding NdHNL was retrieved using the sequences of N-terminal region and trypsin-digested peptide as the query in TBLASTN searches of the N. domestica transcriptome sequences.
Rapid amplification of cDNA ends (RACE)-ready cDNA was synthesized using the SMART RACE cDNA Amplification Kit (Clontech Laboratories, Palo Alto, CA, USA). Based on the partial sequence of cDNA-encoding NdHNL, primers P1–P8 were designed (Table S1). The fragment of 5′-RACE was amplified by PCR with primers P1 and P2 using Tks Gflex DNA polymerase (Takara Bio, Shiga, Japan), and the PCR product was used for nested PCR using primers P3 and P4. The fragment of 3′-RACE was amplified by PCR with primers P5 and P6 in the same manner, and the product was used for nested PCR with primers P7 and P8. The PCR products were purified by NucleoSpin Gel and PCR Clean-up (Macherey-Nagel, Duren, Germany). The DNA sequences were determined using a 3500 Genetic Analyzer (Applied Biosystems, CA, USA). The full-length cDNA sequence was determined using eight independent clones.
Construction of the Escherichia coli expression vector
The NdHNL gene (ndhnl) was amplified by PCR from cDNA using primers P9 and P10 for expression of the original sequence and P10 and P11 for expression with N-terminal his-tag (Table S1). The linear plasmids were amplified with primer P12 and P13 for expression of the original sequence and P13 and P14 for expression with N-terminal his-tag. The inserted DNA was ligated to the plasmid as mentioned above, generating pColdI–ndhnl and pColdI–ndhnl–Nhis. For construction of an expression plasmid with a C-terminal his-tag, the linear DNA was amplified by PCR with primers P15 and P16. The DNA was treated with DpnI at 37°C for 2 h, and self-ligated, generating pColdI–ndhnl–Chis.
Expression of recombinant NdHNL.
E. coli BL21 (DE3) was transformed with pColdI–ndhnl, pColdI–ndhnl–Nhis, or pColdI–ndhnl–Chis and transformants were cultured in Luria–Bertani (LB) medium (1.0% peptone, 1.0% NaCl, and 0.5% yeast extract) containing 100 µg/mL ampicillin at 37°C until the optical density at 600 nm reached 0.5, at which point 0.5 µM isopropyl-β-D-thiogalactopyranoside was added into the culture for induction of NdHNL. Culture growth was continued for another 16 h at 16°C, and cells were harvested by centrifugation.
Other analytical methods
Protein concentration was measured by the method of Bradford [Citation29] using a Bio-Rad protein assay kit (Bio-Rad Laboratories, Hercules, CA, U.S.A.) and bovine serum albumin as the standard protein.
Native- and SDS-PAGE were performed according to the methods of Davis [Citation30] and Laemmli [Citation31], respectively.
Molecular mass of the native enzyme was estimated by gel filtration on Superdex 200 10/300GL (GE Healthcare UK Ltd., Buckinghamshire, UK) using the following marker proteins: glutamate dehydrogenase (290 kDa), lactate dehydrogenase (142 kDa), enolase (67.0 kDa), myokinase (32.0 kDa), and cytochrome c (12.4 kDa). Molecular mass of the denatured enzyme was calculated by SDS-PAGE using phosphorylase b (97.4 kDa), bovine serum albumin (66.2 kDa), ovalbumin (45.0 kDa), carbonic anhydrase (31.0 kDa), and soybean trypsin inhibitor (21.5 kDa).
Carbohydrates in the purified enzyme were analyzed by the phenol/sulfuric acid method according to Hodge and Hofreiter [Citation32]. The reaction mixture, containing 0.5 mL of the purified enzyme or mannose, 0.5 mL of 5% phenol, and 2.5 mL of concentrated sulfuric acid, was mixed and allowed to stand at room temperature for 20 min. Then, absorbance was measured at 490 nm. The presence of carbohydrates in the enzyme was also analyzed by the periodic acid–Schiff (PAS) staining method using a Pierce Glycoprotein Staining Kit (Thermo Fisher Scientific, Waltham, MA, USA).
Bioinformatics analysis was performed using the BLAST program (blast.ncbi.nlm.nih.gov/Blast.cgi) [Citation33], and NetNGlyc 1.0 Server (www.cbs.dtu.dk/services/).
