
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
Site saturation mutagenesis library is a recently developed technique, in which any one out of all amino acid residues in a target region is substituted into other 19 amino acid residues. In this study, we used this technique to increase the thermostability of a GH10 xylanase, XynR, from Bacillus sp. strain TAR-1. We hypothesized that the substrate binding region of XynR is flexible, and that the thermostability of XynR will increase if the flexibility of the substrate binding region is decreased without impairing the substrate binding ability. Site saturation mutagenesis libraries of amino acid residues Tyr43–Lys115 and Ala300–Asn325 of XynR were constructed. By screening 480 clones, S92E was selected as the most thermostable one, exhibiting the residual activity of 80% after heat treatment at 80°C for 15 min in the hydrolysis of Remazol Brilliant Blue-xylan. Our results suggest that this strategy is effective for stabilization of GH10 xylanase.
Abbreviations: DNS: 3,5-dinitrosalicylic acid; RBB-xylan: Remazol Brilliant Blue-xylan
GRAPHICAL ABSTRACT
Site saturation mutagenesis library of GH10 xylanase XynR was constructed, expressed in E. coli expression system, and screened.
In protein engineering, rational design and random mutagenesis are two major strategies. In random mutagenesis, error-prone PCR is the most frequently used to create a mutation. However, it also creates a number of mutations that do not correspond to amino acid substitution including the insertion of termination codon. Site saturation mutagenesis library has recently been devised to overcome this drawback. It can randomize a set of codons at any one out of amino acid residues in a target region and produce a library of variants in which one amino acid residue is substituted into other 19 amino acid residues [Citation1–Citation3]. We previously used this technique to increase the thermostability of Moloney murine leukemia virus (MMLV) reverse transcriptase (RT) and obtained a thermostable variant D200C [Citation4].
Xylanase [EC 3.2.1.8] is a glycoside hydrolase (GH) that catalyzes the hydrolysis of internal β-1,4-linkage of xylan. Xylanase is used in various fields including paper and pulp, food, and biofuel industries [Citation5–Citation7]. In such industries, the reaction is carried out at high temperature and pH conditions so that xylanase with high stability at such conditions has been anticipated [Citation8]. According to the sequence similarities, two main families (GH10 and GH11) have been identified [Citation9]. GH10 xylanase has (β/α)8 TIM barrel fold with a shallow active-site cleft, while GH11 xylanase has a β-jellyroll fold with a deep active-site cleft [Citation10]. Although GH10 and GH11 xylanases have two catalytic Glu residues in the active site, they do not have sequence similarity. Generally, GH10 xylanase is more thermostable than GH11 xylanase [Citation11].
XynR is a thermophilic and alkaline GH10 xylanase, identified in the culture broth of alkaliphilic and thermophilic Bacillus sp. strain TAR-1 isolated from soil sample from Kanagawa, Japan [Citation12]. The molecular mass of XynR is 40 kDa, and the optimum reaction temperature and pH are 70‒75°C and 5.0‒9.5, respectively [Citation12]. In the culture broth of TAR-1, GH11 xylanase was also identified, whose molecular mass is 23 kDa and optimum reaction temperature and pH are 70°C and 5.0‒8.0, respectively [Citation13].
Thermostability of xylanase is important for their wide-range practical use, and screening and engineering of thermostable xylanase have extensively been done. In this study, we attempted to increase the thermostability of XynR using site saturation mutagenesis library. We constructed the library, expressed it in Escherichia coli, and evaluated the thermostability of each clone.
Materials and methods
Materials
Remazol Brilliant Blue-xylan (RBB-xylan) was purchased from Sigma (St. Louis, Mo). Beechwood xylan was obtained from Megazyme (Bray, Ireland). 3,5-Dinitrosalicylic acid (DNS) was obtained from Nacalai Tesque (Kyoto, Japan). The concentration of the XynR expressed in E. coli was determined using Protein Assay CBB Solution (Nacalai Tesque) with bovine serum albumin (Nacalai Tesque) as standard.