Results
Enzyme production
The activity of HNL from the fresh leaves of N. domestica, N. domestica var. leucocarpa, and N. domestica cv. Otafukunanten in MAN synthesis was assayed using the crude enzyme solution. N. domestica exhibited highest enzyme activity (Table S2). Hence, HNL activity of N. domestica was assayed using three types of leaves, young leaves of tender green color, mature leaves of deep green color, and mature leaves of red color. Of these three leaves, young green leaves showed remarkably high activity compared with both of the mature leaves. Furthermore, the specific activity (units per mg of protein) of young leaves was much higher than those of the mature leaves (). Therefore, we purified NdHNL from young leaves of N. domestica.
Table 1. HNL activities detected in the leaves of N. domestica Thunb harvested in several growth phases.
Purification and molecular mass of NdHNL
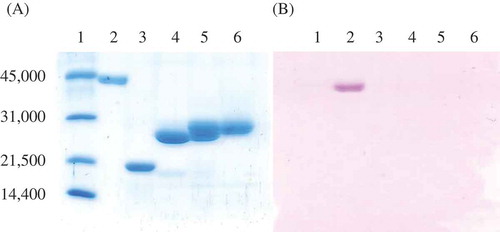
During the purification of NdHNLs, HNL activity was separated into two peaks by GigaCapQ-Toyopearl column chromatography. The first peak showed a molecular mass of 24.9 kDa (NdHNL-S) and the second peak showed a molecular mass of 28.0 kDa (NdHNL-L) on SDS-PAGE (). The molecular mass of native NdHNL-S and -L was estimated to be 46–47 kDa on a Superdex 200 column, indicating that both enzymes may exist in the dimer form with two identical subunits in the native state. The specific activity of the purified NdHNL-S was slightly higher than that of NdHNL-L, but the difference was not significant (NdHNL-S and -L are fractions A and C, respectively, in ).
Table 2. Purification of HNL from N. domestica Thunb.
Figure 1. SDS-PAGE analysis of NdHNL eluates from GigCapQ and PAS staining.
(A) SDS-PAGE analysis. (B) PAS staining. 1, Standard proteins for molecular mass; 2, Positive protein for PAS staining; 3, Negative protein for PAS staining; 4, Fraction A mentioned in (NdHNL-S); 5, Fraction B mentioned in ; and 6, Fraction C mentioned in (NdHNL-L).
Amino acid sequence and other physicochemical properties of NdHNL-Sand -L
The N-terminal and internal amino acid sequences of NdHNL-S and -L were analyzed using mature enzymes and Lys-C-digested peptides. The N-terminal amino acid sequence ESTVKVTPMI was identical in both NdHNLs. The internal peptide sequences GFNFPASLVE and DAIQQFLDXF were also identical in both NdHNLs. The purified enzyme solution of both the enzymes exhibited absorption maximum at 275 nm, with no absorption maxima in the visible region (Fig. S1), indicating that both enzymes do not contain flavin as a cofactor. It was also confirmed that both the enzymes were not glycosylated because yellow and purple colors were not developed in both enzymes by the phenol/sulfuric acid method and PAS staining, respectively ().
Thus, certain physicochemical properties and the N-terminal and certain internal amino acid sequences of NdHNL-S and -L were similar, although the specific activity in MAN synthesis and molecular mass of the subunit were slightly different from each other. Then, further studies were performed on NdHNL-L.
Substrate specificity, stereospecificity, and kinetic values of NdHNL-L
The substrate specificity of NdHNL-L was assayed under standard assay conditions of MAN synthesis at pH 4.0 using 50 mM benzaldehyde and its derivatives. The enzyme exhibited high activity toward benzaldehyde, 4-methoxybenzaldehyde, and 4-methylbenzaldehyde and produced the corresponding R-products with high e.e. Moreover, the enzyme reacted with 2-methylbenzaldehyde and 2,4-dimethylbenzaldehyde and produced the corresponding R-product with high e.e. However, the enzyme activity toward 2-methylbenzaldehyde, 2,4-dimethylbenzaldehyde and 3-methoxybenzaldehyde was very low ().
Table 3. Substrate specificity of HNL-L from N. domestica Thunb.
The kinetic parameters of this enzyme for benzaldehyde were analyzed in (R)-MAN synthesis reaction (). Km and Vmax values were estimated to be 12.5 mM and 1,230 µmol/min/mg of protein, respectively. Thus, it was revealed that NdHN-L had the highest Vmax value among plant HNLs in (R)-MAN synthesis.
Table 4. Kinetic parameters of HNL-L from N. domestica Thunb for benzaldehyde and KCN in MAN synthesis reaction.
Effects of pH and temperature on enzyme activity and stability of NdHNL-L
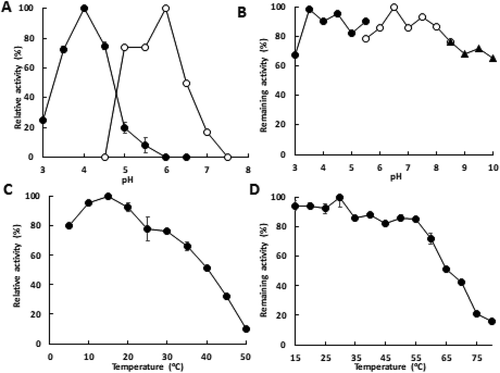
The effects of pH on MAN synthesis and degradation were assayed under standard assay conditions, by varying the reaction pH between 3.0 and 8.0. The enzyme exhibited the highest activity for MAN synthesis at pH 4.0 and no activity over pH 6.0. In contrast, activity for MAN degradation was optimal at pH 6.0, and no activity was observed below pH 4.5 and over pH 7.5 (). When the enzyme was incubated at 30°C for 30 min in the pH range of 3.0–10 without the substrate, the enzyme was stable in the pH range of 3.5–8.0 (). The enzyme exhibited highest activity at 15°C, and the activity at 50°C was < 10% of the highest activity (). Thermal stability was analyzed after the enzyme was incubated at 15–80°C for 30 min at pH 6.0 without the substrate. More than 80% of the original activity was retained after incubation at 55°C (). These results indicated that the enzyme was stable without the substrate but was remarkably unstable by incubation with benzaldehyde and KCN.
Figure 2. Effects of pH and temperature on enzyme activity and stability.
(A) Effect of pH on enzyme activity. Enzyme activities in MAN synthesis (closed circles) and MAN degradation (open circles) were assayed in the pH range of 3.0–8.0. (B) Effect of pH on enzyme stability. pH stability was analyzed by assaying the enzyme activity in MAN synthesis under standard assay conditions by incubating the enzyme at 30°C for 30 min without the substrate in the pH range of 3.0–10.0 using citrate buffer (pH 3.0–5.5; closed circles), phosphate buffer (pH 5.5–8.5; open circles), and glycine–NaOH buffer (pH 8.5–10.0; closed triangles). (C) Effect of temperature on enzyme activity. Enzyme activities in MAN synthesis were assayed under standard assay conditions, except for the reaction temperatures. (D) Effect of temperature on enzyme stability. Thermal stability was analyzed by assaying the enzyme activity in MAN synthesis under standard assay conditions by incubating the enzyme at indicated temperatures at pH 6.0 for 30 min without the substrate. The error bars represent the standard deviation of triplicates.
Effects of compounds and metals on NdHNL-L activity
Effects of different compounds on MAN synthesis were assayed by adding 1 mM carbonyl reagents, chelating reagents, sulfhydryl reagents, or metals. The enzyme activity was strongly inhibited by Cu2+ but not by other metals, chelating agents, carbonyl reagents, and sulfhydryl reagents (Table S3).
Cloning of cDNA encoding NdHNL-L and heterologous production
Using the N-terminal and internal amino acid sequences (TAEELLD, which was obtained from trypsin-digested peptide), cloning of cDNA encoding NdHNL-L was performed as follows: Total RNA was isolated from fresh young leaves of N. domestica Thunb, and the sequence containing the N-terminal and internal amino acid sequences (TAEELLD) was analyzed using the contigs of transcriptome analysis as described in Materials and Methods. Then, the full-length cDNA encoding NdHNL-L, a 729-bp fragment encoding 243 amino acid residues, was cloned by 5′- and 3′-RACE. The predicted molecular mass 26.6 kDa of the open reading frame was similar to that of the NdHNL-L protein estimated by SDS-PAGE. The deduced amino acid sequence of the N-terminal region was identical to that of NdHNL-L. The sequence TAEELLD, which was used for cloning cDNA encoding NdHNL-L, was confirmed in the residue numbers of 73–79. The sequences GFNFPASLVE, DAIQQFLDXF, and RSIANRMSK, which were obtained from Lys-C-digested peptides, were also confirmed in the residue numbers of 13–22, 133–142, and 207–214 of the deduced amino acid sequences, respectively (). These results indicated that the isolated gene was NdHNL-L and it was submitted in the DDBJ nucleotide sequence database (accession LC379213).
Figure 3. Deduced amino acid sequence of NdHNL.