Construction of site saturation mutagenesis library
The wild-type XynR expression plasmid, pET-21b(+)-XynR (Fig. S1) was constructed by inserting the complete XynR gene (GenBank accession no. JF912896.1) (Fig. S2) between NdeI and SalI sites. Three site saturation mutagenesis libraries were constructed using Quikchange HT Protein Engineering System (Agilent Technologies) as follows: Three oligonucleotide sets each targeting Tyr43–Asn81, Met82–Lys115, and Ala300–Asn325 were designed and synthesized by Agilent Technology with a microarray technology. Each oligonucleotide set was amplified by PCR (50 µL) with the corresponding forward and reverse custom primers (0.5 µM). A thermal cycling reaction (25 µL) was performed with pET-21b(+)-XynR (1 ng/µL) as a template and corresponding oligonucleotide set (1 µM) as primers. Amplified products were transfected into E. coli strain BL21(DE3).
Expression of site saturation mutagenesis library
A colony of the transformed E. coli cells was inoculated in LB broth (500 µL) containing 50 µg/mL ampicillin in a 96-well deep plate, and the culture was incubated at 37°C for 12–16 h with shaking. Fifteen µL of each culture was added to 500 µL of LB broth containing 50 µg/mL ampicillin in another 96-well deep plate, and the culture was incubated at 37°C for 2–4 h with shaking until the optimal density of 660 nm (OD660) reached 0.6. Then, 12.5 µL of 5 mM isopropyl-β-D-1-thiogalactopyranoside (IPTG) was added, and the culture was incubated at 37°C for 2 h with shaking. After centrifugation (3,200 × g, 10 min, 4°C), the precipitates were collected and suspended in 100 µL of BugBuster Protein Extraction Reagent (Merck Bioscience, Tokyo, Japan) containing 0.025 units/mL Benzonase (Merck Bioscience) and incubated at room temperature for 20 min. After centrifugation (3,200 × g, 20 min, 4°C), and the supernatants were collected and stored at 4°C before use.
Preparation of S92 variants
For the construction of plasmids, site-directed mutagenesis was carried out using Quikchange method with pET-21b(+)-XynR as a template with the oligonucleotides listed in Table S1 as primers. Transfection and expression were carried out as described above.
Hydrolysis of RBB-xylan
The activity was measured as described previously[Citation14]. Briefly, the reaction was initiated by mixing 5 μL of sample solution and 45 µL of substrate solution (0.5% w/v RBB-xylan in 250 mM Tris-HCl buffer (pH 7.8)) both pre-incubated at 37°C. The reaction solution was incubated at 37°C for 15 min, terminated by adding 50 µL of 99% ethanol, and incubated at 4ºC for 15‒30 min. After centrifugation (3,200 × g, 5 min, 4°C), an aliquot (100 µL) of the supernatants was collected and added to 100 µL of water, and the absorbance at 540 nm (A540) was measured with an EnSight (PerkinElmer, Waltham, MA). Based on the results, relative activity of each clone was calculated by the following terms.
Preparation of purified XynR
Preparation of purified XynR was carried out as described previously [Citation15,Citation16]. Briefly, the overnight culture of the transformants (2.5 mL) was added to 250 mL of LB broth and incubated at 37ºC with shaking. When OD660 reached 0.6–0.8, 0.25 mL of 0.5 M IPTG was added, and growth was continued at 37ºC for 4 h. After centrifugation (10,000 × g, 10 min, 4°C), the cells were suspended with 20 mL of 10 mM carbonate buffer (pH 10) (buffer A) and disrupted by sonication. After centrifugation (20,000 × g, 20 min, 4°C), the supernatant was collected and applied to a column [25 mm (inner diameter) × 120 mm] packed with Toyopearl DEAE-650M gel (Tosoh, Tokyo, Japan) equilibrated with buffer A containing 0.2 M NaCl. The column was washed with 30 mL of buffer A containing 0.2 M NaCl, followed by buffer A containing 0.25 M NaCl. Each fraction (1 mL) was assessed for SDS-PAGE. Active fractions were pooled and concentrated. Purified enzyme solution was stored at 4°C before use.
SDS-PAGE
Samples were mixed with five volumes of the SDS-PAGE sample buffer (0.25 M Tris-HCl buffer (pH 6.8), 50% v/v glycerol, 10% w/v SDS, 5% v/v 2-mercaptoethanol, 0.05% w/v bromophenol blue) and were boiled for 10 min. The solution (10 μL) was applied to 12.5% polyacrylamide gel with a constant current of 40 mA for 40 min. After electrophoresis, gels were stained with 0.25% Coomassie Brilliant Blue R-250, 50% methanol, and 7% acetic acid.
Hydrolysis of beechwood xylan
The activity was measured as described previously [Citation15,Citation16]. Briefly, the reaction was initiated by mixing 20 μL of enzyme solution (1 μM in 10 mM carbonate buffer (pH 10)) and 180 µL of substrate solution (0.25‒1.5% w/v beechwood xylan in 225 mM Tris-HCl buffer (pH 7.8)) both pre-incubated at 37°C. The reaction solution was incubated at 37°C. An aliquot (40 µL) was taken from the reaction mixture at specified times and immediately added to 40 µL of DNS solution (0.5% w/v 3,5-dinitrosalicylic acid, 1.6% w/v NaOH, 30% w/v potassium sodium tartrate). After incubating at 100°C for 10 min and at 4°C for 15 min, 80 µL of the solution was added to 120 µL of water, and A540 was measured with an EnSight. The initial reaction rate was estimated from the time-course for production of reducing sugars, and 1 unit was defined as the amount of enzyme producing 1 µmol of reducing sugar at 37°C per min.
Results and discussion
Construction of the site saturation mutagenesis library
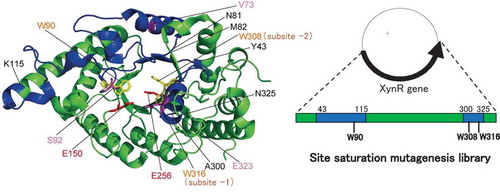
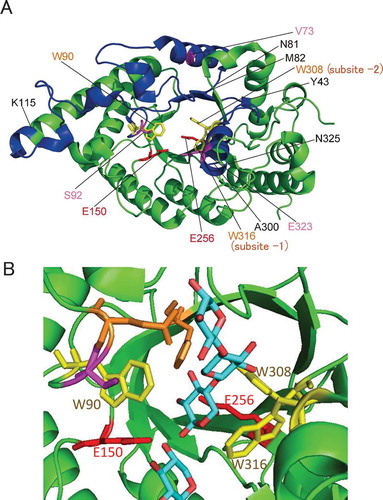
In this paper, the nomenclature for sugar-binding subsite is based on that by Davies et al [Citation17], in which cleavage occurs between −1 and + 1 subsites, and subsites are labelled −1, −2 to -n toward the non-reducing end and + 1, + 2 to + n toward the reducing end. The numbering of amino acid residues is based on that of XynR (Gene bank accessions no. JF912896.1). Studies on various GH10 xylanases subsite mapping have revealed that conserved aromatic amino acid residues, Trp308, Trp316, Tyr194, Trp264, and Trp232 are located at subsite −2, −1, + 1, + 2, and + 3, respectively, making stacking interactions with xylan [Citation18]. In the crystal structure of the complex of xylanase and xylobiose, xylobiose occupied at the subsites −2 and −1 [Citation19]. Based on these evidences, we hypothesized that among the five subsites, those at −2 and −1 are more flexible than others, and that the thermostability of XynR will increase if the flexibility can be decreased by the mutation without impairing the substrate binding ability. This is in contrast to previous studies on themostabilization of GH10 xylanase by stabilizing the N- and C- terminal loops based on the hypothesis that the N- and C- terminal are the most flexible [Citation20,Citation21]. shows multiple sequence alignment of the amino acid sequences of xylanases from Bacillus halodurans S7 (BhS7Xyn) [Citation22], XynR, and xylanase from Geobacillus stearothermophilus (GsXyn) [Citation18]. Twenty two aromatic residues are conserved in the three xylanases, including Trp308, Trp316, Tyr194, Trp264, and Trp232 located at the subsites. Because the crystal structure of XynR has not been determined, we used the whole structure of BhS7Xyn and the active-site structure of the complex of GsXyn and xylopentose ( and ). We noticed that like Trp308 and Trp316, Trp90 protrudes to the subsites −2 and −1 [Citation21]. We thus selected Tyr43–Lys115 that contains Trp90 and Ala300–Asn325 that contains Trp308 and Trp316 as target regions. We constructed three site saturation mutagenesis libraries each corresponding to Tyr43–Asn81, Met82–Lys115, and Ala300–Asn325 ().