The amino acid sequences used for cloning ndhnl are shown in blue color. The amino acid sequences identified by the protein sequence analysis are underlined. The glycosylation sites (NQT and NGT) predicted by NetNGlyc 1.0 Server are shown by double underlines.

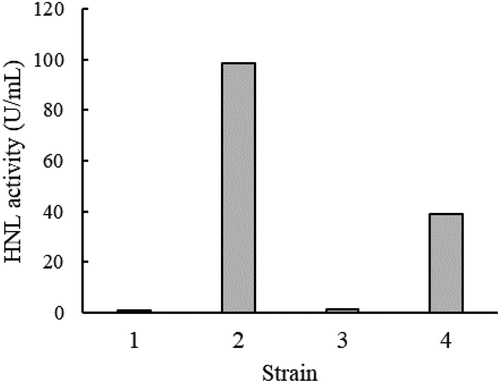
Next, ndhnl was overexpressed in E. coli BL21 (DE3) using pCold system using three plasmids, pColdI–ndhnl, pColdI–ndhnl–Nhis, and pColdI–ndhnl–Chis. Of these plasmids, pColdI–ndhnl and pColdI–ndhnl–Chis were expressed at 16°C, and HNL activities of 19.7 U/mL and 7.76 U/mL of culture, respectively, were obtained, but pColdI–ndhnl–Nhis was not expressed (). The purified recombinant NdHNL-L exhibited specific activity similar to that of the wild-type enzyme. The recombinant NdHNL-L with His-tag at C-terminus was also purified to the homogeneous state by Ni-affinity chromatography, and its specific activity was 733 U/mg of protein.
Figure 4. Production of NdHNL by the E. coli expression system.
The E. coli strains harboring pColdI (1), pColdI–ndhnl (2), pColdI – ndhnl–Nhis (3), and pColdI – ndhnl–Chis (4) were cultured, and the HNL activity of each crude extract was assayed under standard assay conditions of MAN synthesis. The values are means of triplicates.
Discussion
During the last two decades, different types of HNLs have been identified from plants, bacteria, and millipedes [Citation4–Citation23]. In this study, we isolated two novel HNLs, NdHNL-S and -L, from young leaves of N. domestica. Both enzymes enantioselectively catalyzed (R)-MAN synthesis, and the specific activity of NdHNL-S was slightly higher than that of NdHNL-L in (R)-MAN synthesis, but the difference was not significant. Moreover, both the enzymes exhibited different molecular masses, but the N-terminal and certain internal amino acid sequences were identical and carbohydrates were not detected in the enzymes (). Both of them were not flavoproteins. These results indicated that the basic primary structures of NdHNL-L and -S are similar to each other, whereas their molecular masses are different. The multiple forms of HNL were reported in PaHNL (five forms) [Citation16,Citation17], PsHNL (five forms) [Citation14], and PmHNL (at least three forms) [Citation8], while the nature and physiological significance of this heterogeneity are poorly understood. Since these enzymes are glycoproteins, they are presumed to show multiple forms due to the differences in the primary structures and/or carbohydrate side-chains. In the case of NdHNL, the two enzymes with different molecular masses might have slightly different amino acid sequences, because NdHNL does not contain carbohydrates.
NdHNL-L showed the same R-stereospecificity as PmHNL [Citation7], PaHNL [Citation11], PsHNL [Citation18], EjHNL [Citation6], PeHNL [Citation9], AtHNL [Citation16,Citation17], GtHNL [Citation15], CsHNL [Citation4], and SaHNL [Citation4]. Of these R-HNLs, PmHNL, PaHNL, PsHNL, and EjHNL contain FAD, but no FAD-binding motif was recognized in the sequence of NdHNL-L, indicating that NdHNL-L is different from these plant HNLs. Among the non-FAD R-HNLs, such as PeHNL, AtHNL and NdHNL, PeHNL is a monomeric enzyme with a molecular mass of 15–18 kDa [Citation9], whereas NdHNL-L is a dimeric enzyme with a 28-kDa subunit. AtHNL is a dimeric enzyme without carbohydrates, similar to NdHNL, but the specific activity for MAN synthesis is remarkably lower than that of NdHNL, and the enzymatic properties including the primary structure are also different from those of NdHNL (). Thus, NdHNL is unique among plant R-HNLs.
Table 5. Characteristics of R-stereospecific HNLs from plants.