Figure 1. Amino acid sequences of xylanases.
Notes: The ClustalW program was used for multiple sequence alignment of the amino acid sequences of xylanases from Bacillus halodurans S7 (BhS7Xyn, Gene bank accession no. AY687345.1), Bacillus sp. strain TAR-1 (XynR) (JF912896.1), and Geobacillus stearothermophilus (GsXyn) (AAC98140). The asterisk indicates the amino acid residues conserved in BhS7Xyn, XynR, and XynGR40. Two catalytic Glu residues and three Trp residues conserved are boxed with a broken line. The target amino acid sequences of the site saturation mutagenesis library of XynR, Tyr43–Lys115 and Ala300–Asn325, are underlined. Val73, Ser92, and Glu323 of XynR are boxed.

Figure 2. Structures of xylanases.
Notes: The PyMOL program was used to visualize the whole structure of BhS7Xyn (PDB accession no. 2UWF) (A) and the active-site structure of GsXyn complexed with xylopentose (1R87) (B). Two catalytic Glu residues (Glu150 and Glu256) and three Trp residues (Trp90, Trp308, and Trp316) conserved are colored in red and yellow, respectively. Val73, Ser92, and Glu323 are colored in pink. The target regions to be mutated, Tyr43–Lys115 and Ala300–Asn325, are colored in blue, while other regions in green. The amino acid number indicates that corresponding to those of XynR.
Investigation of the quality of the library
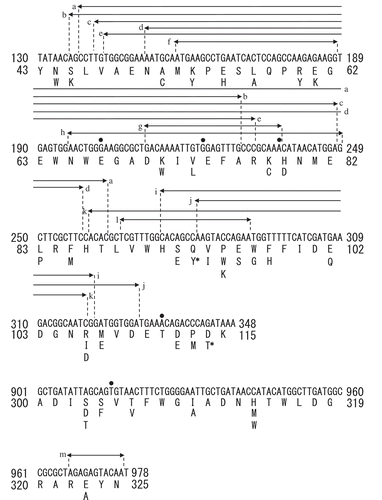
The nucleotide sequences of the XynR genes were analyzed for arbitrarily selected 32, 32, and 30 clones for Tyr43–Asn81, Met82–Lys115, and Ala300–Asn325, respectively. Thirty-five clones (37%) had expected mutations corresponding to one amino acid substitution (). There was no hot spots for mutation, indicating that the library was not biased toward expected mutations. On the other hand, 19 clones (20%) had unexpected mutations (one nucleotide deletion for five clones; two amino acid substation for one clone; and 11‒155 nucleotides deletion for 13 clones) (), 36 clones (38%) had both expected and unexpected mutations, and 4 clones (4%) had no mutation. The success rate (37%) in this study was the same to that (37%) in our previous study [Citation4], indicating that errors occur in oligonucleotides synthesis in the microarray at a fixed frequency. We considered that the success rate of 37% to be high enough and moved to the subsequent screening.
Figure 3. Sequence analysis of the site saturation mutagenesis library of XynR.
Notes: Substitution of amino acid residue is indicated by characters; deletion of one nucleotide is indicated by dot, and deletion of 11‒155 nucleotides is indicated by arrow. a‒m indicates independent clones. 93Y and 114T, to which asterisk is attached, are in the same clone.