NdHNL-L exhibited the highest specific activity in MAN synthesis as compared with that shown by other plant HNLs, whereas the specific activity of NdHNL-L was lower than that of HNL from an invasive millipede C. hualienensis (ChuaHNL, specific activity is 7,420) [Citation24]. Since NdHNL-L was easily produced in E. coli expression system, we concluded that NdHNL-L has suitable properties for applications such as the enzymatic synthesis of (R)-cyanohydrins.
We revealed amino acid sequence of NdHNL-L and compared it with those of other plant HNLs. The deduced amino acid sequence of NdHNL-L showed the highest similarity to that of chalcone flavonone isomerases from plants such as chalcone-flavonone isomerase-like protein from Malus domestica (51% identity, XP_008392235.1), chalcone isomerase from Camellia sinensis (49% identity, ASU87415.1) and chalcone-flavonone isomerase from Actinidia chinensis var.chinensis (48% identity, PSR93060.1), but the sequence identity to that of the known HNLs was less than 20%. Thus, NdHNL-L is a novel R-specific HNL with a unique amino acid sequence. Since it was reported that the chalcone-flavonone isomerase is an “open-faced β-sandwitch fold” [Citation34], NdHNL-L is expected to have a new type of structure among R-specific HNL. The crystal structure analysis of NdHNL-L is now in progress.
Author contributions
Conceived and designed the experiments: KI KF HS YA. Performed the experiments: AK KK NK DM TY. Analyzed the data: KI MD KF YA. Wrote the paper: KI AK YA. Revising the manuscript: KI DM TY YA.
Revised_Suppl-20180525.docx
Download MS Word (68.3 KB)Acknowledgments
We thank A. Ina for assistance in internal amino acid sequence analysis, and D. Zheng for assistance in synthesis of some chemical compounds. This work was supported by ERATO (Exploratory Research for Advanced Technology Program) Asano Active Enzyme Molecule Project of Japan Science and Technology Agency (Grant Numbers JPMJER1102). This research was also supported in part by a grant-in-aid for Scientific Research (S) from The Japan Society for Promotion of Sciences (No. 17H06169) to Y. Asano.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplemental data
supplementry data for this article can be accessed here.
Additional information
Funding
Related Research Data
References
- Kadow D, Voss K, Selmar D, et al. The cyanogenic syndrome in rubber tree Hevea brasiliensis: tissue-damage-dependent activation of linamarase and hydroxynitrile lyase accelerates hydrogen cyanide release. Ann Bot. 2012;109:1253–1262.
- Matthews BR, Gountzos H, Jackson WR, et al. Synthesis of threo-3-aryl-2,3-dihydroxypropanoic acid derivatives with high optical purity. Tetrahedron Lett. 1989;30:5157–5158.
- Brown RFC, Jackson WR, McCarthy TD. The synthesis of homochiral naturally occurring hydroxy amides. Tetrahedron: Asymmetry. 1993;4:205–206.
- Asano Y, Tamura K, Doi N, et al. Screening for new hydroxynitrilases from plants. Biosci Biotechnol Biochem. 2005;69:2349–2357.
- Dadashipour M, Asano Y. Hydroxynitrile lyases: insights into biochemistry, discovery and engineering. ACS Catal. 2011;1:1121–1149.
- Ueatrongchit T, Kayo A, Komeda H, et al. Purification and characterization of a novel (R)-hydroxynitrile lyase from Eriobotrya japonica (loquat). Biosci Biotech Biochem. 2008;72:1513–1522.
- Nanda S, Kato Y, Asano Y. A new (R)-hydroxynitrile lyase from Prunus mume:asymmetric synthesis of cyanohydrins. Tetrahedron. 2005;61:10908–10916.
- Fukuta Y, Nanda S, Kato Y, et al. Characterization of a new (R)-hydroxynitrile lyase from the Japanese apricot Prunus mume and cDNA cloning and secretory expression of one of the isozymes in Pichia pastoris, Biosci. Biotech Biochem. 2011;75:214–220.
- Ueatrongchit T, Tamura K, Ohmiya T, et al. Hydroxynitrile lyase from Passiflora edulis: purification, characteristics and application in asymmetric synthesis of (R)-mandelonitrile. Enzyme Microb Technol. 2010; 46:456–465.
- Motojima F, Nuylert A, Asano Y. The crystal structure and catalytic mechanism of hydroxynitrile lyase from passion fruit, Passiflora edulis, FEBS J. 2018;2:313–324.
- Lauble H, Muller K, Schindelin H, et al. Crystallization and preliminary X-ray diffraction studies of mandelonitrile lyase from almonds Proteins: Struct Funct Bioinf. 1994;19:343–347.
- Seely MK, Criddle RS, Conn EE. The metabolism of aromatic compounds in higher plants VIII. On the requirement of hydroxynitril lyase for flavin. J Biol Chem. 1966;241:4456–4462.
- Jorns MS. Studies on the kinetics of cyanohydrin synthesis and cleavage by the flavoenzyme oxynitrilase. Biochim Biophys Acta. 1980;613:203–209.
- Hochuli E. A rapid purification of D-oxynitrilase from almond meal by affinity chromatography. Helv Chim Acta. 1983;66:489–493.
- Hajnal I, Lyskowski A, Hanefeld U, et al. Biochemical and structural characterization of a novel bacterial manganese-dependent hydroxynitrile lyase. FEBS J. 2013;280:5815–5828.
- Guterl J-K, Andexer JN, Sehl T, et al. Uneven twins: comparison of two enantiocomplementary hydroxynitrile lyases with α/β-hydrolase fold. J Biotechnol. 2009;141:166–173.
- Andexer J, Von Langermann J, Mell A, et al. R-selective hydroxynitrile lyase from Arabidopsis thaliana with an α/β-hydrolase fold. Angew Chem Int Ed. 2007;46:8679–8681.
- Yemm RS, Poulton JE. Isolation and characterization of multiple forms of mandelonitrile lyase from mature black cherry (Pwnus serofina Ehrh) seeds. Arch Biochem Biophys. 1986;247:440–445.
- Hanefeld U, Straathof AJ, Heijnen JJ. Study of the (S)-hydroxynitrile lyase from Hevea brasiliensis: mechanistic implications. Biochim Biophys Acta (BBA)-Protein Structure Molecular Enzymology. 1999;1432:185–193.
- Hughes J, Decarvalho J, Hughes MA. Purification, characterization, and cloning of α-hydroxynitrile lyase from Cassava (Manihot esculenta Crantz). Arch Biochem Biophys. 1994;311:496–502.
- Semba H, Dobashi Y, Matsui T. Expression of hydroxynitrile lyase from Manihot esculenta in yeast and its application in (S)-mandelonitrile production using an immobilized enzyme reactor. Biosci Biotech Biochem. 2008;72:1457–1463.
- Lauble H, Miehlich B, Förster S, et al. Crystal structure of hydroxynitrile lyase from Sorghum bicolor in complex with the inhibitor benzoic acid: a novel cyanogenic enzyme. Biochemistry. 2002;41:12043–12050.
- Dadashipour M, Yamazaki M, Momonoi K, et al. S-Selective hydroxynitrile lyase from a plant Baliospermum montanum: molecular characterization of recombinant enzyme. J Biotechnol. 2011;153:100–110.
- Dadashipour M, Ishida Y, Yamamoto K, et al. Discovery and molecular and biocatalytic properties of hydroxynitrile lyase from an invasive millipede Chamberlinius hualienensis. Proc Natl Acad Sci USA. 2015;112:10605–10610.
- Nakano S, Dadashipour M, Asano Y. Structural and functional analysis of hydroxynitrile lyase from Baliospermum montanum with crystal structure, molecular dynamics and enzyme kinetics. Biochim Biophys Acta Proteins and Proteomics. 2014;1844:2059–2067.
- Trummler K, Wajant H. Molecular cloning of acetone cyanohydrin lyase from flax (Linum usitatissimum): definition of a novel class of hydroxynitrile lyases. J Biol Chem. 1997;272:4770–4774.
- Lanfranchi E, Pavkov‐Keller T, Koehler E-M, et al. Enzyme discovery beyond homology: a unique hydroxynitrile lyase in the Bet v1 superfamily. Sci Rep. 2017;7:46738.
- Grabherr MG, Haas BJ, Yassour M, et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 2011;29:644–652.
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254.
- Davis BJ. Disc electrophoresis II Method and application to human serum proteins. Ann NY Acad Sci. 1964;121:404–427.
- Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685.
- Hodge JE, Hofreiter BT. Determination of reducing sugars and carbohydrates. In: Whistler RL, Jnb M, eds. Methods in carbohydrate chemistry 1. New York: Academic Press; 1962. p. 380–389.
- Johnson M, Zaretskaya I, Raytselis Y, et al. NCBI BLAST: a better web interface. Nucleic Acids Res. 2008;36:W5–9.
- Joseph MJ, Marianne EB, Richard AD, et al. Structure and mechanism of the evolutionarily unique plant enzyme chalcone isomerase. Nat Struct. Biol. 2000;7:786–791.