Expression of the site saturation mutagenesis library and screening of thermostable XynR variants
The wild-type XynR (WT) and each 480 clone (192 for Tyr43–Asn81 and Met82–Lys115, and 96 for Ala300–Asn325) were expressed in E. coli using a 96-well deep plate. We set the number of clones to be examined as 480. Considering the success rate of 37%, this number (480) covered about 10% of all variants (19 amino acids × 99 residues = 1,881 clones). Soluble fractions of the E. coli cells were prepared for WT and the 480 clones. We first investigated their activities. shows the distribution for the number of clones with the same relative activity (activity compared to that of WT) for 480 clones. The relative activity reflects not only the specific activity but also the amount of activity. The distribution profile exhibited the valley-shaped one with > 90% of 178 clones (37%) and 0‒5% of 111 clones (23%), indicating that a number of mutations abolished or decreased the activity. We selected 223 clones that exhibited 70% or more relative activity.
Table 1. Distribution of clones with the same activity or stability.
To compare the thermostabilities of the clones accurately, we made two-stage selection. We first investigated the thermostabilities of WT and the 223 clones at 70ºC for 15 min. WT exhibited the residual activity (activity compared to that before heat treatment) of 36%. shows the distribution for the number of clones with the same residual activity for the 223 clones. One hundred eighty-one clone exhibited less than 36% residual activity, indicating that a number of mutations decreased the thermostability. We selected 42 clones that exhibited 36% or more residual activity. We investigated the thermostabilities of WT and the 42 clones at 80ºC for 15 min. WT exhibited the residual activity of 20%. also shows the distribution for the 42 clones. We selected three clones which exhibited higher residual activity than the other 39 clones.
Sequence analysis showed that these three clones had the V73L, S92E, and E323A mutations, respectively. The residual activities after the treatment at 70 and 80ºC for 15 min were 67 and 43%, respectively, for V73L, 69 and 67%, respectively, for S92E, and 67 and 49%, respectively, for E323A. The residual activity after heat treatment at 80ºC for 15 min of WT was 14 ± 5% (), and those of V73L, S92E, and E323A were 34 ± 3, 65 ± 5, and 39 ± 3%, respectively (), indicating that S92E was the most thermostable. In our understanding, introduction of disulfide bridge, increase of surface charge, stabilization of hydrophobic core, and introduction of salt bridge are the most frequent strategy to increase protein stability by site-directed mutagenesis [Citation23,Citation24]. Val73 lies in the α-helix with the side chain buried in the interior of the α-helix (). Therefore, the mutation V73L is thought to pack the amino acid side chain at the central part, probably by stabilizing hydrophobic core. Ser92 lies in the loop in the active site, and is close to Trp90 that protrudes to the subsites −2 and −1 (). Therefore, the mutation S92E is thought to decrease the flexibility of the substrate-binding region, probably by introducing salt bridge. Glu323 lies in the α-helix with the side chain protruding to Trp316 at the subsite −1 (). Therefore, the mutation E323A, like S92E, is to decrease the flexibility of the substrate-binding region, by decreasing surface charge or stabilizing hydrophobic core.
Figure 4. Thermostability of XynR variants.
Notes: Residual activities of WT (A), V73L, S92E, and E323A (B), and S92X (C) are shown. Hydrolysis reaction of RBB-xylan was carried out at 37ºC for 15 min with E. coli expression product of WT which received heat treatment at 60, 70 or 80ºC for 15 min (A) or V73L, S92E, and E323A (B) or S92X (C) which received heat treatment at 80ºC for 15 min. Residual activity indicates the value compared to that before heat treatment.Error bars indicate SD values for three-times measurements.
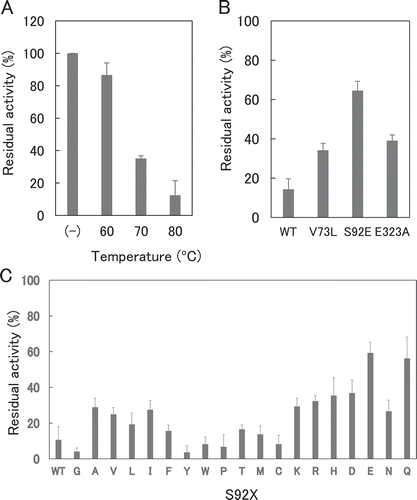
To obtain more thermostable variants, we performed saturation mutation analysis of Ser92. WT and 19 single variants at amino acid position 92 were expressed in E. coli, and cellular soluble fractions were prepared. The residual activities of WT and the 19 variants after 15-min heat treatment at 80ºC were in the range of 0‒75% with that of S92E the highest (). We thus selected S92E as the most thermostable variant. These results suggest that the thermostability of GH10 xylanase is governed by the flexibility in the active site to some extent. It is of note that replacement of amino acid residue with Glu residue sometimes increase protein stability, and that this is explained by the formation of hydrogen bonds and salt bridges. To address this issue, crystallographic analysis is required, which is currently under way.
Characterization of the thermostable variant S92E
WT and S92E were expressed in the E. coli cells using a flask and purified from the cells by the anion exchange column chromatography. Figure S3 shows the SDS-PAGE of the purified preparations. WT and S92E yielded a single band with a molecular mass of 40 kDa.
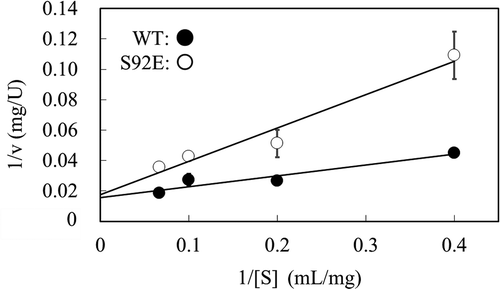
To characterize S92E, the steady-state kinetic analysis of WT and S92E was carried out. In the hydrolysis of RBB-xylan, the A540 value that corresponds to the amount of solubilized RBB in 50% ethanol is not precisely proportional to the number of cleavage by xylanase. Therefore, beechwood xylan was used as a substrate, and the resulting reducing ends were quantified using DNS. shows the dependence on the substrate concentration of the initial reaction rate (v) at pH 7.8 at 37ºC. Linear relationships were observed between the reciprocal of v and the reciprocal of the substrate concentration in the Lineweaver-Burk plot, and the kcat and Km values were determined separately. The kcat values of WT and S92E were almost the same (42.5 s−1 and 38.0 s−1, respectively), while the Km value of S92E (12.5 mg/mL) was 280% of that of WT (4.5 mg/mL). Therefore, the enhancement of thermostability in the S92E mutant could be explained by the stabilization of the xylan-binding region and the consequent decrease of the binding ability to xylan. This applies to the trade-off between activity and stability in enzymes [Citation23]. Considering that enzyme reaction is usually done under high substrate concentrations in industry, we think that increase in Km value, unlike decrease in kcat value, does not much lower the value of variant enzyme for industrial use. As for the pH-dependency of S92E on activity, the introduced negative charge might suppress the protonation of catalytically important residues, leading to the increase of acidic and/or alkaline pKa values in the pH-activity profile, as previously reported with subtilisin BPN’ [Citation25], although the microenvironment of the active site is complex, and prediction of electrostatic interaction is difficult.
Figure 5. Steady-state kinetic analysis of thermostable XynR variant.
Notes: Hydrolysis reaction of beechwood xylan was carried out at 37ºC with the purified enzyme preparation of WT or S92E (0.1 µM). Lineweaver-Burk plot is shown. One unit is defined as the amount of enzyme that produces 1 µmol of reducing sugar per min. Error bars indicate SD values for three-times measurements.
Thermostabilization of xylanase using a site saturation mutagenesis library
Various protein engineering strategies have been carried out to increase the thermostabilities of GH10 and GH11 xylanases. In GH10 xylanase, the thermostablilities were first increased by the mutation of active-site aromatic residues [Citation26,Citation27]. The N- or C-terminal flexible loop is a main target for the mutation. The thermostabilities were increased by stabilizing these terminal loops by rational design [Citation21,Citation28] or random mutagenesis [Citation29]. Another target is the external α-helix. The thermostability was increased by replacing amino acid residues into proline or glutamate [Citation30]. In GH11 xylanase, the thermostabilities were increased by stabilizing the N-terminal loop [Citation31–Citation33] or introducing Arg[Citation34] on the molecular surface. It should be noted that in GH11 xylanase, alkaliphily was also improved by engineering of surface charges [Citation16,Citation35].
In this study, we used site saturation mutagenesis library with the substrate-binding site as mutational target. We think that the N-terminal and C-terminal loops and the external α-helix are also attractive as a target. One of the problems in this method is that the success rates of the mutation are limited, which are 37% in this and our previous studies [Citation4] although it is theoretically 100%. The increase in the success rate of the mutation is an important future subject.
In conclusion, the thermostability of GH10 xylanase, XynR, from Bacillus sp. strain TAR-1 was increased using a site saturation mutagenesis library. Various enzymes have been stabilized by combining multiple stabilizing mutations. We think that combination of the thermostabilizing mutations identified by the screening of the site saturation mutagenesis library with the thermostabilizing mutations identified previously is a promising strategy for further stabilization of GH10.
Author contribution
K.N., Y.K., and K.Y. designed research; K.N. and Y.K. performed research; K.N., Y.K., K.K., T.T., R.Y., S.N., and K.Y. analyzed data; N.K., Y.K., and K.Y. wrote the manuscript.
SFig180526.pdf
Download PDF (241.2 KB)BBB_STable_180526-h.docx
Download MS Word (51.7 KB)Acknowledgments
We appreciate Mr. Tongyang Li and Ms. Tomomi Yamasaki of Kyoto University for their contributions to this work.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
supplemental material for this article can be accessed here
Additional information
Funding
Related Research Data
References
- Reetz MT, Carballeira JD. Iterative saturation mutagenesis (ISM) for rapid directed evolution of functional enzymes. Nat Protoc. 2007;2:891−903.
- Reetz MT, Prasad S, Carballeira JD, et al. Iterative saturation mutagenesis accelerates laboratory evolution of enzyme stereoselectivity: rigorous comparison with traditional methods. J Am Chem Soc. 2010;132:9144−9152.
- Cheng F, Xu J-M, Xiang C, et al. Simple-MSSM: a simple and efficient method for simultaneous multi-site saturation mutagenesis. Biotechnol Lett. 2017;39:567–575.
- Katano Y, Li T, Baba M, et al. Generation of thermostable moloney murine leukemia virus reverse transcriptase variants using site saturation mutagenesis library and cell-free protein expression system. Biosci Biotechnol Biochem. 2017;81:2339–2345.
- Kumar D, Kumar SS, Kumar J, et al. Xylanases and their industrial applications: a review. Biochem Cell Arch. 2017;17:353–360.
- Goswami GK, Rawat S. Microbial xylanases and their applications: a review. Int J Res Aca Rev. 2015;3:436–450.
- Kalim B, Böhringer N, Ali N, et al. Xylanases–from microbial origin to industrial application. Brit Biotechnol J. 2015;7:1–20.
- Horikoshi K. Alkaliphiles. Tokyo (Japan): Kodansha; 1999.
- Glycoside hydrolase family classification [Internet]. Available from http://www.cazy.org/glycoside-hydrolases.html
- Linares-Pasten JA, Aronsson A, Karlsson EN. Structural considerations on the use of endo-xylanases for the production of prebiotic xylooligosaccharides from biomass. Curr Protein Pept Sci. 2018;19:48–67.
- Liu L, Sun X, Yan P, et al. Non-structured amino-acid impact on GH11 differs from GH10 xylanase. PLoS ONE. 2012;7:e45762.
- Nakamura S, Nakai R, Wakabayashi K, et al. Thermophilic alkaline xylanase from newly isolated alkaliphilic and thermophilic Bacillus spstrain TAR-1. Biosci Biotechnol Biochem. 1994;58:78–81.
- Takahashi H, Nakai R, Nakamura S. Purification and partial characterization of a basic xylanase produced by thermoalkaliphilic Bacillus spstrain TAR-1. Biosci Biotechnol Biochem. 2000;64:887–890.
- Biely P, Mislovicová D, Toman R. Soluble chromogenic substrates for the assay of endo-1,4-beta-xylanases and endo-1,4-beta-glucanases. Anal Biochem. 1985;144:142–146.
- Nakamura S, Wakabayashi K, Nakai R, et al. Purification and some properties of an alkaline xylanase from alkaliphilic Bacillus spstrain 41M-1. Appl Environ Microbiol. 1993;59:2311–2316.
- Umemoto H, Ihsanawati, Inami M, et al. Improvement of alkaliphily of Bacillus alkaline xylanase by introducing amino acid substitutions both on catalytic cleft and protein surface. Biosci Biotechnol Biochem. 2009;73:965–967.
- Suganuma T, Matsuno R, Ohnishi M, et al. A study of the mechanism of action of Taka-amylase A1 on linear oligosaccharides by product analysis and computer simulation. J Biochem. 1978;84:293–316.
- Zolotnitsky G, Cogan U, Adir N, et al. Mapping glycoside hydrolase substrate subsites by isothermal titration calorimetry. Proc Natl Acad Sci USA. 2004;101:11275–11280.
- Zheng Y, Li Y, Liu W, et al. Structural insight into potential cold adaptation mechanism through a psychrophilic glycoside hydrolase family 10 endo-β-1,4-xylanase. J Struct Biol. 2016;193:206–211.
- Kamondi S, Szilágyi A, Barna L, et al. Engineering the thermostability of a TIM-barrel enzyme by rational family shuffling. Biochem Biophys Res Commun. 2008;374:725–730.
- Liu L, Zhang G, Zhang Z, et al. Terminal amino acids disturb xylanase thermostability and activity. J Biol Chem. 2011;286:44710–44715.
- Mamo G, Thunnissen M, Hatti-Kaul R, et al. An alkaline active xylanase: insights into mechanisms of high pH catalytic adaptation. Biochimie. 2009;91:1187–1196.
- Shoichet BK, Baase WA, Kuroki R, et al. A relationship between protein stability and protein function. Proc Natl Acad Sci USA. 1995;92:452–456.
- Baba M, Kakue R, Leucht C, et al. Further increase in thermostability of Moloney murine leukemia virus reverse transcriptase by mutational combination. Protein Eng Des Sel. 2017;30:551–557.
- Thomas PG, Russell AJ, Fersht AR. Tailoring the pH dependence of enzyme catalysis using protein engineering. Nature. 1985;318:375–376.
- Moreau A, Shareck F, Kluepfel D, et al. Increase in catalytic activity and thermostability of the xylanase A of Streptomyces lividans 1326 by site-specific mutagenesis. Enzyme Microb Technol. 1994;16:420–424.
- Roberge M, Shareck F, Morosoli R, et al. Site-directed mutagenesis study of a conserved residue in family 10 glycanases: histidine 86 of xylanase A from Streptomyces lividans. Protein Eng. 1998;11:399–404.
- Bhardwaj A, Leelavathi S, Mazumdar-Leighton S, et al. The critical role of partially exposed N-terminal valine residue in stabilizing GH10 xylanase from Bacillus sp. NG-27 under poly-extreme conditions. PLoS One. 2008;26:e3063.
- Song L, Tsang A, Sylvestre M. Engineering a thermostable fungal GH10 xylanase, importance of N-terminal amino acids. Biotechnol Bioeng. 2015;202:1081–1091.
- Wang K, Luo H, Tian J, et al Thermostability improvement of a Streptomyces xylanase by introducing proline and glutamic acid residues. Appl Environ Microbiol. 2014;80:2158–2165.
- Georis J, De Lemos Esteves F, Lamotte-Brasseur J, et al. An additional aromatic interaction improves the thermostability and thermophilicity of a mesophilic family 11 xylanase: structural basis and molecular study. Protein Sci. 2000;9:466–475.
- Dumon C, Varvak A, Wall MA, et al. Engineering hyperthermostability into a GH11 xylanase is mediated by subtle changes to protein structure. J Biol Chem. 2008;283:22557–22564.
- Watanabe M, Fukada H, Ishikawa K. Construction of thermophilic xylanase and its structural analysis. Biochem. 2016;55:4399–4409.
- Umemoto H, Ihsanawati, Inami M, et al. Improvement of alkaline xylanase by introducing amino acid substitutions both on catalytic cleft and protein surface. Biosci Biotechnol Biochem. 2009;73:965–967.
- Umemoto H, Yazawa R, Takakura J, et al. Analysis of functional domains and improvement of alkaliphily of an alkaline xylanase on the basis of its three-dimensional structure. J Appl Glycosci. 2010;57:145–150.